Synthesis and Neuroprotective Action of Optically Pure Neoechinulin A and Its Analogs

Abstract
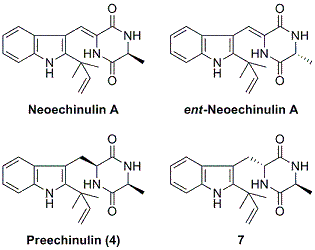
:
1. Introduction

2. Results and Discussion
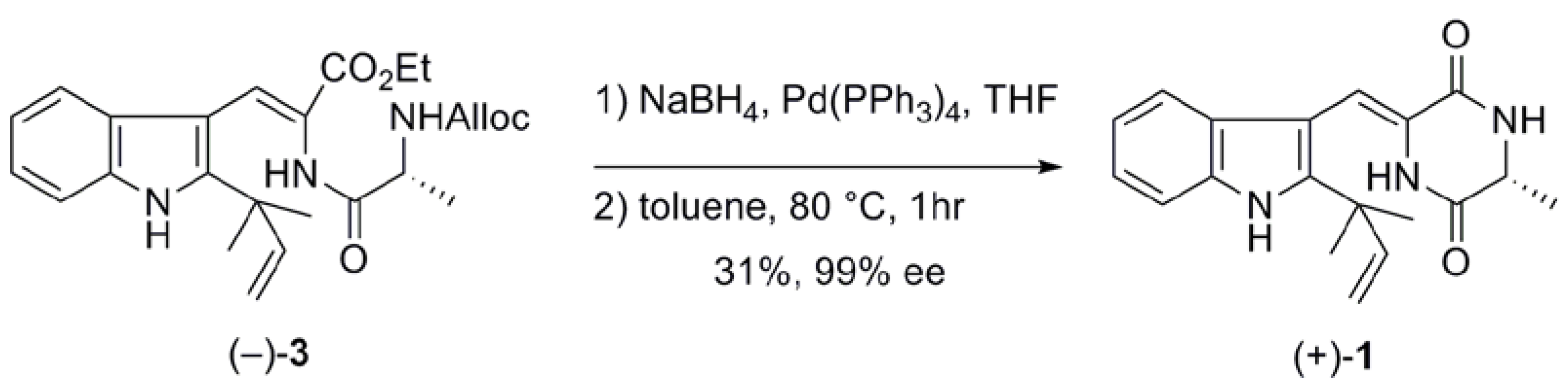
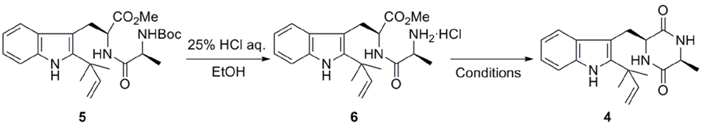
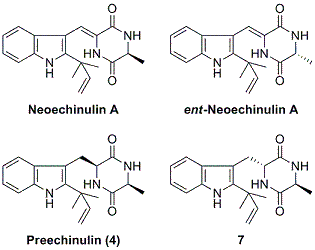
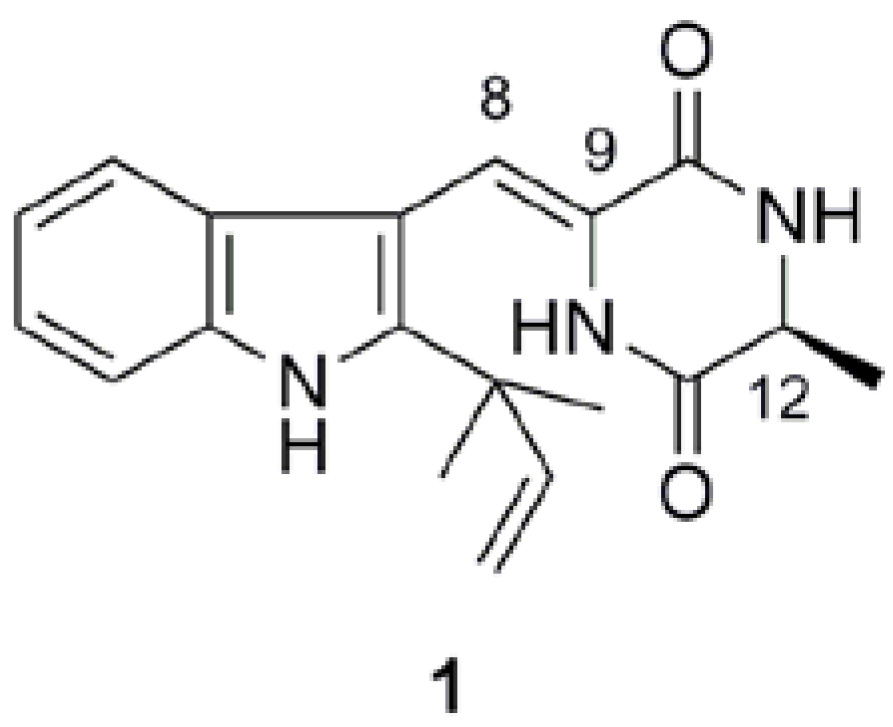
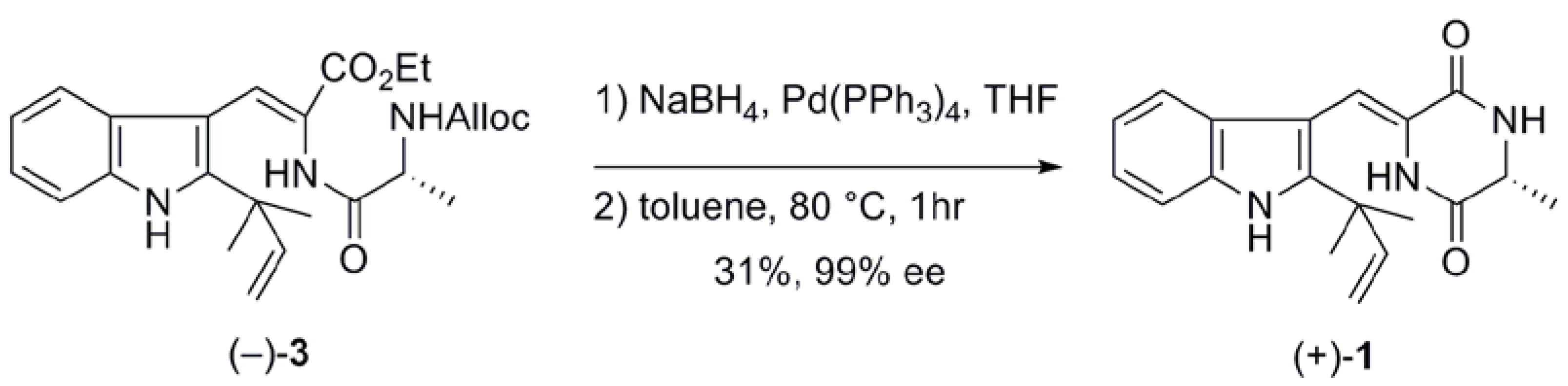
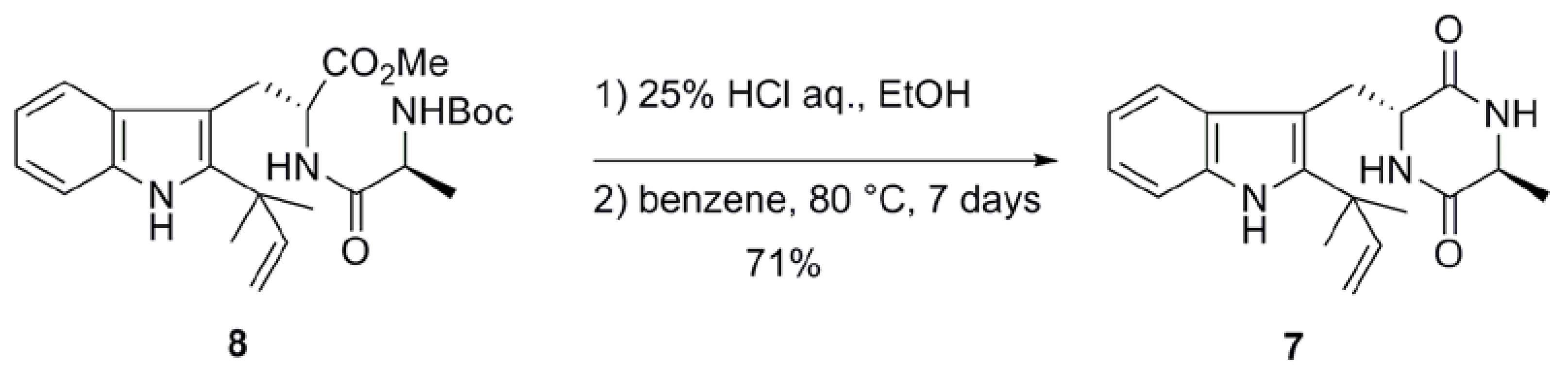
2.1. Preparation of neoechinulin A and its analogs via intramolecular cyclization

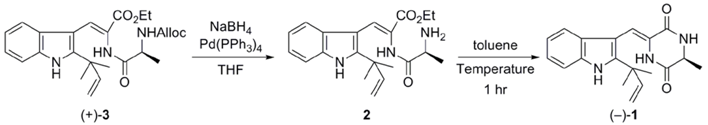
| Entry | Temperature (ºC) | Yield (%) a | ee (%) b |
|---|---|---|---|
| 1 | 110 | 43 | 81 |
| 2c | 80 | 58 | 95 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Temperature (ºC) | Time (days) | Yield (%)a |
|---|---|---|---|---|
| 1b | benzene | 80 | 7 | 73 |
| 2 | toluene | 110 | 1 | 83 |
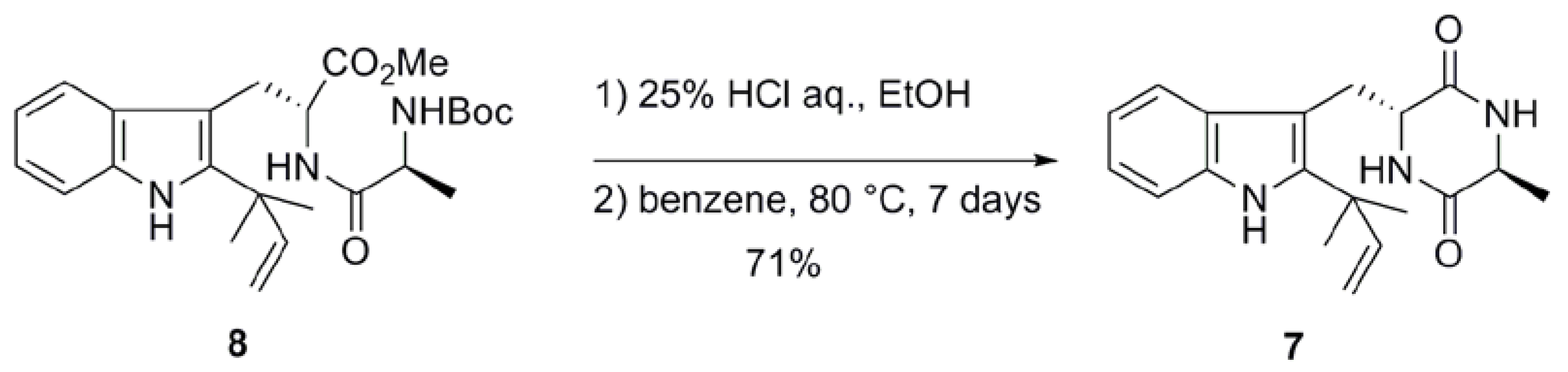
2.2. Biological and chemical properties of optical isomers of neoechinulin A and preechinulin
| Compound | Viability (%) | Relative protection (% vs. (+)-1) | Reference |
|---|---|---|---|
| vehicle | 31.4 ± 2.0 | 0 | [4] |
| (–)-1 | 89.6 ± 11.2b | 113.9 | |
| (+)-1 | 82.5 ± 11.6b | 100.0 | |
| vehicle | 9.7 ± 13.2 | 0 | [5] |
| (–)-1 | 49.7 ± 8.3b | 100 | |
| 4 | 7.0 ± 7.0 | –6.8 | |
| 7 | 12.5 ± 10.2 | 7.1 |
3. Experimental Section
General
4. Conclusions
Acknowledgements
References
- Martins, M.B.; Carvalho, I. Diketopiperazines: biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar]
- Pettit, G.R.; Hogan, F.; Xu, J.P.; Tan, R.; Nogawa, T.; Cichacz, Z.; Pettit, R.K.; Du, J.; Ye, Q.H.; Cragg, G.M.; Herald, C.L.; Hoard, M.S.; Goswami, A.; Searcy, J.; Tackett, L.; Doubek, D.L.; Williams, L.; Hooper, J.N.; Schmidt, J.M.; Chapuis, J.C.; Tackett, D.N.; Craciunescu, F. Antineoplastic agents. 536. New sources of naturally occurring cancer cell growth inhibitors from marine organisms, terrestrial plants, and microorganisms. J. Nat. Prod. 2008, 71, 438–444. [Google Scholar] [PubMed]
- Maruyama, K; Ohushi, T.; Yoshida, K.; Shibata, Y.; Sugawara, F.; Arai, T. Protective properties of neoechinulin A against SIN-1-induced neuronal cell death. J. Biochem. 2004, 136, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, K.; Aoki, T.; Shibata, Y.; Kamisuki, S.; Sugawara, F.; Kuramochi, K.; Nakazaki, A.; Kobayashi, S.; Kuroiwa, K.; Watanabe, N.; Arai, T. Structure-activity relationships of neoechinulin A analogues with cytoprotection against peroxynitrite-induced PC12 cell death. J. Antibiot. 2007, 60, 614–621. [Google Scholar]
- Kuramochi, K.; Ohnishi, K.; Fujieda, S.; Nakajima, M.; Saitoh, Y.; Watanabe, N.; Takeuchi, T.; Nakazaki, A.; Sugawara, F.; Arai, T.; Kobayashi, S. Synthesis and biological activities of neoechinulin A derivatives: new aspects of structure-activity relationships for neoechinulin A. Chem. Pharm. Bull. 2008, 56, 1738–1743. [Google Scholar]
- Kajimura, Y.; Aoki, T.; Kuramochi, K.; Kobayashi, S.; Sugawara, F.; Watanabe, N.; Arai, T. Neoechinulin A protects PC12 cells against MPP+-induced cytotoxicity. J. Antibiot. 2008, 61, 330–333. [Google Scholar]
- Aoki, T.; Kamisuki, S.; Kimoto, M.; Ohnishi, K.; Takakusagi, Y.; Kuracmohi, K.; Takeda, Y.; Nakazaki, A.; Kuroiwa, K.; Ohuchi, T.; Sugawara, F.; Arai, T.; Kobayashi, S. Total synthesis of (–)-neoechinulin A. Synlett 2006, 677–680. [Google Scholar]
- Kuramochi, K.; Aoki, T.; Nakazaki, A.; Kamisuki, S.; Takeno, M.; Ohnishi, K.; Kimoto, K.; Watanabe, N.; Kamakura, T.; Arai, T.; Sugawara, F.; Kobayashi, S. Synthesis of neoechinulin A and its derivatives. Synthesis 2008, 3810–3818. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aoki, T.; Ohnishi, K.; Kimoto, M.; Fujieda, S.; Kuramochi, K.; Takeuchi, T.; Nakazaki, A.; Watanabe, N.; Sugawara, F.; Arai, T.; et al. Synthesis and Neuroprotective Action of Optically Pure Neoechinulin A and Its Analogs. Pharmaceuticals 2010, 3, 1063-1069. https://doi.org/10.3390/ph3041063
Aoki T, Ohnishi K, Kimoto M, Fujieda S, Kuramochi K, Takeuchi T, Nakazaki A, Watanabe N, Sugawara F, Arai T, et al. Synthesis and Neuroprotective Action of Optically Pure Neoechinulin A and Its Analogs. Pharmaceuticals. 2010; 3(4):1063-1069. https://doi.org/10.3390/ph3041063
Chicago/Turabian StyleAoki, Toshiaki, Kensuke Ohnishi, Masaaki Kimoto, Satoshi Fujieda, Kouji Kuramochi, Toshifumi Takeuchi, Atsuo Nakazaki, Nobuo Watanabe, Fumio Sugawara, Takao Arai, and et al. 2010. "Synthesis and Neuroprotective Action of Optically Pure Neoechinulin A and Its Analogs" Pharmaceuticals 3, no. 4: 1063-1069. https://doi.org/10.3390/ph3041063
APA StyleAoki, T., Ohnishi, K., Kimoto, M., Fujieda, S., Kuramochi, K., Takeuchi, T., Nakazaki, A., Watanabe, N., Sugawara, F., Arai, T., & Kobayashi, S. (2010). Synthesis and Neuroprotective Action of Optically Pure Neoechinulin A and Its Analogs. Pharmaceuticals, 3(4), 1063-1069. https://doi.org/10.3390/ph3041063