
Beyond the Limit: MYC Mediates Tumor Immune Escape



Abstract
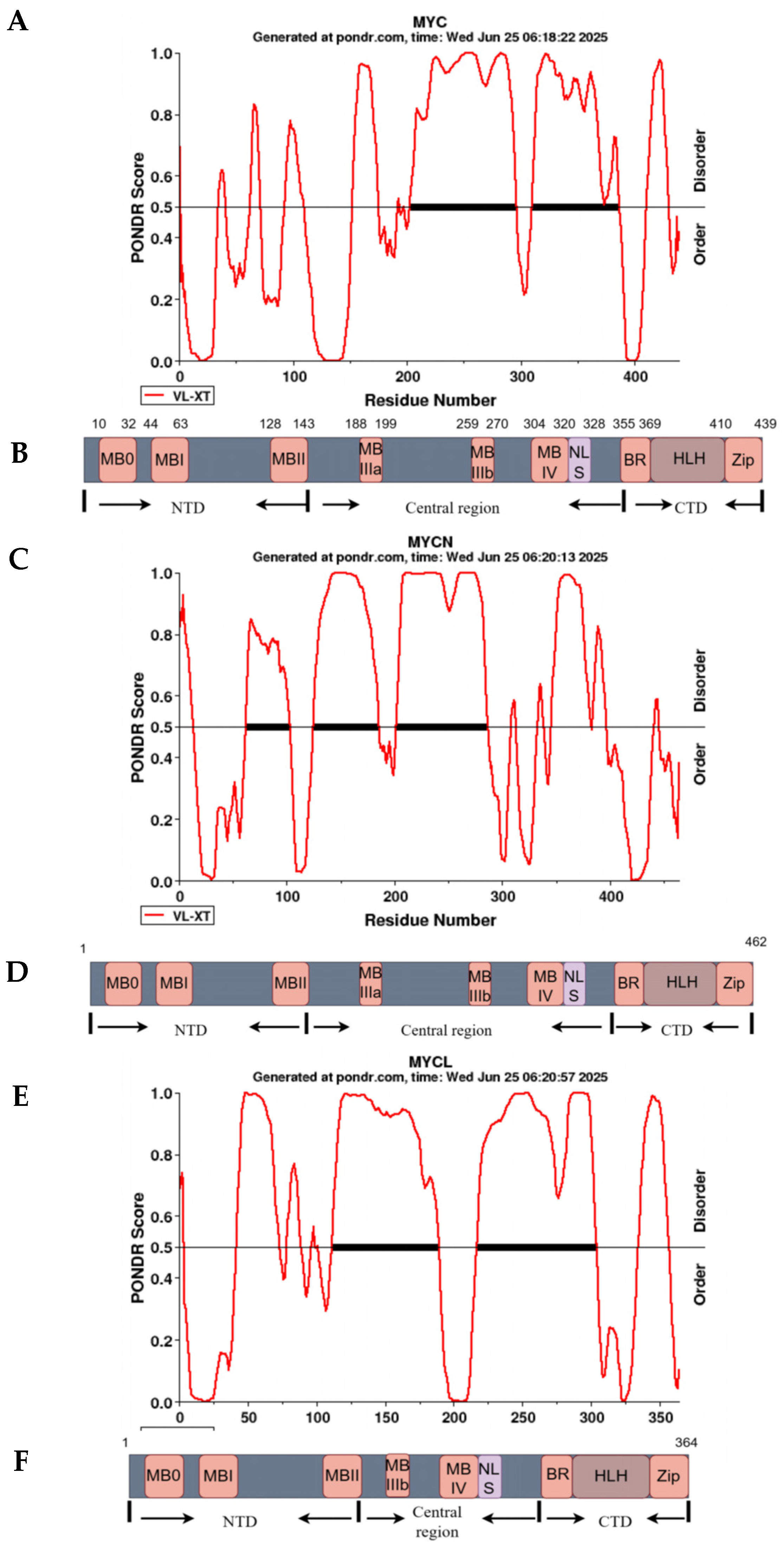
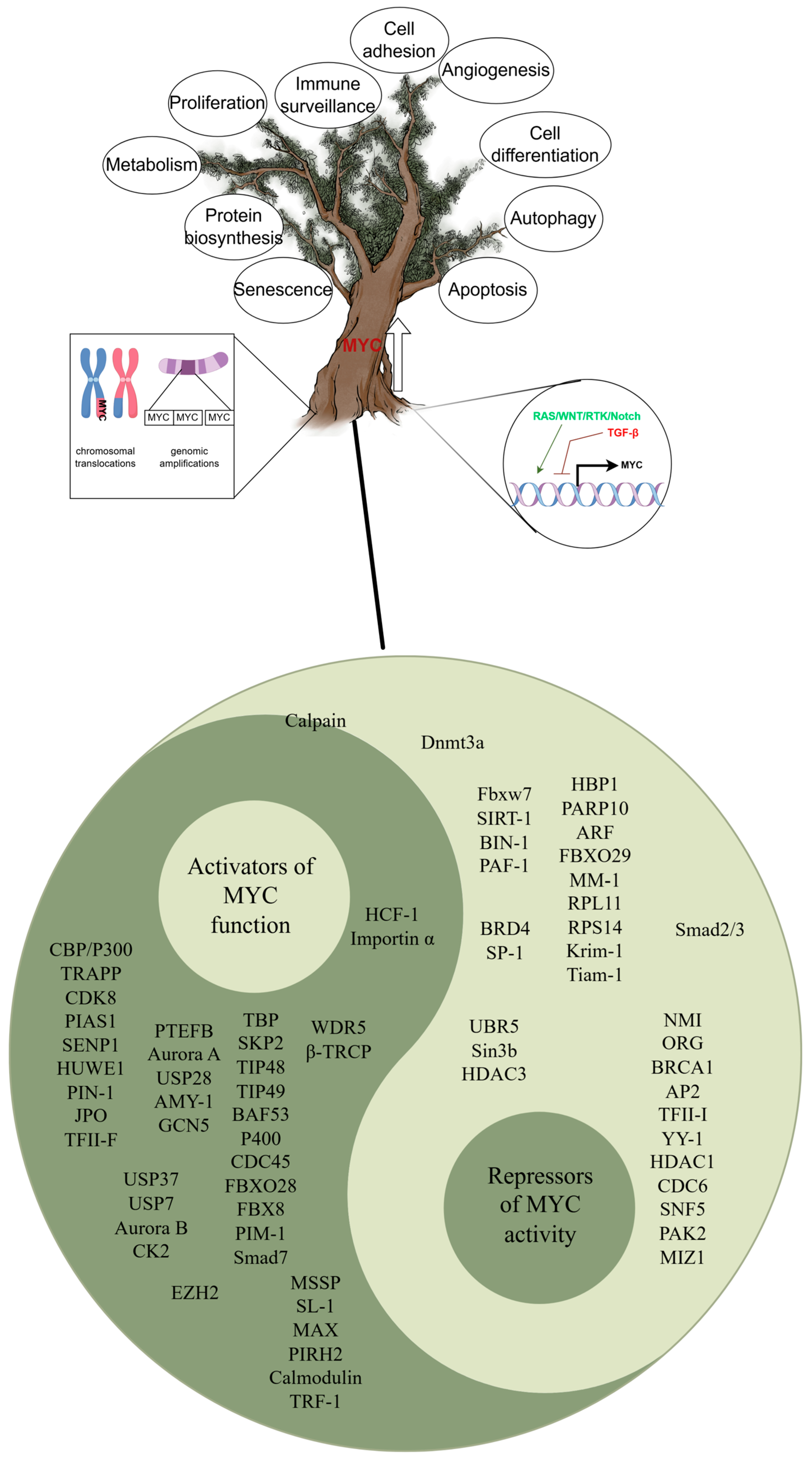
1. MYC
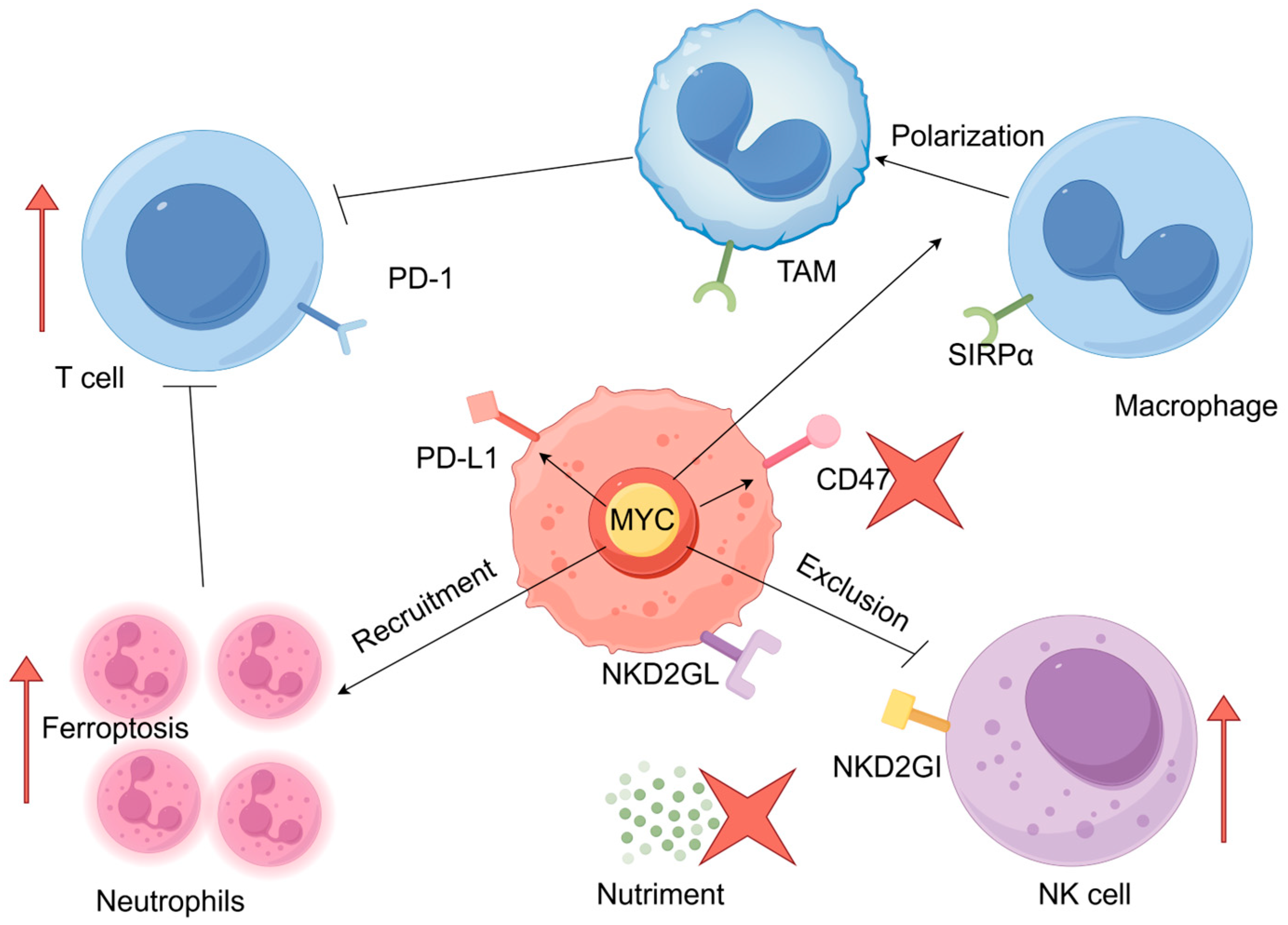
2. MYC and Tumor Immune Escape
2.1. MYC Mediates the Escape of Tumor Cells from T-Cell Immunity
2.2. MYC Mediates the Escape of Tumor Cells from Macrophage Immunity
2.3. MYC May Mediate Immune Escape of Tumor Cells by Inducing Neutrophil Infiltration
2.4. MYC Mediates the Escape of Tumor Cells from Natural Killer (NK) Cells Immunity
3. Strategies Combining the Targeting of MYC and Immunotherapy
3.1. Inhibition of MYC Combined with Cell Therapy
3.2. Inhibition of MYC Combined with Monoclonal Antibodies
3.3. Inhibition of MYC Combined with Adjustment for Nutritional Intake
4. Future Directions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategies | Representative Target | Promising Therapeutic Agents | References |
|---|---|---|---|
| MYC-MAX dimerization disruption | MYC-MAX | Omomyc | [98] |
| KSI-3716 | [121] | ||
| MYCi361/MYCi975 | [36] | ||
| VPC-70067 | [122] | ||
| 10058-F4 | [123] | ||
| IIA6B17 | [124] | ||
| 10074-G5 | [125] | ||
| MYCMI-6 | [126] | ||
| H1 peptide | [127] | ||
| MI1-PD | [128] | ||
| KJ-Pyr-9 | [129] | ||
| Mycro3 | [130] | ||
| Disrupt the binding of MYC-DNA | MYC-DNA | ME47 | [131] |
| Transcriptional and translational suppression | CDK7 | SNS-032 | [132] |
| CDK9 | AZD4573 | [133] | |
| BRD4 | JQ-1 | [134] | |
| Targeting interacting proteins | AURKB | AZD-1152 | [19,135] |
| WDR5 | C6 | [136] | |
| EZH2 | Tazemetostat | [137] | |
| Targeting the upstream signaling pathways | RAS | RMC-7977 | [138] |
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef]
- Tansey, W.P. Mammalian MYC proteins and cancer. New J. Sci. 2014, 2014, 1–27. [Google Scholar] [CrossRef]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Sollazzo, M.; Paglia, S.; Grifoni, D. MYC, cell competition, and cell death in cancer: The inseparable triad. Genes 2017, 8, 120. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Williamson, D.; Lu, Y.J.; Gordon, T.; Sciot, R.; Kelsey, A.; Fisher, C.; Poremba, C.; Anderson, J.; Pritchard-Jones, K.; Shipley, J. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J. Clin. Oncol. 2005, 23, 880–888. [Google Scholar] [CrossRef]
- Williams, R.D.; Al-Saadi, R.; Natrajan, R.; Mackay, A.; Chagtai, T.; Little, S.; Hing, S.N.; Fenwick, K.; Ashworth, A.; Grundy, P.; et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer 2011, 50, 982–995. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc–mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The expanding world of N-MYC–driven tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Hatakeyama, K.; Nagashima, T.; Watanabe, Y.; Kanto, K.; Doi, Y.; Ide, T.; Shimoda, Y.; Tanabe, T.; Ohnami, S.; et al. Integrated analysis of gene expression and copy number identified potential cancer driver genes with amplification-dependent overexpression in 1,454 solid tumors. Sci. Rep. 2017, 7, 641. [Google Scholar] [CrossRef]
- Boutros, P.C.; Fraser, M.; Harding, N.J.; de Borja, R.; Trudel, D.; Lalonde, E.; Meng, A.; Hennings-Yeomans, P.H.; McPherson, A.; Sabelnykova, V.Y.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef]
- Thege, F.I.; Rupani, D.N.; Barathi, B.B.; Manning, S.L.; Maitra, A.; Rhim, A.D.; Wörmann, S.M. A Programmable In Vivo CRISPR Activation Model Elucidates the Oncogenic and Immunosuppressive Functions of MYC in Lung Adenocarcinoma. Cancer Res. 2022, 82, 2761–2776. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC deregulation in primary human cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; et al. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis. Cancer Cell 2020, 37, 200–215.e5. [Google Scholar] [CrossRef]
- Tu, W.B.; Shiah, Y.J.; Lourenco, C.; Mullen, P.J.; Dingar, D.; Redel, C.; Tamachi, A.; Ba-Alawi, W.; Aman, A.; Al-Awar, R.; et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell 2018, 34, 579–595.e8. [Google Scholar] [CrossRef]
- Qu, A.; Jiang, C.; Cai, Y.; Kim, J.H.; Tanaka, N.; Ward, J.M.; Shah, Y.M.; Gonzalez, F.J. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J. Hepatol. 2014, 60, 331–338. [Google Scholar] [CrossRef]
- Qin, N.; Paisana, E.; Langini, M.; Picard, D.; Malzkorn, B.; Custódia, C.; Cascão, R.; Meyer, F.D.; Blümel, L.; Göbbels, S.; et al. Intratumoral heterogeneity of MYC drives medulloblastoma metastasis and angiogenesis. Neuro Oncol. 2022, 24, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Gwynne, W.D.; Suk, Y.; Custers, S.; Mikolajewicz, N.; Chan, J.K.; Zador, Z.; Chafe, S.C.; Zhai, K.; Escudero, L.; Zhang, C.; et al. Cancer-selective metabolic vulnerabilities in MYC-amplified medulloblastoma. Cancer Cell 2022, 40, 1488–1502.e7. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Wu, C.H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Lourenco, C.; Kalkat, M.; Houlahan, K.E.; De Melo, J.; Longo, J.; Done, S.J.; Boutros, P.C.; Penn, L.Z. Modeling the MYC-driven normal-to-tumor switch in breast cancer. Dis. Model. Mech. 2019, 12, dmm038083. [Google Scholar] [CrossRef] [PubMed]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; d’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2017, 36, 1911–1924. [Google Scholar] [CrossRef]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc reveals new mechanisms to inhibit the MYC oncogene. Mol. Cell Biol. 2019, 39, e00248-19. [Google Scholar] [CrossRef]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef]
- English, I.A.; Sears, R.C. Deconstructing Pancreatic Adenocarcinoma by Targeting the Conductor, MYC. Cancer Discov. 2020, 10, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Zhu, J.; Wang, Z.; Peng, Y.; Zeng, L. Comprehensive pan-cancer analysis reveals ENC1 as a promising prognostic biomarker for tumor microenvironment and therapeutic responses. Sci. Rep. 2024, 14, 25331. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Liu, J.; Tong, J. Long Non-coding RNA MIAT Knockdown Prevents the Formation of Intracranial Aneurysm by Downregulating ENC1 via MYC. Front. Physiol. 2021, 11, 572605. [Google Scholar] [CrossRef]
- Duan, W.W.; Yang, L.T.; Liu, J.; Dai, Z.Y.; Wang, Z.Y.; Zhang, H.; Zhang, X.; Liang, X.S.; Luo, P.; Zhang, J.; et al. A TGF-β signaling-related lncRNA signature for prediction of glioma prognosis, immune microenvironment, and immunotherapy response. CNS Neurosci. Ther. 2024, 30, e14489. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- MYC Promotes Tumorigenesis via Activation of CD47 and PD-L1. Cancer Discov. 2016, 6, 472. [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. [Google Scholar] [CrossRef]
- Tsushima, F.; Yao, S.; Shin, T.; Flies, A.; Flies, S.; Xu, H.; Tamada, K.; Pardoll, D.M.; Chen, L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood 2007, 110, 180–185. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, costimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T-cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Melero, I.; de Miguel Luken, M.; de Velasco, G.; Garralda, E.; Martín-Liberal, J.; Joerger, M.; Alonso, G.; Goebeler, M.E.; Schuler, M.; König, D.; et al. Neutralizing GDF-15 can overcome anti-PD-1 and anti-PD-L1 resistance in solid tumors. Nature 2025, 637, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Haake, M.; Haack, B.; Schäfer, T.; Harter, P.N.; Mattavelli, G.; Eiring, P.; Vashist, N.; Wedekink, F.; Genssler, S.; Fischer, B.; et al. Tumor-derived GDF-15 blocks LFA-1 dependent T-cell recruitment and suppresses responses to anti-PD-1 treatment. Nat. Commun. 2023, 14, 4253. [Google Scholar] [CrossRef]
- Li, S.; Ma, Y.M.; Zheng, P.S.; Zhang, P. GDF15 promotes the proliferation of cervical cancer cells by phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J. Exp. Clin. Cancer Res. 2018, 37, 80. [Google Scholar] [CrossRef]
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Kamijo, H.; Miyagaki, T.; Takahashi-Shishido, N.; Nakajima, R.; Oka, T.; Suga, H.; Sugaya, M.; Sato, S. Thrombospondin-1 promotes tumor progression in cutaneous T-cell lymphoma via CD47. Leukemia 2020, 34, 845–856. [Google Scholar] [CrossRef]
- Morris, S.M., Jr. Enzymes of arginine metabolism. J. Nutr. 2004, 134, 2743S–2767S. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine Signaling and Cancer Metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef] [PubMed]
- Keshet, R.; Lee, J.S.; Adler, L.; Iraqi, M.; Ariav, Y.; Lim, L.Q.J.; Lerner, S.; Rabinovich, S.; Oren, R.; Katzir, R.; et al. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat. Cancer 2020, 1, 894–908. [Google Scholar] [CrossRef]
- Kuo, M.T.; Long, Y.; Tsai, W.B.; Li, Y.Y.; Chen, H.H.W.; Feun, L.G.; Savaraj, N. Collaboration Between RSK-EphA2 and Gas6-Axl RTK Signaling in Arginine Starvation Response That Confers Resistance to EGFR Inhibitors. Transl. Oncol. 2020, 13, 355–364. [Google Scholar] [CrossRef]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 2018, 174, e1522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, Z.; Du, X.; Lin, X.; Liang, Z.M.; Chen, S.; Sun, Y.; Wang, Y.; Na, Z.; Wu, Z.; et al. Cancer cell-derived arginine fuels polyamine biosynthesis in tumor-associated macrophages to promote immune evasion. Cancer Cell 2025, 43, 1045–1060.e7. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Wang, W.; Xia, X.; Mao, L.; Wang, S. The CCAAT/Enhancer-Binding Protein Family: Its Roles in MDSC Expansion and Function. Front. Immunol. 2019, 10, 1804. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Beury, D.W.; Parker, K.H.; Horn, L.A. Survival of the fittest: How myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol. Immunother. 2020, 69, 215–221. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, X.; Ding, T.W.; Gong, Z. Enhanced angiogenesis, hypoxia and neutrophil recruitment during Myc-induced liver tumorigenesis in zebrafish. Sci. Rep. 2016, 6, 31952. [Google Scholar] [CrossRef]
- Hosoi, F.; Izumi, H.; Kawahara, A.; Murakami, Y.; Kinoshita, H.; Kage, M.; Nishio, K.; Kohno, K.; Kuwano, M.; Ono, M. N-myc downstream regulated gene 1/Cap43 suppresses tumor growth and angiogenesis of pancreatic cancer through attenuation of inhibitor of kappaB kinase beta expression. Cancer Res. 2009, 69, 4983–4991. [Google Scholar] [CrossRef]
- Camargo, S.; Moskowitz, O.; Giladi, A.; Levinson, M.; Balaban, R.; Gola, S.; Raizman, A.; Lipczyc, K.; Richter, A.; Keren-Khadmy, N.; et al. Neutrophils physically interact with tumor cells to form a signaling niche promoting breast cancer aggressiveness. Nat. Cancer 2025, 6, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, L.; He, D.; Xie, H.; Chen, J.; Yeh, C.R.; Chang, L.S.; Yeh, S.; Chang, C. Infiltrating neutrophils promote renal cell carcinoma (RCC) proliferation via modulating androgen receptor (AR) → c-Myc signals. Cancer Lett. 2015, 368, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Hashimoto, A.; Markosyan, N.; Tyurin, V.A.; Tyurina, Y.Y.; Kar, G.; Fu, S.; Sehgal, M.; Garcia-Gerique, L.; Kossenkov, A.; et al. Ferroptosis of tumor neutrophils causes immune suppression in cancer. Nature 2022, 612, 338–346. [Google Scholar] [CrossRef]
- Zeng, W.; Zhang, R.; Huang, P.; Chen, M.; Chen, H.; Zeng, X.; Liu, J.; Zhang, J.; Huang, D.; Lao, L. Ferroptotic Neutrophils Induce Immunosuppression and Chemoresistance in Breast Cancer. Cancer Res. 2025, 85, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, C.; Zhao, C.; Li, T.; Ma, L.; Jiang, J.; Duan, Z.; Si, Q.; Chuang, T.H.; Xiang, R.; et al. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduct. Target. Ther. 2024, 9, 64. [Google Scholar] [CrossRef]
- Jin, Y.; Qiu, J.; Lu, X.; Li, G. C-MYC Inhibited Ferroptosis and Promoted Immune Evasion in Ovarian Cancer Cells through NCOA4 Mediated Ferritin Autophagy. Cells 2022, 11, 4127. [Google Scholar] [CrossRef]
- Lin, J.F.; Hu, P.S.; Wang, Y.Y.; Tan, Y.T.; Yu, K.; Liao, K.; Wu, Q.N.; Li, T.; Meng, Q.; Lin, J.Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 54. [Google Scholar] [CrossRef]
- Chen, X.J.; Guo, C.H.; Yang, Y.; Wang, Z.C.; Liang, Y.Y.; Cai, Y.Q.; Cui, X.F.; Fan, L.S.; Wang, W. HPV16 integration regulates ferroptosis resistance via the c-Myc/miR-142-5p/HOXA5/SLC7A11 axis during cervical carcinogenesis. Cell Biosci. 2024, 14, 129. [Google Scholar] [CrossRef]
- Vokshi, B.H.; Davidson, G.; Tawanaie Pour Sedehi, N.; Helleux, A.; Rippinger, M.; Haller, A.R.; Gantzer, J.; Thouvenin, J.; Baltzinger, P.; Bouarich, R.; et al. SMARCB1 regulates a TFCP2L1-MYC transcriptional switch promoting renal medullary carcinoma transformation and ferroptosis resistance. Nat. Commun. 2023, 14, 3034. [Google Scholar] [CrossRef]
- Alborzinia, H.; Flórez, A.F.; Kreth, S.; Brückner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat. Cancer 2022, 3, 471–485. [Google Scholar] [CrossRef]
- Sun, N.; Wang, J.; Qin, J.; Ma, S.; Luan, J.; Hou, G.; Zhang, W.; Gao, M. Oncogenic RTKs sensitize cancer cells to ferroptosis via c-Myc mediated upregulation of ACSL4. Cell Death Dis. 2024, 15, 861. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A.; Cybulska, P.; Mager, K.L.; Koplev, S.; Cast, O.; Couturier, D.L.; Memon, D.; Selenica, P.; Nikolovski, I.; Mazaheri, Y.; et al. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Teo, J.M.N.; Chen, Z.; Chen, W.; Tan, R.J.Y.; Cao, Q.; Chu, Y.; Ma, D.; Chen, L.; Yu, H.; Lam, K.H.; et al. Tumor-associated neutrophils attenuate the immunosensitivity of hepatocellular carcinoma. J. Exp. Med. 2025, 222, e20241442. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Baird, A.M.; Leonard, J.; Naicker, K.M.; Kilmartin, L.; O’Byrne, K.J.; Gray, S.G. IL-23 is pro-proliferative, epigenetically regulated and modulated by chemotherapy in non-small cell lung cancer. Lung Cancer 2013, 79, 83–90. [Google Scholar] [CrossRef]
- Teng, M.W.; Andrews, D.M.; McLaughlin, N.; von Scheidt, B.; Ngiow, S.F.; Möller, A.; Hill, G.R.; Iwakura, Y.; Oft, M.; Smyth, M.J. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 8328–8333. [Google Scholar] [CrossRef]
- Sorrentino, C.; Ciummo, S.L.; D’Antonio, L.; Fieni, C.; Lanuti, P.; Turdo, A.; Todaro, M.; Di Carlo, E. Interleukin-30 feeds breast cancer stem cells via CXCL10 and IL23 autocrine loops and shapes immune contexture and host outcome. J. Immunother. Cancer 2021, 9, e002966. [Google Scholar] [CrossRef]
- Cho, K.M.; Kim, M.S.; Jung, H.J.; Choi, E.J.; Kim, T.S. Mst1-Deficiency Induces Hyperactivation of Monocyte-Derived Dendritic Cells via Akt1/c-myc Pathway. Front. Immunol. 2019, 10, 2142. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, X.; Li, H.; Liu, Y.; Cui, Y.; Tian, L.; Cheng, Y. c-Myc Targets HDAC3 to Suppress NKG2DL Expression and Innate Immune Response in N-Type SCLC through Histone Deacetylation. Cancers 2022, 14, 457. [Google Scholar] [CrossRef]
- Lam, A.R.; Bert, N.L.; Ho, S.S.; Shen, Y.J.; Tang, L.F.; Xiong, G.M.; Croxford, J.L.; Koo, C.X.; Ishii, K.J.; Akira, S.; et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014, 74, 2193–2203. [Google Scholar] [CrossRef]
- Muthalagu, N.; Monteverde, T.; Raffo-Iraolagoitia, X.; Wiesheu, R.; Whyte, D.; Hedley, A.; Laing, S.; Kruspig, B.; Upstill-Goddard, R.; Shaw, R.; et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2020, 10, 872–887. [Google Scholar] [CrossRef]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef]
- Xu, Y.; Poggio, M.; Jin, H.Y.; Shi, Z.; Forester, C.M.; Wang, Y.; Stumpf, C.R.; Xue, L.; Devericks, E.; So, L.; et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 2019, 25, 301–311. [Google Scholar] [CrossRef]
- Vartuli, R.L.; Zhou, H.; Zhang, L.; Powers, R.K.; Klarquist, J.; Rudra, P.; Vincent, M.Y.; Ghosh, D.; Costello, J.C.; Kedl, R.M.; et al. Eya3 promotes breast tumor-associated immune suppression via threonine phosphatase-mediated PD-L1 upregulation. J. Clin. Investig. 2018, 128, 2535–2550. [Google Scholar] [CrossRef]
- Good, C.R.; Aznar, M.A.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, A.K.; Ma, Y.; et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021, 184, 6081–6100.e26. [Google Scholar] [CrossRef]
- Kim, M.; Singh, M.; Lee, B.K.; Hibbs, M.; Richardson, K.; Ellies, L.; Wintle, L.; Stuart, L.M.; Wang, J.Y.; Voon, D.C.; et al. A MYC-ZNF148-ID1/3 regulatory axis modulating cancer stem cell traits in aggressive breast cancer. Oncogenesis 2022, 11, 60. [Google Scholar] [CrossRef]
- Dong, H.; Hu, J.; Wang, L.; Qi, M.; Lu, N.; Tan, X.; Yang, M.; Bai, X.; Zhan, X.; Han, B. SOX4 is activated by C-MYC in prostate cancer. Med. Oncol. 2019, 36, 92. [Google Scholar] [CrossRef]
- Marin, D.; Li, Y.; Basar, R.; Rafei, H.; Daher, M.; Dou, J.; Mohanty, V.; Dede, M.; Nieto, Y.; Uprety, N.; et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+ B-cell tumors: A phase 1/2 trial. Nat. Med. 2024, 30, 772–784. [Google Scholar] [CrossRef]
- Luo, J.; Guo, M.; Huang, M.; Liu, Y.; Qian, Y.; Liu, Q.; Cao, X. Neoleukin-2/15-armored CAR-NK cells sustain superior therapeutic efficacy in solid tumors via c-Myc/NRF1 activation. Signal Transduct. Target. Ther. 2025, 10, 78. [Google Scholar] [CrossRef]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Dong, H.; Adams, N.M.; Xu, Y.; Cao, J.; Allan, D.S.J.; Carlyle, J.R.; Chen, X.; Sun, J.C.; Glimcher, L.H. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 2019, 20, 865–878. [Google Scholar] [CrossRef]
- Schuler, M.; Cuppens, K.; Plönes, T.; Wiesweg, M.; Du Pont, B.; Hegedus, B.; Köster, J.; Mairinger, F.; Darwiche, K.; Paschen, A.; et al. Neoadjuvant nivolumab with or without relatlimab in resectable non-small cell lung cancer: A randomized phase 2 trial. Nat. Med. 2024, 30, 1602–1611. [Google Scholar] [CrossRef]
- Psyrri, A.; Fayette, J.; Harrington, K.; Gillison, M.; Ahn, M.J.; Takahashi, S.; Weiss, J.; Machiels, J.P.; Baxi, S.; Vasilyev, A.; et al. Durvalumab with or without tremelimumab versus the EXTREME regimen as first-line treatment for recurrent or metastatic squamous cell carcinoma of the head and neck: KESTREL, a randomized, open-label, phase III study. Ann. Oncol. 2023, 34, 262–274. [Google Scholar] [CrossRef]
- Cheng, W.; Kang, K.; Zhao, A.; Wu, Y. Dual blockade immunotherapy targeting PD-1/PD-L1 and CTLA-4 in lung cancer. J. Hematol. Oncol. 2024, 17, 54. [Google Scholar] [CrossRef]
- Xu, D.; Wang, H.; Bao, Q.; Jin, K.; Liu, M.; Liu, W.; Yan, X.; Wang, L.; Zhang, Y.; Wang, G.; et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 plus lenvatinib in advanced unresectable or metastatic hepatocellular carcinoma: A phase II trial. Nat. Commun. 2025, 16, 1443. [Google Scholar] [CrossRef]
- Li, Q.; Liu, J.; Zhang, Q.; Ouyang, Q.; Zhang, Y.; Liu, Q.; Sun, T.; Ye, F.; Zhang, B.; Xia, S.; et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 in combination with nab-paclitaxel in first-line treatment of metastatic triple-negative breast cancer: A multicenter phase II trial. Nat. Commun. 2024, 15, 1015. [Google Scholar] [CrossRef]
- Galle, P.; Finn, R.S.; Mitchell, C.R.; Ndirangu, K.; Ramji, Z.; Redhead, G.S.; Pinato, D.J. Treatment-emergent antidrug antibodies related to PD-1, PD-L1, or CTLA-4 inhibitors across tumor types: A systematic review. J. Immunother. Cancer 2024, 12, e008266. [Google Scholar] [CrossRef]
- Fan, Y.; Song, S.; Li, Y.; Dhar, S.S.; Jin, J.; Yoshimura, K.; Yao, X.; Wang, R.; Scott, A.W.; Pizzi, M.P.; et al. Galectin-3 Cooperates with CD47 to Suppress Phagocytosis and T-cell Immunity in Gastric Cancer Peritoneal Metastases. Cancer Res. 2023, 83, 3726–3738. [Google Scholar] [CrossRef]
- Gimple, R.C.; Yang, K.; Halbert, M.E.; Agnihotri, S.; Rich, J.N. Brain cancer stem cells: Resilience through adaptive plasticity and hierarchical heterogeneity. Nat. Rev. Cancer 2022, 22, 497–514. [Google Scholar] [CrossRef]
- Wang, W.; Zou, W. Amino Acids and Their Transporters in T-Cell Immunity and Cancer Therapy. Mol. Cell 2020, 80, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Wu, X.; Wu, Q.; Chatoff, A.; Megill, E.; Gao, J.; Huang, T.; Duan, T.; Yang, K.; Jin, C.; et al. Lysine catabolism reprograms tumor immunity through histone crotonylation. Nature 2023, 617, 818–826. [Google Scholar] [CrossRef]
- Cao, T.; Zhang, W.; Wang, Q.; Wang, C.; Ma, W.; Zhang, C.; Ge, M.; Tian, M.; Yu, J.; Jiao, A.; et al. Cancer SLC6A6-mediated taurine uptake transactivates immune checkpoint genes and induces exhaustion in CD8+ T cells. Cell 2024, 187, 2288–2304.e27. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Guo, Y.; Zhang, S.; Wang, X.; Teng, Y.; Jin, Q.; Jin, Q.; Shen, W.; Wang, R. Solanine Represses Gastric Cancer Growth by Mediating Autophagy Through AAMDC/MYC/ATF4/Sesn2 Signaling Pathway. Drug Des. Dev. Ther. 2023, 17, 389–402. [Google Scholar] [CrossRef]
- Calhoon, D.; Sang, L.; Ji, F.; Bezwada, D.; Hsu, S.C.; Cai, F.; Kim, N.; Basu, A.; Wu, R.; Pimentel, A.; et al. Glycosaminoglycan-driven lipoprotein uptake protects tumours from ferroptosis. Nature 2025. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Omodei, M.S.; Chimicoviaki, J.; Buttros, D.A.B.; Almeida-Filho, B.S.; Carvalho-Pessoa, C.P.; Carvalho-Pessoa, E.; Vespoli, H.L.; Nahas, E.A.P. Vitamin D Supplementation Improves Pathological Complete Response in Breast Cancer Patients Undergoing Neoadjuvant Chemotherapy: A Randomized Clinical Trial. Nutr. Cancer 2025, 77, 648–657. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Soucek, L. MYC in cancer: From undruggable target to clinical trials. Nat. Rev. Drug Discov. 2025, 24, 445–457. [Google Scholar] [CrossRef]
- Boyd, S.R.; Chamakuri, S.; Trostle, A.J.; Chen, H.; Liu, Z.; Jian, A.; Wang, J.; Malovannaya, A.; Young, D.W. MYC-Targeting PROTACs Lead to Bimodal Degradation and N-Terminal Truncation. ACS Chem. Biol. 2025, 20, 896–906. [Google Scholar] [CrossRef]
- Ma, J.; Li, L.; Ma, B.; Liu, T.; Wang, Z.; Ye, Q.; Peng, Y.; Wang, B.; Chen, Y.; Xu, S.; et al. MYC induces CDK4/6 inhibitors resistance by promoting pRB1 degradation. Nat. Commun. 2024, 15, 1871. [Google Scholar] [CrossRef]
- Weber, L.I.; Hartl, M. Strategies to target the cancer driver MYC in tumor cells. Front. Oncol. 2023, 13, 1142111. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.C.; Ahn, K.O.; Yang, C.H. Small-molecule inhibitors of c-Myc transcriptional factor suppress proliferation and induce apoptosis of promyelocytic leukemia cell via cell cycle arrest. Mol. Biosyst. 2010, 6, 1503–1509. [Google Scholar] [CrossRef]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef]
- Guo, J.; Parise, R.A.; Joseph, E.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 2009, 63, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Vogt, P.K.; Boger, D.L.; Lunec, J. Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAX dimerization. Oncol. Rep. 2008, 19, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064. [Google Scholar] [CrossRef]
- Draeger, L.J.; Mullen, G.P. Interaction of the bHLH-zip domain of c-Myc with H1-type peptides. Characterization of helicity in the H1 peptides by NMR. J. Biol. Chem. 1994, 269, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Soodgupta, D.; Pan, D.; Cui, G.; Senpan, A.; Yang, X.; Lu, L.; Weilbaecher, K.N.; Prochownik, E.V.; Lanza, G.M.; Tomasson, M.H. Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol. Cancer Ther. 2015, 14, 1286–1294. [Google Scholar] [CrossRef]
- Hart, J.R.; Garner, A.L.; Yu, J.; Ito, Y.; Sun, M.; Ueno, L.; Rhee, J.K.; Baksh, M.M.; Stefan, E.; Hartl, M.; et al. Inhibitor of MYC identified in a Kröhnke pyridine library. Proc. Natl. Acad. Sci. USA 2014, 111, 12556–12561. [Google Scholar] [CrossRef]
- Stellas, D.; Szabolcs, M.; Koul, S.; Li, Z.; Polyzos, A.; Anagnostopoulos, C.; Cournia, Z.; Tamvakopoulos, C.; Klinakis, A.; Efstratiadis, A. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. J. Natl. Cancer Inst. 2014, 106, dju320. [Google Scholar] [CrossRef]
- Lustig, L.C.; Dingar, D.; Tu, W.B.; Lourenco, C.; Kalkat, M.; Inamoto, I.; Ponzielli, R.; Chan, W.C.W.; Shin, J.A.; Penn, L.Z. Inhibiting MYC binding to the E-box DNA motif by ME47 decreases tumour xenograft growth. Oncogene 2017, 36, 6830–6837. [Google Scholar] [CrossRef]
- Zeng, H.; Yang, H.; Song, Y.; Fang, D.; Chen, L.; Zhao, Z.; Wang, C.; Xie, S. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 suppresses tumor growth and metastasis in esophageal squamous cell carcinoma. Cell Death Dis. 2021, 12, 1048. [Google Scholar] [CrossRef] [PubMed]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; San Martin, M.; Pop-Damkov, P.; Su, N.; Roamio Franklin, V.N.; Sekhar Reddy Chilamakuri, C.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26, 922–934. [Google Scholar] [CrossRef]
- Wang, X.; Yu, B.; Cao, B.; Zhou, J.; Deng, Y.; Wang, Z.; Jin, G. A chemical conjugation of JQ-1 and a TLR7 agonist induces tumoricidal effects in a murine model of melanoma via enhanced immunomodulation. Int. J. Cancer 2021, 148, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Boss, D.S.; Witteveen, P.O.; van der Sar, J.; Lolkema, M.P.; Voest, E.E.; Stockman, P.K.; Ataman, O.; Wilson, D.; Das, S.; Schellens, J.H. Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Ann. Oncol. 2011, 22, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.R.; Adams, C.M.; Wang, J.; Weissmiller, A.M.; Creighton, J.; Lorey, S.L.; Liu, Q.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. USA 2019, 116, 25260–25268. [Google Scholar] [CrossRef]
- Straining, R.; Eighmy, W. Tazemetostat: EZH2 Inhibitor. J. Adv. Pract. Oncol. 2022, 13, 158–163. [Google Scholar] [CrossRef]
- Wasko, U.N.; Jiang, J.; Dalton, T.C.; Curiel-Garcia, A.; Edwards, A.C.; Wang, Y.; Lee, B.; Orlen, M.; Tian, S.; Stalnecker, C.A.; et al. Tumour-selective activity of RAS-GTP inhibition in pancreatic cancer. Nature 2024, 629, 927–936. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Z.; Ming, S.; Luan, X.; Sun, Z.; Zhang, W. Beyond the Limit: MYC Mediates Tumor Immune Escape. Pharmaceuticals 2025, 18, 978. https://doi.org/10.3390/ph18070978
Hong Z, Ming S, Luan X, Sun Z, Zhang W. Beyond the Limit: MYC Mediates Tumor Immune Escape. Pharmaceuticals. 2025; 18(7):978. https://doi.org/10.3390/ph18070978
Chicago/Turabian StyleHong, Zhongyang, Sitong Ming, Xin Luan, Zhe Sun, and Weidong Zhang. 2025. "Beyond the Limit: MYC Mediates Tumor Immune Escape" Pharmaceuticals 18, no. 7: 978. https://doi.org/10.3390/ph18070978
APA StyleHong, Z., Ming, S., Luan, X., Sun, Z., & Zhang, W. (2025). Beyond the Limit: MYC Mediates Tumor Immune Escape. Pharmaceuticals, 18(7), 978. https://doi.org/10.3390/ph18070978