Endogenous Matrix-Derived Inhibitors of Angiogenesis

Abstract
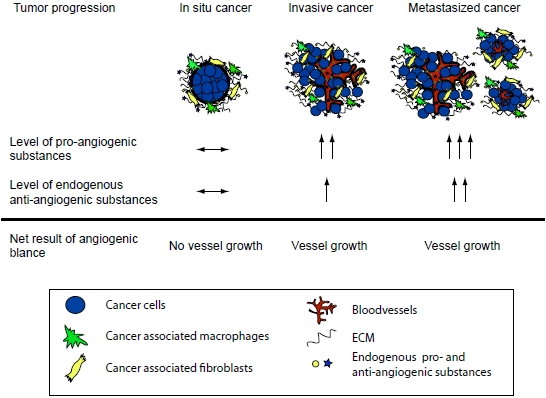
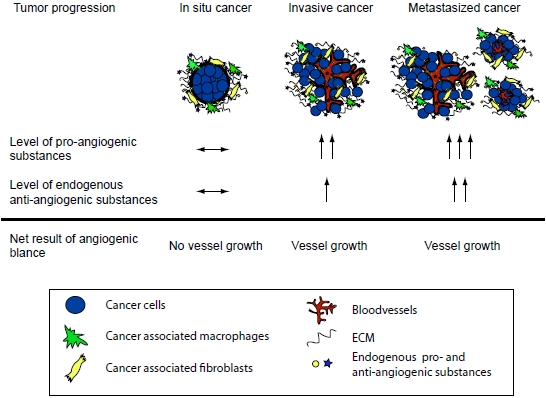
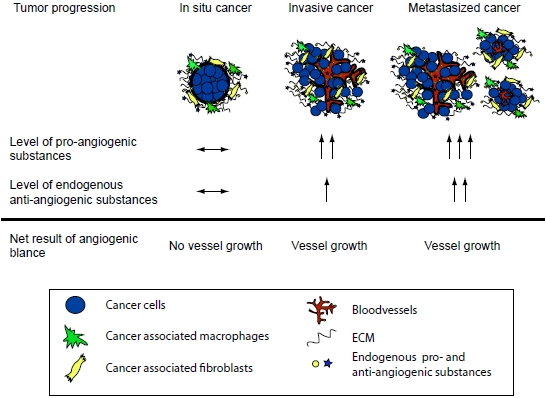
:
{kind=link}
{kind=link}
{kind=link}
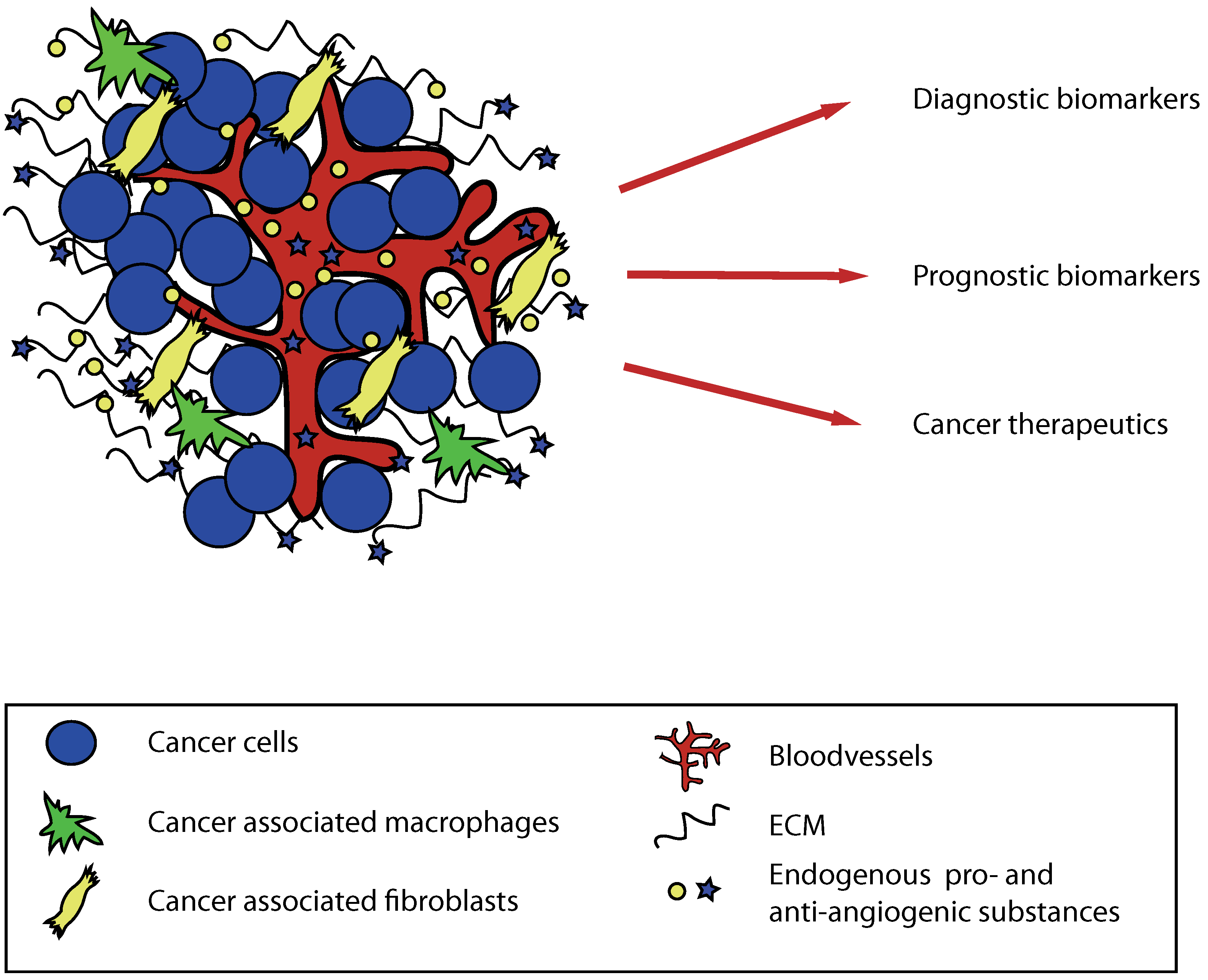{kind=link}
1. Introduction
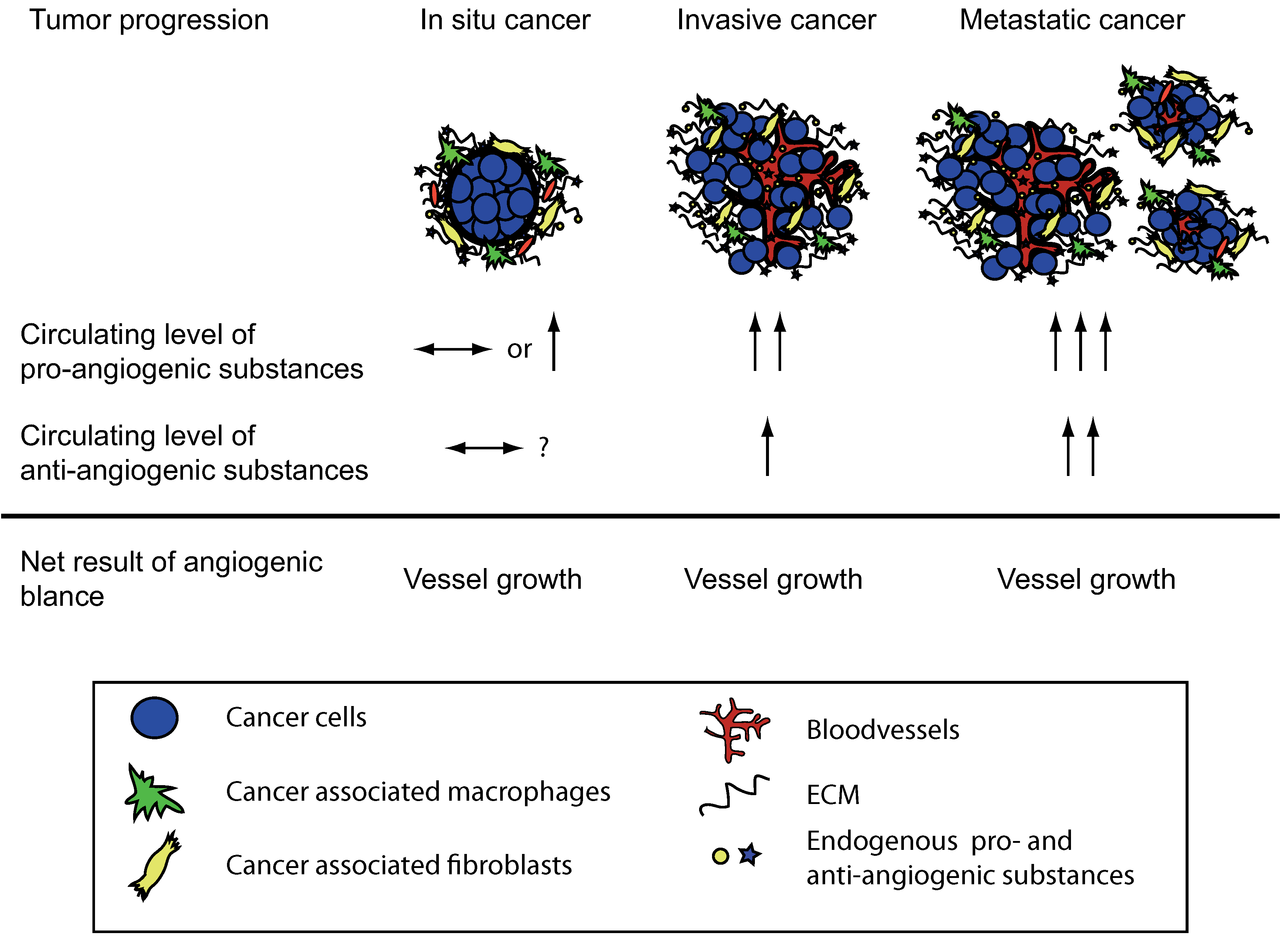
2. Type XVIII Collagen Derived Endostatin
2.1. Cellular source and effects—in vitro and in vivo studies
2.2. Role in human cancer diagnostics and therapy
3. Type IV Collagen Derived Anti-Angiogenic Substances
3.1. Tumstatin
3.2. Arresten
3.3. Canstatin
3.4. Tetrastatin, pentastatin and hexastatin
4. Conclusions
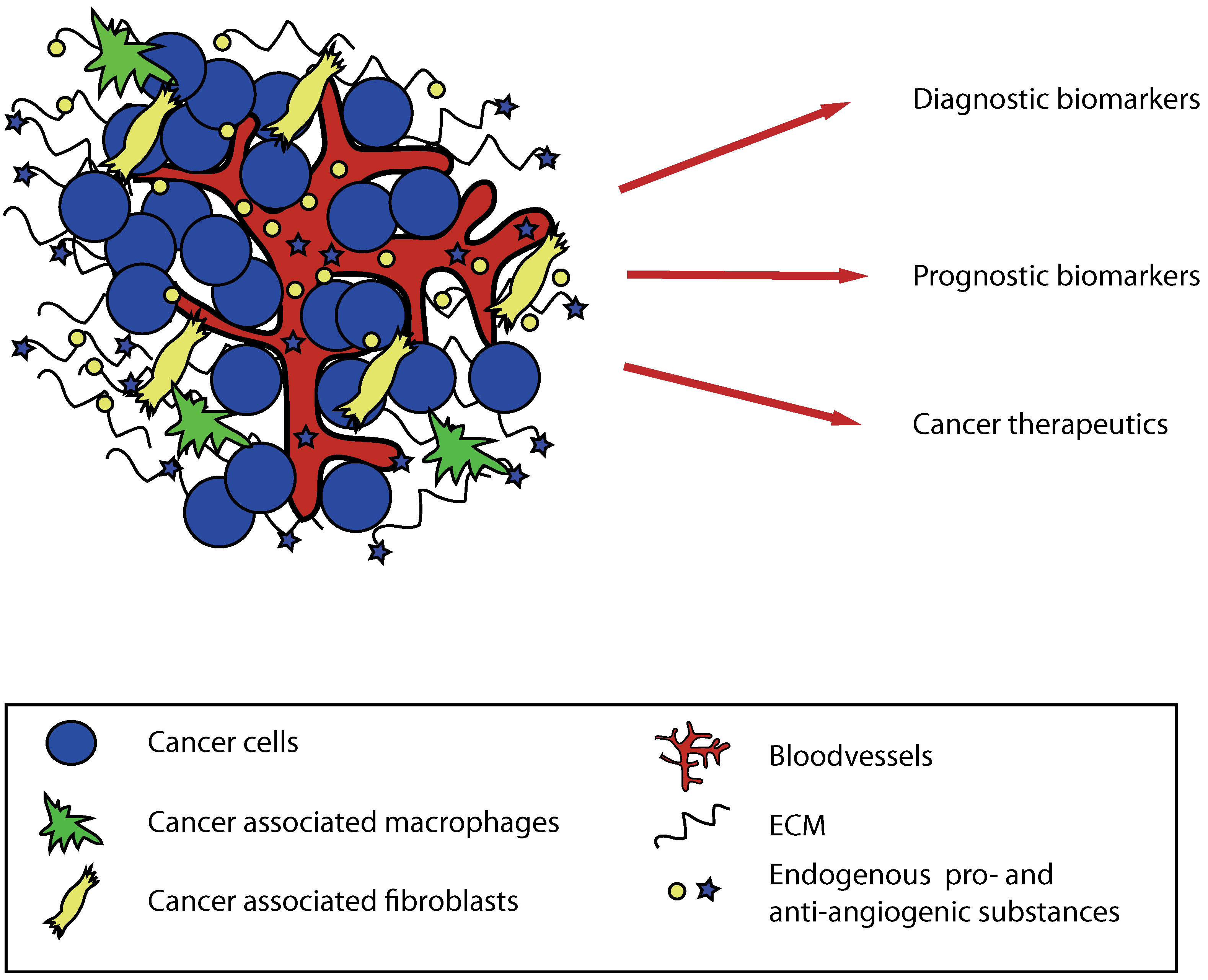
Acknowledgements
References
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar]
- Kunz-Schughart, L.A.; Knuechel, R. Tumor-associated fibroblasts (part I): Active stromal participants in tumor development and progression? Histol. Histopathol. 2002, 17, 599–621. [Google Scholar]
- Elenbaas, B.; Weinberg, R.A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 2001, 264, 169–184. [Google Scholar]
- Pietras, K.; Ostman, A. Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res. 316, 1324–1331.
- Jain, R.K. Determinants of tumor blood flow: a review. Cancer Res. 1988, 48, 2641–2658. [Google Scholar]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar]
- Sund, M.; Zeisberg, M.; Kalluri, R. Endogenous stimulators and inhibitors of angiogenesis in gastrointestinal cancers: basic science to clinical application. Gastroenterology 2005, 129, 2076–2091. [Google Scholar]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar]
- Nyberg, P.; Salo, T.; Kalluri, R. Tumor microenvironment and angiogenesis. Front. Biosci. 2008, 13, 6537–6553. [Google Scholar]
- Jain, R.K. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology (Williston Park) 2005, 19, 7–16. [Google Scholar]
- Kalluri, R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar]
- Ohlund, D.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Expression pattern and circulating levels of endostatin in patients with pancreas cancer. Int. J. Cancer 2008, 122, 2805–2810. [Google Scholar]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar]
- Cornelius, L.A.; Nehring, L.C.; Harding, E.; Bolanowski, M.; Welgus, H.G.; Kobayashi, D.K.; Pierce, R.A.; Shapiro, S.D. Matrix metalloproteinases generate angiostatin: effects on neovascularization. J. Immunol. 1998, 161, 6845–6852. [Google Scholar]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 2004, 114, 623–633. [Google Scholar]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar]
- Benouchan, M.; Colombo, B.M. Anti-angiogenic strategies for cancer therapy (Review). Int. J. Oncol. 2005, 27, 563–571. [Google Scholar]
- Heath, V.L.; Bicknell, R. Anticancer strategies involving the vasculature. Nat. Rev. Clin. Oncol. 2009, 6, 395–404. [Google Scholar]
- Abdelrahim, M.; Konduri, S.; Basha, R.; Philip, P.A.; Baker, C.H. Angiogenesis: An update and potential drug approaches. Int. J. Oncol. 2010, 36, 5–18. [Google Scholar]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar]
- Cao, Y. Angiogenesis: What can it offer for future medicine? Exp. Cell Res. 2010, 316, 1304–1308. [Google Scholar]
- Cao, Y. Angiogenesis in malignancy. Semin. Cancer Biol. 2009, 19, 277–278. [Google Scholar]
- Jain, R.K.; Duda, D.G.; Willett, C.G.; Sahani, D.V.; Zhu, A.X.; Loeffler, J.S.; Batchelor, T.T.; Sorensen, A.G. Biomarkers of response and resistance to antiangiogenic therapy. Nat. Rev. Clin. Oncol. 2009, 6, 327–338. [Google Scholar]
- Sorensen, A.G.; Batchelor, T.T.; Zhang, W.T.; Chen, P.J.; Yeo, P.; Wang, M.; Jennings, D.; Wen, P.Y.; Lahdenranta, J.; Ancukiewicz, M.; di Tomaso, E.; Duda, D.G.; Jain, R.K. A "vascular normalization index" as potential mechanistic biomarker to predict survival after a single dose of cediranib in recurrent glioblastoma patients. Cancer Res. 2009, 69, 5296–5300. [Google Scholar]
- Timpl, R.; Wiedemann, H.; van Delden, V.; Furthmayr, H.; Kuhn, K. A network model for the organization of type IV collagen molecules in basement membranes. Eur. J. Biochem. 1981, 120, 203–211. [Google Scholar]
- Kuhn, K.; Wiedemann, H.; Timpl, R.; Risteli, J.; Dieringer, H.; Voss, T.; Glanville, R.W. Macromolecular structure of basement membrane collagens. FEBS Lett. 1981, 125, 123–128. [Google Scholar]
- Rehn, M.; Pihlajaniemi, T. Alpha 1(XVIII), a collagen chain with frequent interruptions in the collagenous sequence, a distinct tissue distribution, and homology with type XV collagen. Proc. Natl. Acad. Sci. USA 1994, 91, 4234–4238. [Google Scholar]
- Saarela, J.; Rehn, M.; Oikarinen, A.; Autio-Harmainen, H.; Pihlajaniemi, T. The short and long forms of type XVIII collagen show clear tissue specificities in their expression and location in basement membrane zones in humans. Am. J. Pathol. 1998, 153, 611–626. [Google Scholar]
- Wen, W.; Moses, M.A.; Wiederschain, D.; Arbiser, J.L.; Folkman, J. The generation of endostatin is mediated by elastase. Cancer Res. 1999, 59, 6052–6056. [Google Scholar]
- Felbor, U.; Dreier, L.; Bryant, R.A.; Ploegh, H.L.; Olsen, B.R.; Mothes, W. Secreted cathepsin L generates endostatin from collagen XVIII. EMBO J. 2000, 19, 1187–1194. [Google Scholar]
- Lin, H.C.; Chang, J.H.; Jain, S.; Gabison, E.E.; Kure, T.; Kato, T.; Fukai, N.; Azar, D.T. Matrilysin cleavage of corneal collagen type XVIII NC1 domain and generation of a 28-kDa fragment. Invest. Ophthalmol. Vis. Sci. 2001, 42, 2517–2524. [Google Scholar]
- Herbst, R.S.; Hess, K.R.; Tran, H.T.; Tseng, J.E.; Mullani, N.A.; Charnsangavej, C.; Madden, T.; Davis, D.W.; McConkey, D.J.; O'Reilly, M.S.; Ellis, L.M.; Pluda, J.; Hong, W.K.; Abbruzzese, J.L. Phase I study of recombinant human endostatin in patients with advanced solid tumors. J. Clin. Oncol. 2002, 20, 3792–3803. [Google Scholar]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar]
- Klement, G.L.; Yip, T.T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; Almog, N.; Kieran, M.W.; Folkman, J. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar]
- Sasaki, T.; Hohenester, E.; Timpl, R. Structure and function of collagen-derived endostatin inhibitors of angiogenesis. IUBMB Life 2002, 53, 77–84. [Google Scholar]
- Hohenester, E.; Sasaki, T.; Olsen, B.R.; Timpl, R. Crystal structure of the angiogenesis inhibitor endostatin at 1.5 A resolution. EMBO J. 1998, 17, 1656–1664. [Google Scholar]
- Hohenester, E.; Sasaki, T.; Mann, K.; Timpl, R. Variable zinc coordination in endostatin. J. Mol. Biol. 2000, 297, 1–6. [Google Scholar]
- Sasaki, T.; Larsson, H.; Kreuger, J.; Salmivirta, M.; Claesson-Welsh, L.; Lindahl, U.; Hohenester, E.; Timpl, R. Structural basis and potential role of heparin/heparan sulfate binding to the angiogenesis inhibitor endostatin. EMBO J. 1999, 18, 6240–6248. [Google Scholar]
- Sasaki, T.; Larsson, H.; Tisi, D.; Claesson-Welsh, L.; Hohenester, E.; Timpl, R. Endostatins derived from collagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J. Mol. Biol. 2000, 301, 1179–1190. [Google Scholar]
- Sasaki, T.; Fukai, N.; Mann, K.; Gohring, W.; Olsen, B.R.; Timpl, R. Structure, function and tissue forms of the C-terminal globular domain of collagen XVIII containing the angiogenesis inhibitor endostatin. EMBO J. 1998, 17, 4249–4256. [Google Scholar]
- Karumanchi, S.A.; Jha, V.; Ramchandran, R.; Karihaloo, A.; Tsiokas, L.; Chan, B.; Dhanabal, M.; Hanai, J.I.; Venkataraman, G.; Shriver, Z.; Keiser, N.; Kalluri, R.; Zeng, H.; Mukhopadhyay, D.; Chen, R.L.; Lander, A.D.; Hagihara, K.; Yamaguchi, Y.; Sasisekharan, R.; Cantley, L.; Sukhatme, V.P. Cell surface glypicans are low-affinity endostatin receptors. Mol. Cell 2001, 7, 811–822. [Google Scholar]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar]
- Yamaguchi, N.; Anand-Apte, B.; Lee, M.; Sasaki, T.; Fukai, N.; Shapiro, R.; Que, I.; Lowik, C.; Timpl, R.; Olsen, B.R. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. Embo J. 1999, 18, 4414–4423. [Google Scholar]
- O'Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar]
- Dhanabal, M.; Ramchandran, R.; Waterman, M.J.; Lu, H.; Knebelmann, B.; Segal, M.; Sukhatme, V.P. Endostatin induces endothelial cell apoptosis. J. Biol. Chem. 1999, 274, 11721–11726. [Google Scholar]
- Dhanabal, M.; Volk, R.; Ramchandran, R.; Simons, M.; Sukhatme, V.P. Cloning, expression, and in vitro activity of human endostatin. Biochem. Biophys. Res. Commun. 1999, 258, 345–352. [Google Scholar]
- Wickstrom, S.A.; Alitalo, K.; Keski-Oja, J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002, 62, 5580–5589. [Google Scholar]
- Wickstrom, S.A.; Alitalo, K.; Keski-Oja, J. An endostatin-derived peptide interacts with integrins and regulates actin cytoskeleton and migration of endothelial cells. J. Biol. Chem. 2004, 279, 20178–20185. [Google Scholar]
- Kim, Y.M.; Hwang, S.; Pyun, B.J.; Kim, T.Y.; Lee, S.T.; Gho, Y.S.; Kwon, Y.G. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J. Biol. Chem. 2002, 277, 27872–27879. [Google Scholar]
- Kim, Y.M.; Jang, J.W.; Lee, O.H.; Yeon, J.; Choi, E.Y.; Kim, K.W.; Lee, S.T.; Kwon, Y.G. Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res. 2000, 60, 5410–5413. [Google Scholar]
- Fukai, N.; Eklund, L.; Marneros, A.G.; Oh, S.P.; Keene, D.R.; Tamarkin, L.; Niemela, M.; Ilves, M.; Li, E.; Pihlajaniemi, T.; Olsen, B.R. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 2002, 21, 1535–1544. [Google Scholar]
- Sertie, A.L.; Sossi, V.; Camargo, A.A.; Zatz, M.; Brahe, C.; Passos-Bueno, M.R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 2000, 9, 2051–2058. [Google Scholar]
- Li, Q.; Olsen, B.R. Increased angiogenic response in aortic explants of collagen XVIII/endostatin-null mice. Am. J. Pathol. 2004, 165, 415–424. [Google Scholar]
- Sund, M.; Hamano, Y.; Sugimoto, H.; Sudhakar, A.; Soubasakos, M.; Yerramalla, U.; Benjamin, L.E.; Lawler, J.; Kieran, M.; Shah, A.; Kalluri, R. Function of endogenous inhibitors of angiogenesis as endothelium-specific tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 2934–2939. [Google Scholar]
- Brideau, G.; Makinen, M.J.; Elamaa, H.; Tu, H.; Nilsson, G.; Alitalo, K.; Pihlajaniemi, T.; Heljasvaara, R. Endostatin overexpression inhibits lymphangiogenesis and lymph node metastasis in mice. Cancer Res. 2007, 67, 11528–11535. [Google Scholar]
- Folkman, J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar]
- Bono, P.; Teerenhovi, L.; Joensuu, H. Elevated serum endostatin is associated with poor outcome in patients with non-Hodgkin lymphoma. Cancer 2003, 97, 2767–2775. [Google Scholar]
- Feldman, A.L.; Pak, H.; Yang, J.C.; Alexander, H.R., Jr.; Libutti, S.K. Serum endostatin levels are elevated in patients with soft tissue sarcoma. Cancer 2001, 91, 1525–1529. [Google Scholar]
- Feldman, A.L.; Alexander, H.R., Jr.; Yang, J.C.; Linehan, W.M.; Eyler, R.A.; Miller, M.S.; Steinberg, S.M.; Libutti, S.K. Prospective analysis of circulating endostatin levels in patients with renal cell carcinoma. Cancer 2002, 95, 1637–1643. [Google Scholar]
- Feldman, A.L.; Alexander, H.R., Jr.; Bartlett, D.L.; Kranda, K.C.; Miller, M.S.; Costouros, N.G.; Choyke, P.L.; Libutti, S.K. A prospective analysis of plasma endostatin levels in colorectal cancer patients with liver metastases. Ann. Surg. Oncol. 2001, 8, 741–745. [Google Scholar]
- Kuroi, K.; Toi, M. Circulating angiogenesis regulators in cancer patients. Int. J. Biol. Markers 2001, 16, 5–26. [Google Scholar]
- Iizasa, T.; Chang, H.; Suzuki, M.; Otsuji, M.; Yokoi, S.; Chiyo, M.; Motohashi, S.; Yasufuku, K.; Sekine, Y.; Iyoda, A.; Shibuya, K.; Hiroshima, K.; Fujisawa, T. Overexpression of collagen XVIII is associated with poor outcome and elevated levels of circulating serum endostatin in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 5361–5366. [Google Scholar]
- Eder, J.P., Jr.; Supko, J.G.; Clark, J.W.; Puchalski, T.A.; Garcia-Carbonero, R.; Ryan, D.P.; Shulman, L.N.; Proper, J.; Kirvan, M.; Rattner, B.; Connors, S.; Keogan, M.T.; Janicek, M.J.; Fogler, W.E.; Schnipper, L.; Kinchla, N.; Sidor, C.; Phillips, E.; Folkman, J.; Kufe, D.W. Phase I clinical trial of recombinant human endostatin administered as a short intravenous infusion repeated daily. J. Clin. Oncol. 2002, 20, 3772–3784. [Google Scholar]
- Thomas, J.P.; Arzoomanian, R.Z.; Alberti, D.; Marnocha, R.; Lee, F.; Friedl, A.; Tutsch, K.; Dresen, A.; Geiger, P.; Pluda, J.; Fogler, W.; Schiller, J.H.; Wilding, G. Phase I pharmacokinetic and pharmacodynamic study of recombinant human endostatin in patients with advanced solid tumors. J. Clin. Oncol. 2003, 21, 223–231. [Google Scholar]
- Twombly, R. First clinical trials of endostatin yield lukewarm results. J. Natl. Cancer Inst. 2002, 94, 1520–1521. [Google Scholar]
- Ryan, D.P.; Penson, R.T.; Ahmed, S.; Chabner, B.A.; Lynch, T.J., Jr. Reality testing in cancer treatment: the phase I trial of endostatin. Oncologist 1999, 4, 501–508. [Google Scholar]
- Dhanabal, M.; Ramchandran, R.; Volk, R.; Stillman, I.E.; Lombardo, M.; Iruela-Arispe, M.L.; Simons, M.; Sukhatme, V.P. Endostatin: yeast production, mutants, and antitumor effect in renal cell carcinoma. Cancer Res. 1999, 59, 189–197. [Google Scholar]
- Jiang, L.P.; Zou, C.; Yuan, X.; Luo, W.; Wen, Y.; Chen, Y. N-terminal modification increases the stability of the recombinant human endostatin in vitro. Biotechnol. Appl. Biochem. 2009, 54, 113–120. [Google Scholar]
- Kulke, M.H.; Bergsland, E.K.; Ryan, D.P.; Enzinger, P.C.; Lynch, T.J.; Zhu, A.X.; Meyerhardt, J.A.; Heymach, J.V.; Fogler, W.E.; Sidor, C.; Michelini, A.; Kinsella, K.; Venook, A.P.; Fuchs, C.S. Phase II study of recombinant human endostatin in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2006, 24, 3555–3561. [Google Scholar]
- Lu, N.; Ling, Y.; Gao, Y.; Chen, Y.; Mu, R.; Qi, Q.; Liu, W.; Zhang, H.; Gu, H.; Wang, S.; Yang, Y.; Guo, Q. Endostar suppresses invasion through downregulating the expression of matrix metalloproteinase-2/9 in MDA-MB-435 human breast cancer cells. Exp. Biol. Med. (Maywood) 2008, 233, 1013–1020. [Google Scholar]
- Sun, L.; Ye, H.Y.; Zhang, Y.H.; Guan, Y.S.; Wu, H. Epidermal growth factor receptor antibody plus recombinant human endostatin in treatment of hepatic metastases after remnant gastric cancer resection. World J. Gastroenterol. 2007, 13, 6115–6118. [Google Scholar]
- Wang, J.; Huang, C.; Wei, X.Y.; Qi, D.L.; Gong, L.Q.; Mu, H.Y.; Yao, Q.; Li, K. Changes of activated circulating endothelial cells and survivin in patients with non-small cell lung cancer after antiangiogenesis therapy. Chin. Med. J. (Engl) 2008, 121, 2234–2240. [Google Scholar] [PubMed]
- Maeshima, Y.; Colorado, P.C.; Torre, A.; Holthaus, K.A.; Grunkemeyer, J.A.; Ericksen, M.B.; Hopfer, H.; Xiao, Y.; Stillman, I.E.; Kalluri, R. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J. Biol. Chem. 2000, 275, 21340–21348. [Google Scholar]
- Pasco, S.; Brassart, B.; Ramont, L.; Maquart, F.X.; Monboisse, J.C. Control of melanoma cell invasion by type IV collagen. Cancer Detect. Prev. 2005, 29, 260–266. [Google Scholar]
- Hudson, B.G.; Reeders, S.T.; Tryggvason, K. Type IV collagen: structure, gene organization, and role in human disease. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J. Biol. Chem. 1993, 268, 26033–26036. [Google Scholar] [PubMed]
- Maeshima, Y.; Sudhakar, A.; Lively, J.C.; Ueki, K.; Kharbanda, S.; Kahn, C.R.; Sonenberg, N.; Hynes, R.O.; Kalluri, R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science 2002, 295, 140–143. [Google Scholar]
- Maeshima, Y.; Manfredi, M.; Reimer, C.; Holthaus, K.A.; Hopfer, H.; Chandamuri, B.R.; Kharbanda, S.; Kalluri, R. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J. Biol. Chem. 2001, 276, 15240–15248. [Google Scholar]
- Eikesdal, H.P.; Sugimoto, H.; Birrane, G.; Maeshima, Y.; Cooke, V.G.; Kieran, M.; Kalluri, R. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor activity. Proc. Natl. Acad. Sci. USA 2008, 105, 15040–15045. [Google Scholar]
- Maeshima, Y.; Yerramalla, U.L.; Dhanabal, M.; Holthaus, K.A.; Barbashov, S.; Kharbanda, S.; Reimer, C.; Manfredi, M.; Dickerson, W.M.; Kalluri, R. Extracellular matrix-derived peptide binds to alpha(v)beta(3) integrin and inhibits angiogenesis. J. Biol. Chem. 2001, 276, 31959–31968. [Google Scholar]
- Maeshima, Y.; Colorado, P.C.; Kalluri, R. Two RGD-independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J. Biol. Chem. 2000, 275, 23745–23750. [Google Scholar]
- Petitclerc, E.; Boutaud, A.; Prestayko, A.; Xu, J.; Sado, Y.; Ninomiya, Y.; Sarras, M.P., Jr.; Hudson, B.G.; Brooks, P.C. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 2000, 275, 8051–8061. [Google Scholar] [PubMed]
- Han, J.; Ohno, N.; Pasco, S.; Monboisse, J.C.; Borel, J.P.; Kefalides, N.A. A cell binding domain from the alpha3 chain of type IV collagen inhibits proliferation of melanoma cells. J. Biol. Chem. 1997, 272, 20395–20401. [Google Scholar]
- Kawaguchi, T.; Yamashita, Y.; Kanamori, M.; Endersby, R.; Bankiewicz, K.S.; Baker, S.J.; Bergers, G.; Pieper, R.O. The PTEN/Akt pathway dictates the direct alphaVbeta3-dependent growth-inhibitory action of an active fragment of tumstatin in glioma cells in vitro and in vivo. Cancer Res. 2006, 66, 11331–11340. [Google Scholar]
- Chung, I.S.; Son, Y.I.; Ko, Y.J.; Baek, C.H.; Cho, J.K.; Jeong, H.S. Peritumor injections of purified tumstatin delay tumor growth and lymphatic metastasis in an orthotopic oral squamous cell carcinoma model. Oral. Oncol. 2008, 44, 1118–1126. [Google Scholar]
- Gu, Q.; Zhang, T.; Luo, J.; Wang, F. Expression, purification, and bioactivity of human tumstatin from Escherichia coli. Protein Expr. Purif. 2006, 47, 461–466. [Google Scholar]
- Eikesdal, H.P.; Kalluri, R. Drug resistance associated with antiangiogenesis therapy. Semin. Cancer Biol. 2009, 19, 310–317. [Google Scholar]
- Luo, Y.Q.; Wang, L.H.; Yi, Q.; Jiao, B.H. Expression of soluble, biologically active recombinant human tumstatin in Escherichia coli. Clin. Exp. Med. 2008, 8, 37–42. [Google Scholar]
- Clamp, A.R.; Jayson, G.C. The clinical potential of antiangiogenic fragments of extracellular matrix proteins. Br. J. Cancer 2005, 93, 967–972. [Google Scholar]
- Slajpah, M.; Gorinsek, B.; Berginc, G.; Vizjak, A.; Ferluga, D.; Hvala, A.; Meglic, A.; Jaksa, I.; Furlan, P.; Gregoric, A.; Kaplan-Pavlovcic, S.; Ravnik-Glavac, M.; Glavac, D. Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int. 2007, 71, 1287–1295. [Google Scholar]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar]
- Colorado, P.C.; Torre, A.; Kamphaus, G.; Maeshima, Y.; Hopfer, H.; Takahashi, K.; Volk, R.; Zamborsky, E.D.; Herman, S.; Sarkar, P.K.; Ericksen, M.B.; Dhanabal, M.; Simons, M.; Post, M.; Kufe, D.W.; Weichselbaum, R.R.; Sukhatme, V.P.; Kalluri, R. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000, 60, 2520–2526. [Google Scholar]
- Boosani, C.S.; Sudhakar, A. Cloning, purification, and characterization of a non-collagenous anti-angiogenic protein domain from human alpha1 type IV collagen expressed in Sf9 cells. Protein Expr. Purif. 2006, 49, 211–218. [Google Scholar]
- Sudhakar, A.; Nyberg, P.; Keshamouni, V.G.; Mannam, A.P.; Li, J.; Sugimoto, H.; Cosgrove, D.; Kalluri, R. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J. Clin. Invest. 2005, 115, 2801–2810. [Google Scholar]
- Nyberg, P.; Xie, L.; Sugimoto, H.; Colorado, P.; Sund, M.; Holthaus, K.; Sudhakar, A.; Salo, T.; Kalluri, R. Characterization of the anti-angiogenic properties of arresten, an alpha1beta1 integrin-dependent collagen-derived tumor suppressor. Exp. Cell Res. 2008, 314, 3292–3305. [Google Scholar]
- Boosani, C.S.; Nalabothula, N.; Munugalavadla, V.; Cosgrove, D.; Keshamoun, V.G.; Sheibani, N.; Sudhakar, A. FAK and p38-MAP kinase-dependent activation of apoptosis and caspase-3 in retinal endothelial cells by alpha1(IV)NC1. Invest. Ophthalmol. Vis. Sci. 2009, 50, 4567–4575. [Google Scholar]
- Netzer, K.O.; Suzuki, K.; Itoh, Y.; Hudson, B.G.; Khalifah, R.G. Comparative analysis of the noncollagenous NC1 domain of type IV collagen: identification of structural features important for assembly, function, and pathogenesis. Protein Sci. 1998, 7, 1340–1351. [Google Scholar]
- Than, M.E.; Henrich, S.; Huber, R.; Ries, A.; Mann, K.; Kuhn, K.; Timpl, R.; Bourenkov, G.P.; Bartunik, H.D.; Bode, W. The 9-A crystal structure of the noncollagenous (NC1) domain of human placenta collagen IV shows stabilization via a novel type of covalent Met-Lys cross-link. Proc. Natl. Acad. Sci. USA 2002, 99, 6607–6612. [Google Scholar]
- Sundaramoorthy, M.; Meiyappan, M.; Todd, P.; Hudson, B.G. Crystal structure of NC1 domains. Structural basis for type IV collagen assembly in basement membranes. J. Biol. Chem. 2002, 277, 31142–31153. [Google Scholar] [PubMed]
- Kajimura, D.; Takahashi, S.; Yoshikawa, K.; Hattori, S.; Sado, Y.; Imamura, Y.; Hayashi, T. Non-helical type IV collagen polypeptides in human placenta. Biochem. Biophys. Res. Commun. 2004, 314, 11–16. [Google Scholar]
- Senger, D.R.; Claffey, K.P.; Benes, J.E.; Perruzzi, C.A.; Sergiou, A.P.; Detmar, M. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc. Natl. Acad. Sci. USA 1997, 94, 13612–13617. [Google Scholar]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar]
- He, G.A.; Luo, J.X.; Zhang, T.Y.; Wang, F.Y.; Li, R.F. Canstatin-N fragment inhibits in vitro endothelial cell proliferation and suppresses in vivo tumor growth. Biochem. Biophys. Res. Commun. 2003, 312, 801–805. [Google Scholar]
- He, G.A.; Luo, J.X.; Zhang, T.Y.; Hu, Z.S.; Wang, F.Y. The C-terminal domain of canstatin suppresses in vivo tumor growth associated with proliferation of endothelial cells. Biochem. Biophys. Res. Commun. 2004, 318, 354–360. [Google Scholar]
- Panka, D.J.; Mier, J.W. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J. Biol. Chem. 2003, 278, 37632–37636. [Google Scholar]
- Hou, W.H.; Wang, T.Y.; Yuan, B.M.; Chai, Y.R.; Jia, Y.L.; Tian, F.; Wang, J.M.; Xue, L.X. Recombinant mouse canstatin inhibits chicken embryo chorioallantoic membrane angiogenesis and endothelial cell proliferation. Acta. Biochim. Biophys. Sin. (Shanghai) 2004, 36, 845–850. [Google Scholar]
- Magnon, C.; Galaup, A.; Mullan, B.; Rouffiac, V.; Bouquet, C.; Bidart, J.M.; Griscelli, F.; Opolon, P.; Perricaudet, M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Res. 2005, 65, 4353–4361. [Google Scholar]
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.P. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J. Biol. Chem. 2006, 281, 6020–6029. [Google Scholar]
- Lindahl, C.; Simonsson, M.; Bergh, A.; Thysell, E.; Antti, H.; Sund, M.; Wikstrom, P. Increased levels of macrophage-secreted cathepsin S during prostate cancer progression in TRAMP mice and patients. Cancer Genomics Proteomics 2009, 6, 149–159. [Google Scholar]
- He, X.P.; Li, Z.S.; Zhu, R.M.; Tu, Z.X.; Gao, J.; Pan, X.; Gong, Y.F.; Jin, J.; Man, X.H.; Wu, H.Y.; Xu, A.F. Effects of recombinant human canstatin protein in the treatment of pancreatic cancer. World J. Gastroenterol. 2006, 12, 6652–6657. [Google Scholar]
- Magnon, C.; Opolon, P.; Ricard, M.; Connault, E.; Ardouin, P.; Galaup, A.; Metivier, D.; Bidart, J.M.; Germain, S.; Perricaudet, M.; Schlumberger, M. Radiation and inhibition of angiogenesis by canstatin synergize to induce HIF-1alpha-mediated tumor apoptotic switch. J. Clin. Invest. 2007, 117, 1844–1855. [Google Scholar]
- Magnon, C.; Opolon, P.; Connault, E.; Mir, L.M.; Perricaudet, M.; Martel-Renoir, D. Canstatin gene electrotransfer combined with radiotherapy: preclinical trials for cancer treatment. Gene Ther. 2008, 15, 1436–1445. [Google Scholar]
- Wang, W.B.; Zhou, Y.L.; Heng, D.F.; Miao, C.H.; Cao, Y.L. Combination of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and canstatin gene suppression therapy on breast tumor xenograft growth in mice. Breast Cancer Res. Treat. 2008, 110, 283–295. [Google Scholar]
- Lee, J.M.; Jeon, H.B.; Sohn, B.H.; Chung, I.S. Functional expression of recombinant canstatin in stably transformed Drosophila melanogaster S2 cells. Protein Expr. Purif. 2007, 52, 258–264. [Google Scholar]
- Mundel, T.M.; Yliniemi, A.M.; Maeshima, Y.; Sugimoto, H.; Kieran, M.; Kalluri, R. Type IV collagen alpha6 chain-derived noncollagenous domain 1 (alpha6(IV)NC1) inhibits angiogenesis and tumor growth. Int. J. Cancer 2008, 122, 1738–1744. [Google Scholar]
- Karagiannis, E.D.; Popel, A.S. Identification of novel short peptides derived from the alpha 4, alpha 5, and alpha 6 fibrils of type IV collagen with anti-angiogenic properties. Biochem. Biophys. Res. Commun. 2007, 354, 434–439. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sund, M.; Nyberg, P.; Eikesdal, H.P. Endogenous Matrix-Derived Inhibitors of Angiogenesis. Pharmaceuticals 2010, 3, 3021-3039. https://doi.org/10.3390/ph3103021
Sund M, Nyberg P, Eikesdal HP. Endogenous Matrix-Derived Inhibitors of Angiogenesis. Pharmaceuticals. 2010; 3(10):3021-3039. https://doi.org/10.3390/ph3103021
Chicago/Turabian StyleSund, Malin, Pia Nyberg, and Hans Petter Eikesdal. 2010. "Endogenous Matrix-Derived Inhibitors of Angiogenesis" Pharmaceuticals 3, no. 10: 3021-3039. https://doi.org/10.3390/ph3103021
APA StyleSund, M., Nyberg, P., & Eikesdal, H. P. (2010). Endogenous Matrix-Derived Inhibitors of Angiogenesis. Pharmaceuticals, 3(10), 3021-3039. https://doi.org/10.3390/ph3103021