

Supercomputing Multi-Ligand Modeling, Simulation, Wavelet Analysis and Surface Plasmon Resonance to Develop Novel Combination Drugs: A Case Study of Arbidol and Baicalein Against Main Protease of SARS-CoV-2

 ,
,  , , , , and
, , , , and
Abstract

1. Introduction
2. Results
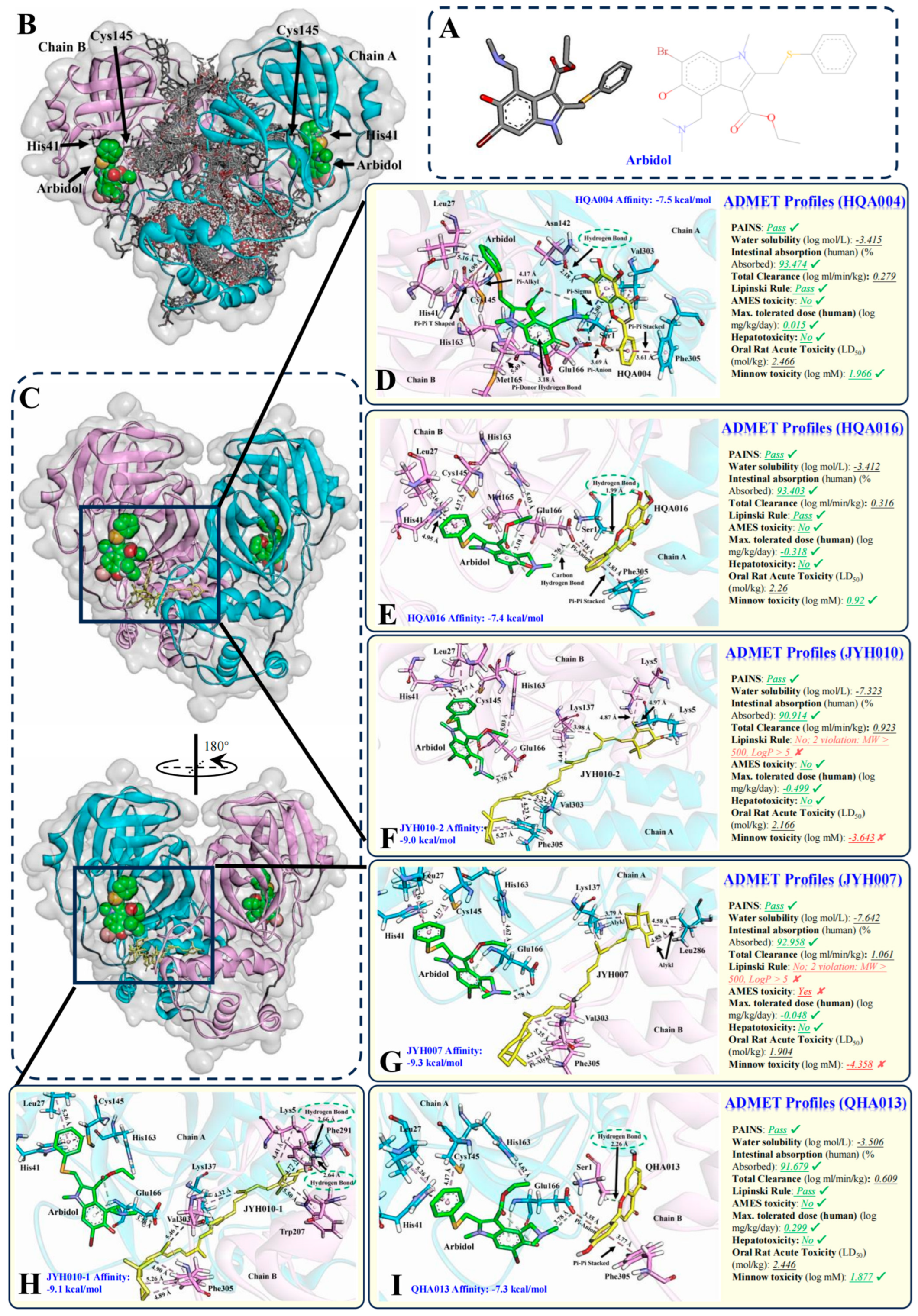
2.1. Five Herbal Compounds Identified from Toujie Quwen Granule Through Multi-Ligand Docking May Have Synergistic Effects with Arbidol
2.2. HQA004, HQA016 and QHA013 Passed the Pharmacokinetic and Toxicity Screening
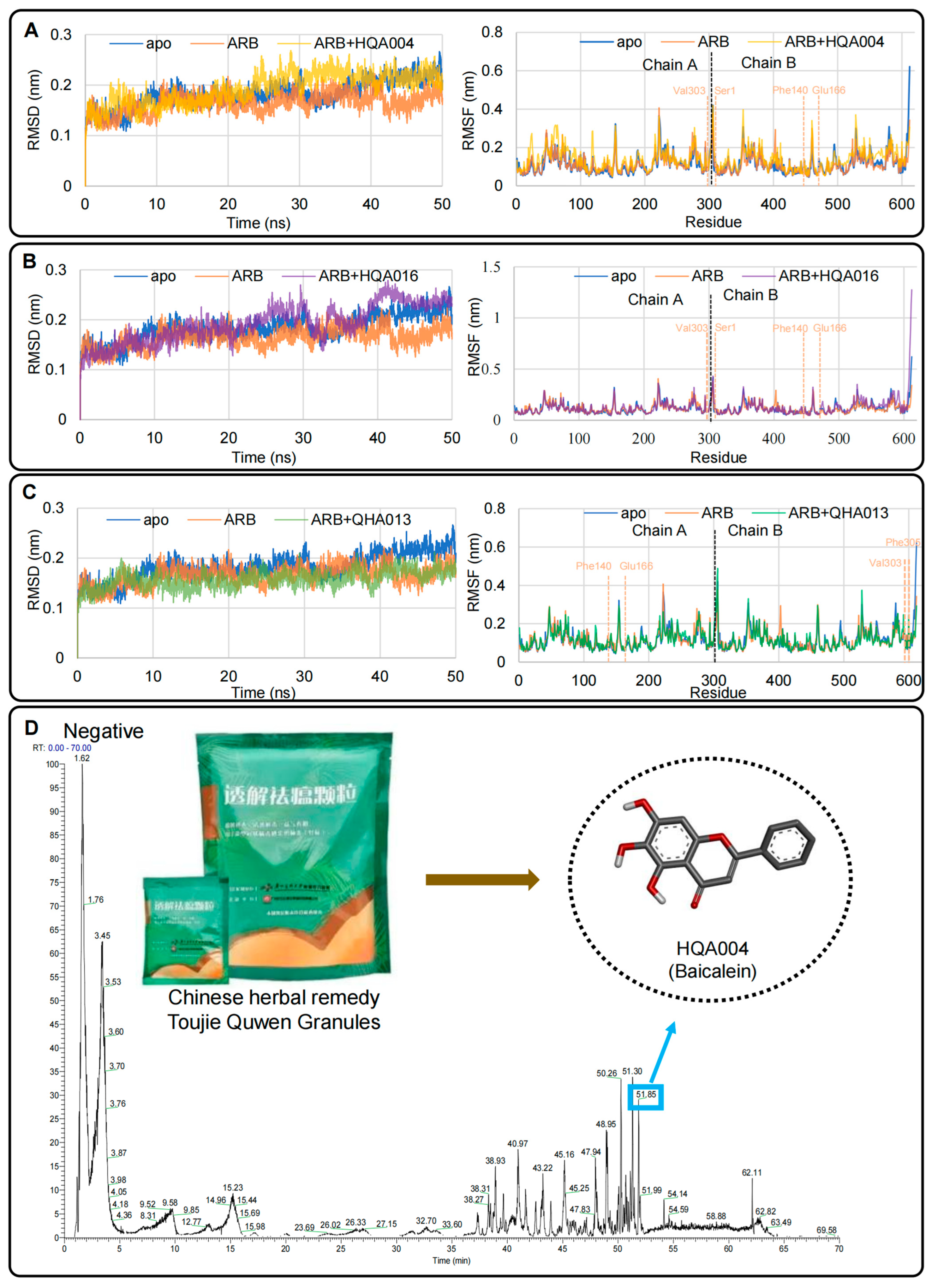
2.3. Preliminary Molecular Dynamics Simulations Indicated Potential Synergistic Effects of HQA004, HQA016, and QHA013 with Arbidol Within 50 ns
2.4. Untargeted Metabolomics Confirmed HQA004 (Baicalein) Was the Only Active Component Identified from the Mixture of Toujie Quwen Granules Among the Three Candidates
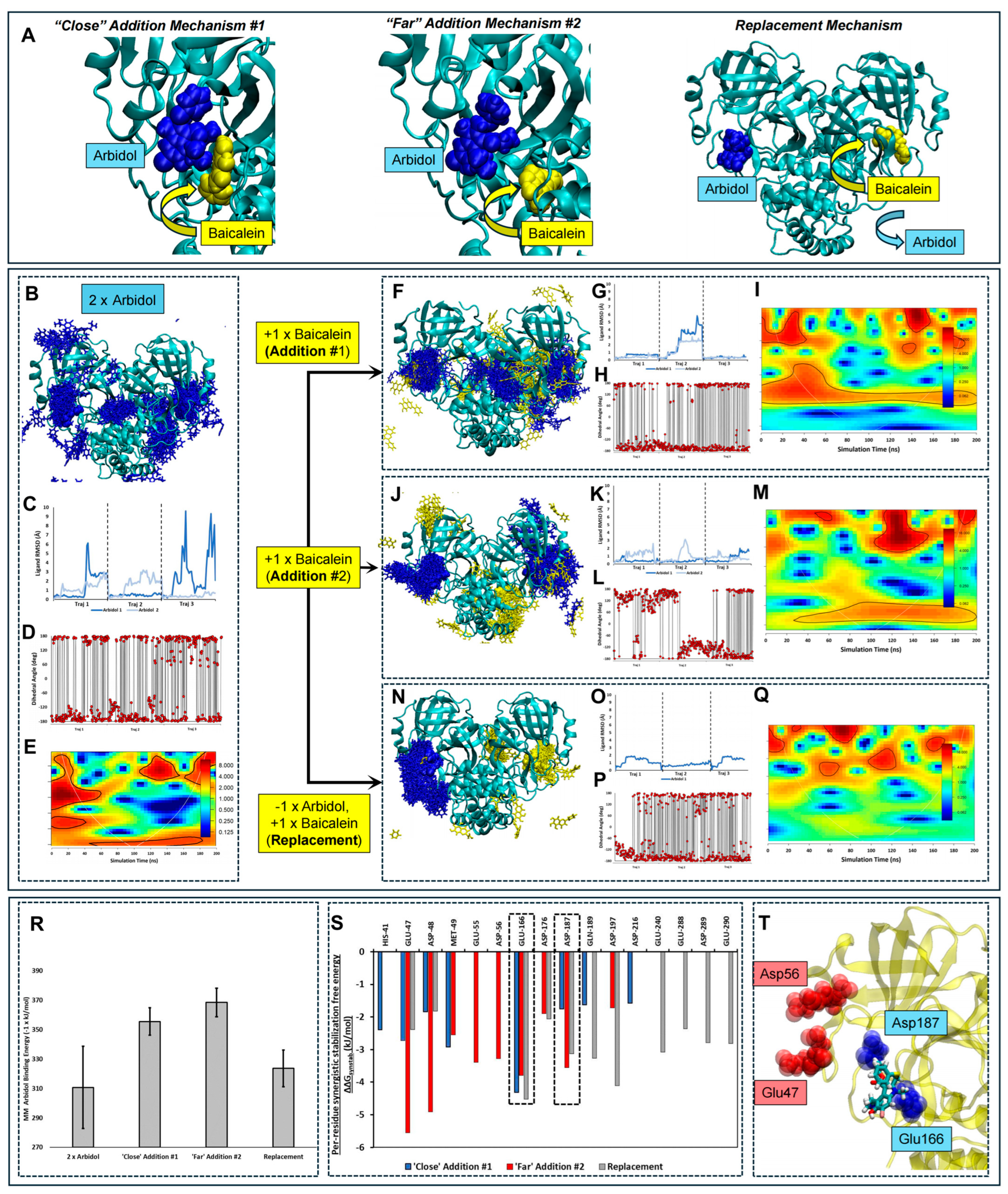
2.5. Extended Molecular Dynamics Simulations Elucidated Synergism Between Herbal Compound HQA004 (Baicalein) and Arbidol: Proposed Synergistic Mechanisms
- -
- Figure 3A (left) shows “Close” Addition Mechanism #1, where HQA004 is initially docked in close proximity to ARB, forming several inter-atomic contacts. This pose is adapted directly from the automated docking study described above. This pose corresponds to a 2:1 stoichiometric ratio of ARB to HQA004.
- -
- Figure 3A (center) shows “Far” Addition Mechanism #2, where HQA004 is initially docked at a slightly more distant position compared to the first proposed mechanism. This mechanism was explored to predict whether a greater synergism could be achieved if a supporting ligand was close enough to produce additional stability to the binding site while avoiding direct close-contact atomic clashes, which may paradoxically destabilize the main ARB ligand. Our previous manuscript on a similar approach suggested that a nominally synergistic ligand may interact repulsively with the main ligand [8,17]. This pose also corresponds to a 2:1 stoichiometric ratio of ARB to HQA004.
- -
- Figure 3A (right) shows the Replacement Mechanism where ARB and HQA004 bind to the two known active sites on separate monomers; thus, the supporting ligand is proposed to displace a previously bound ARB. This pose enables the exploration of the impact of HQA004 binding at an opposing monomer on the stability of ARB. This pose corresponds to a 1:1 stoichiometric ratio of ARB to HQA004.
2.6. HQA004 (Baicalein) Stabilizes the Binding of Arbidol by Reducing Its Diffusion and Intramolecular Conformational Shifts
2.7. A 1:1 Ratio of HQA004 and Arbidol Decouples the Movements Between Monomers and Results in Higher Frequency Structural Oscillation
2.8. HQA004 (Baicalein) Promotes Arbidol Interaction with Acidic Mpro Residues in a Binding Position-Dependent Manner
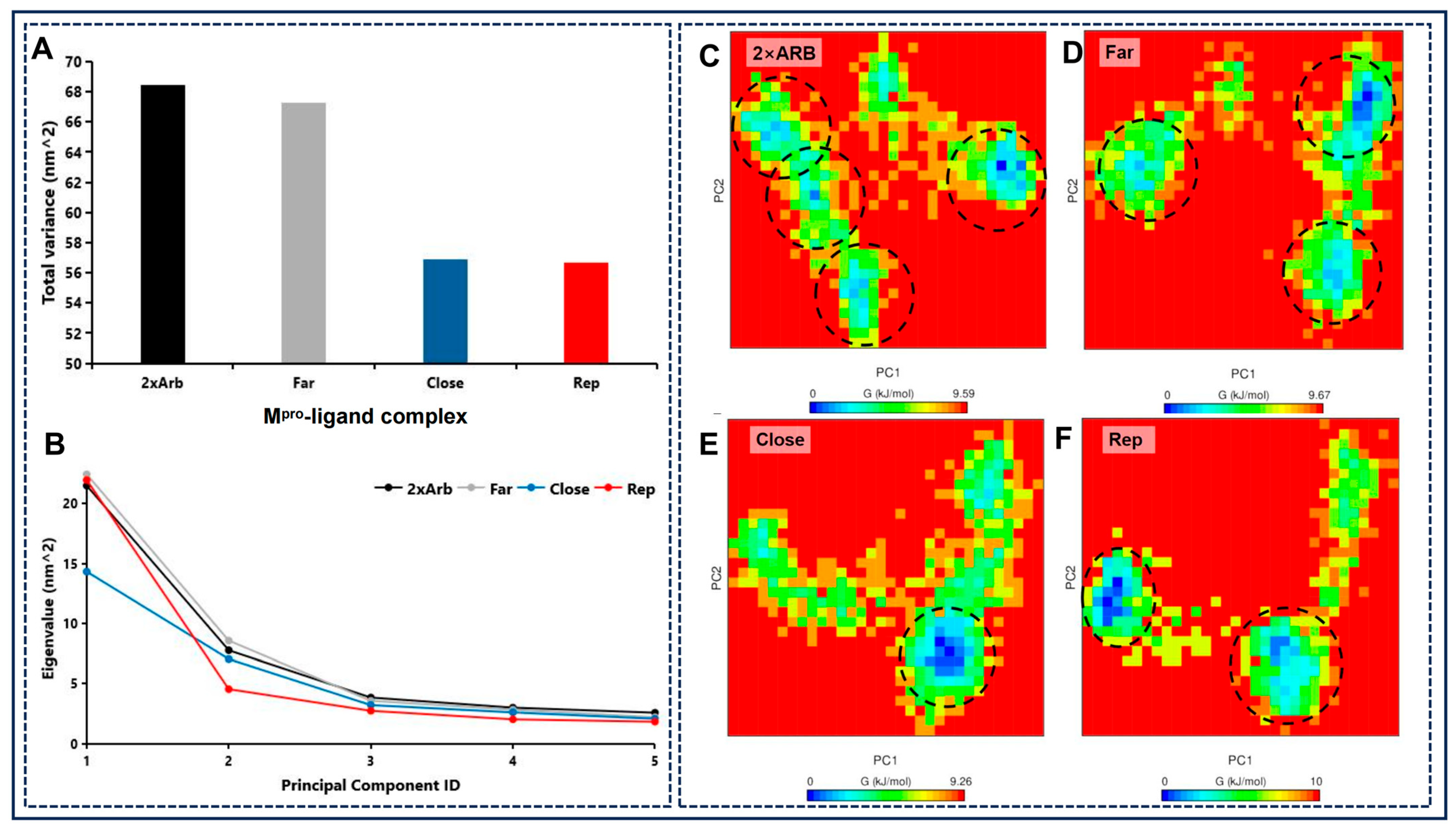
2.9. Principal Component Analysis and Free Energy Landscape Analyses Reveal HQA004 (Baicalein) Binding Enhances Mpro Dimer Rigidity and Stability
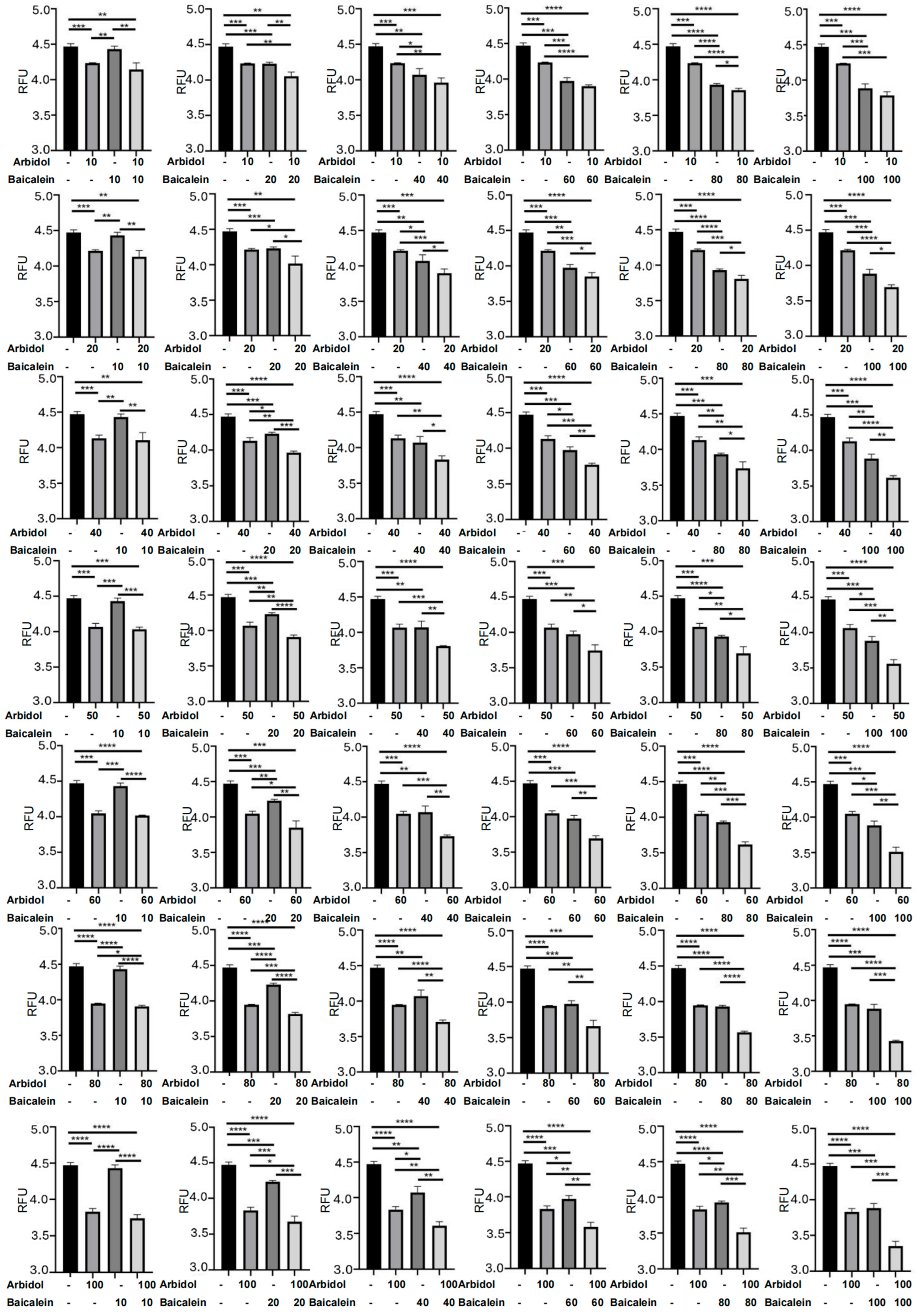
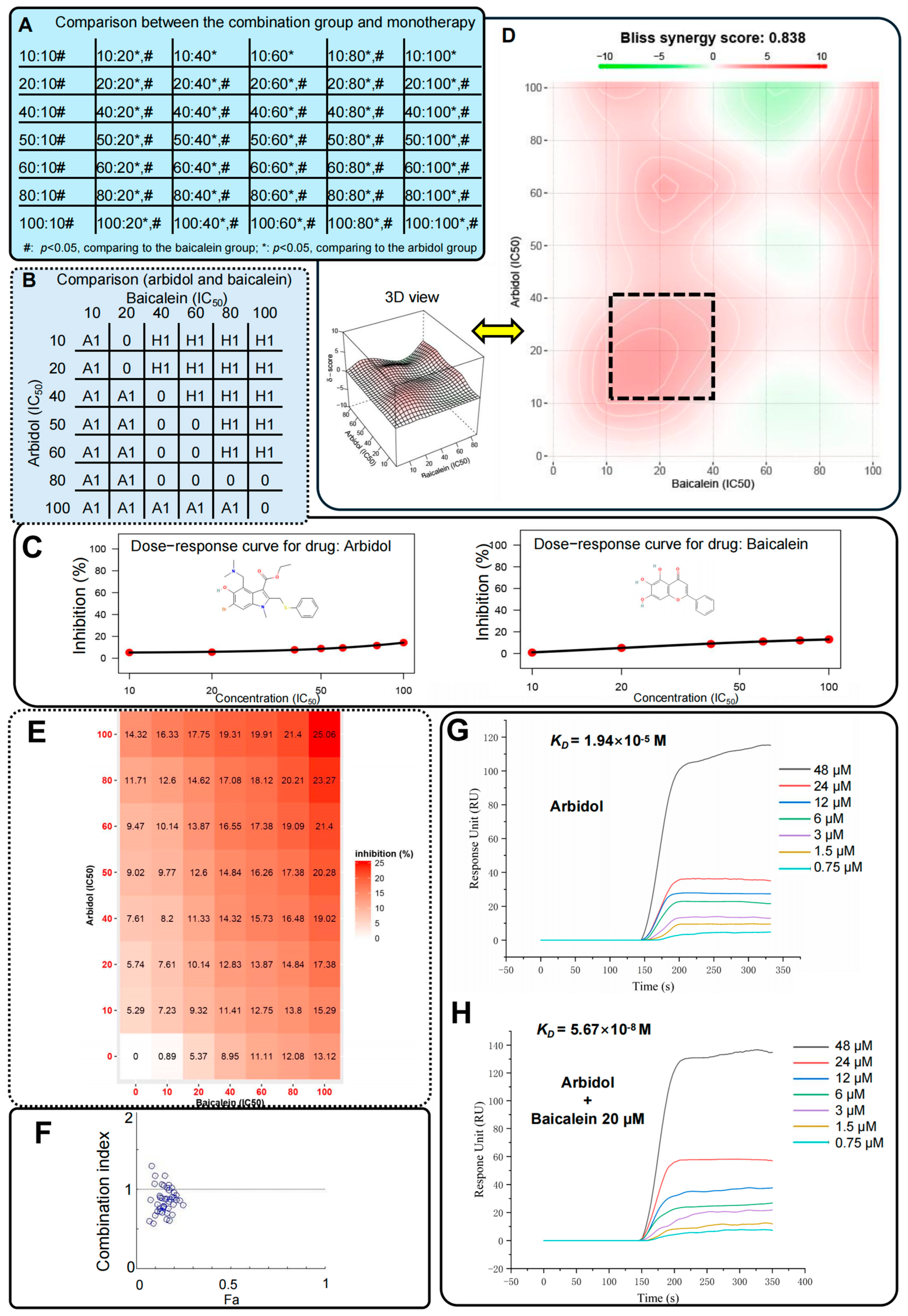
2.10. Luciferase Assays and Surface Plasmon Resonance Approach Clearly Reflected the Synergistic Effects of Arbidol and HQA004 (Baicalein) for Multiple Combination Ratios
3. Discussion
4. Materials and Methods
4.1. Database Search to Identify Compounds from Toujie Quwen Granules
4.2. Acquisition of Structures of Identified Ligands
4.3. Structural Preparation of the Main Protease of SARS-CoV-2
4.4. Multiple Ligands Docking to Investigate Cooperativity Between Arbidol and Compounds from Toujie Quwen Granule
4.5. Virtual Pharmacokinetic and Toxicity Prediction
4.6. Metabolomics Profiling to Analyze the Active Components of Toujie Quwen Granules
4.7. Molecular Dynamics Simulations and Analyses
4.8. Luciferase Assays
4.9. Synergistic Effects Evaluation
4.10. Surface Plasmon Resonance Approaches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADME/T | Absorption, distribution, metabolism, excretion, and toxicity |
| ARB | Arbidol |
| COVID-19 | Coronavirus disease |
| CI | Combination index |
| fa | Fraction affected |
| ΔΔGsynstab | Synergistic stabilization energy |
| H-bonds | Hydrogen bonds |
| MD | Molecular dynamics |
| MM-PBSA | Molecular Mechanics Poisson-Boltzmann Surface Area |
| Mpro | Main protease |
| PAINS | Pan-Assay Interference Compounds |
| RMSD | Root mean square deviation of backbone Cɑ atoms |
| RMSF | Root mean square fluctuation |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SMMM | Structure-based multi-ligand molecular modeling |
| TCM | Traditional Chinese medicine |
| TCMSP | Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform |
| TQG | Toujie Quwen Granules |
References
- Collier, R. Reducing the “pill burden”. CMAJ 2012, 184, E117–E118. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jia, W.; Yang, J.; Cheng, C.; Olaleye, O.E. Multi-compound and drug-combination pharmacokinetic research on Chinese herbal medicines. Acta Pharmacol. Sin. 2022, 43, 3080–3095. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Curran, M.P. Amlodipine/atorvastatin: A review of its use in the treatment of hypertension and dyslipidaemia and the prevention of cardiovascular disease. Drugs 2012, 70, 191–213. [Google Scholar] [CrossRef]
- Diener, H.C.; Jansen, J.P.; Reches, A.; Pascual, J.; Pitei, D.; Steiner, T.J. Efficacy, tolerability and safety of oral eletriptan and ergotamine plus caffeine (Cafergot) in the acute treatment of migraine: A multicentre, randomised, double-blind, placebo-controlled comparison. Eur. Neurol. 2002, 47, 99–107. [Google Scholar] [CrossRef]
- Wen, W.; Chen, C.; Tang, J.; Wang, C.; Zhou, M.; Cheng, Y.; Zhou, X.; Wu, Q.; Zhang, X.; Feng, Z.; et al. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: A meta-analysis. Ann. Med. 2022, 54, 516–523. [Google Scholar] [CrossRef]
- Li, H.; Komori, A.; Li, M.; Chen, X.; Yang, A.W.H.; Sun, X.; Liu, Y.; Hung, A.; Zhao, X.; Zhou, L. Multi-ligand molecular docking, simulation, free energy calculations and wavelet analysis of the synergistic effects between natural compounds baicalein and cubebin for the inhibition of the main protease of SARS-CoV-2. J. Mol. Liq. 2023, 374, 121253. [Google Scholar] [CrossRef]
- Ni, L.; Chen, L.; Huang, X.; Han, C.; Xu, J.; Zhang, H.; Luan, X.; Zhao, Y.; Xu, J.; Yuan, W.; et al. Combating COVID-19 with integrated traditional Chinese and Western medicine in China. Acta Pharm. Sin. B 2020, 10, 1149–1162. [Google Scholar] [CrossRef]
- Wang, W.Y.; Zhou, H.; Wang, Y.F.; Sang, B.S.; Liu, L. Current policies and measures on the development of traditional Chinese medicine in China. Pharmacol. Res. 2021, 163, 105187. [Google Scholar] [CrossRef]
- Li, H.; Hung, A.; Li, M.; Lu, L.; Yang, A.W.H. Phytochemistry, pharmacodynamics, and pharmacokinetics of a classic Chinese herbal formula Danggui Beimu Kushen Wan: A review. Phytother. Res. 2021, 35, 3673–3689. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Wang, Y.P.; Xie, Y.L.; Tian, G.H.; Zhang, X.Y.; Shi, N.N.; Yang, K.H.; Sun, X.; Chen, Y.L.; Wu, D.R.; et al. Research on the development methodology for clinical practice guidelines for organic integration of traditional Chinese and Western medicine. Mil. Med. Res. 2023, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Li, Z.; Chen, L.; Zeng, J.; Zhao, S.; Tang, Y.; Huang, B.; Guan, H.; Chen, Y.; Feng, Y.; et al. The comparative effects of oral Chinese patent medicines combined with western medicine in stable angina: A systematic review and network meta-analysis of 179 trials. Front. Pharmacol. 2022, 13, 918689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, H.; Zhen, D.; Fu, D.; Zhao, M.; Wei, C.; Bai, X. Comparative clinical-related outcomes of Chinese patent medicines for cardiac hypertrophy: A systematic review and network meta-analysis of randomized clinical trials. Front. Pharmacol. 2023, 14, 963099. [Google Scholar] [CrossRef]
- Li, J.; McKay, K.T.; Remington, J.M.; Schneebeli, S.T. A computational study of cooperative binding to multiple SARS-CoV-2 proteins. Sci. Rep. 2021, 11, 16307. [Google Scholar] [CrossRef]
- Mattio, L.M.; Marengo, M.; Parravicini, C.; Eberini, I.; Dallavalle, S.; Bonomi, F.; Iametti, S.; Pinto, A. Inhibition of pancreatic α-amylase by resveratrol derivatives: Biological activity and molecular modelling evidence for cooperativity between Viniferin Enantiomers. Molecules 2019, 24, 3225. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, X.; Chen, X.; You, Y.; Yang, S.; Wu, C.; Chen, M.; Li, M.; Komori, A.; Liu, Y.; et al. Structure-based multi-ligand molecular modeling to predict the synergistic effects of limonin and obacunone from Simiao pill against nitric oxide synthase 3 associated with hyperuricemia. Precis. Med. Res. 2023, 5, 12–17. [Google Scholar] [CrossRef]
- National Computational Infrastructure. HPC System. Available online: https://nci.org.au/our-systems/hpc-systems (accessed on 18 February 2022).
- Heidari, Z.; Roe, D.R.; Galindo-Murillo, R.; Ghasemi, J.B.; Cheatham, T.E., III. Using wavelet analysis to assist in identification of significant events in molecular dynamics simulations. J. Chem. Inf. Model. 2016, 56, 1282–1291. [Google Scholar] [CrossRef]
- Zheng, J. SARS-CoV-2: An emerging coronavirus that causes a global threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 21 June 2025).
- Baj, J.; Karakuła-Juchnowicz, H.; Teresiński, G.; Buszewicz, G.; Ciesielka, M.; Sitarz, R.; Forma, A.; Karakuła, K.; Flieger, W.; Portincasa, P.; et al. COVID-19: Specific and non-specific clinical manifestations and symptoms: The current state of knowledge. J. Clin. Med. 2020, 9, 1753. [Google Scholar] [CrossRef]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Patidar, R.; Younis, K.; Desai, P.; Hosein, Z.; Padda, I.; Mangat, J.; Altaf, M. Comorbidity and its impact on patients with COVID-19. SN Compr. Clin. Med. 2020, 2, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.C.; Silva, E.N.; Soares, L.M.; Scodeler, G.C.; Santos, A.S.; Corsetti, P.P.; Prudêncio, C.R.; de Almeida, L.A. Different drug approaches to COVID-19 treatment worldwide: An update of new drugs and drugs repositioning to fight against the novel coronavirus. Ther. Adv. Vaccines Immunother. 2022, 10, 25151355221144845. [Google Scholar] [CrossRef] [PubMed]
- Gudima, G.; Kofiadi, I.; Shilovskiy, I.; Kudlay, D.; Khaitov, M. Antiviral therapy of COVID-19. Int. J. Mol. Sci. 2023, 24, 8867. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Kang, Y.; Shi, Y.; Xu, S. Arbidol: The current demand, strategies, and antiviral mechanisms. Immun. Inflamm. Dis. 2023, 11, e984. [Google Scholar] [CrossRef]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. SARS-CoV-2: Recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr. Med. Chem. 2020, 27, 4536–4541. [Google Scholar] [CrossRef]
- Gao, W.; Chen, S.; Wang, K.; Chen, R.; Guo, Q.; Lu, J.; Wu, X.; He, Y.; Yan, Q.; Wang, S.; et al. Clinical features and efficacy of antiviral drug, arbidol in 220 nonemergency COVID-19 patients from East-West-Lake Shelter Hospital in Wuhan: A retrospective case series. Virol. J. 2020, 17, 162. [Google Scholar] [CrossRef]
- Nojomi, M.; Yassin, Z.; Keyvani, H.; Makiani, M.J.; Roham, M.; Laali, A.; Dehghan, N.; Navaei, M.; Ranjbar, M. Effect of Arbidol (Umifenovir) on COVID-19: A randomized controlled trial. BMC Infect. Dis. 2020, 20, 954. [Google Scholar] [CrossRef]
- Amani, B.; Amani, B.; Zareei, S.; Zareei, M. Efficacy and safety of arbidol (umifenovir) in patients with COVID-19: A systematic review and meta-analysis. Immun. Inflamm. Dis. 2021, 9, 1197–1208. [Google Scholar] [CrossRef]
- Li, Y.; Xie, Z.; Lin, W.; Cai, W.; Wen, C.; Guan, Y.; Mo, X.; Wang, J.; Wang, Y.; Peng, P.; et al. Efficacy and safety of lopinavir/ritonavir or arbidol in adult patients with mild/moderate COVID-19: An exploratory randomized controlled trial. Medicine 2020, 1, 105–113.e4. [Google Scholar] [CrossRef]
- Ye, M.; Luo, G.; Ye, D.; She, M.; Sun, N.; Lu, Y.J.; Zheng, J. Network pharmacology, molecular docking integrated surface plasmon resonance technology reveals the mechanism of Toujie Quwen Granules against coronavirus disease 2019 pneumonia. Phytomedicine 2021, 85, 153401. [Google Scholar] [CrossRef]
- Banerjee, S.; Baidya, S.K.; Adhikari, N.; Ghosh, B.; Jha, T. Glycyrrhizin as a promising kryptonite against SARS-CoV-2: Clinical, experimental, and theoretical evidences. J. Mol. Struct. 2023, 1275, 134642. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, W.J.; Ni, Y.S.; Li, M.S.; Chen, J.K.; Liu, X.H.; Tan, X.H.; Li, J.Q. Therapeutic mechanism of Toujie Quwen granules in COVID-19 based on network pharmacology. BioData Min. 2020, 13, 15. [Google Scholar] [CrossRef]
- Fu, X.; Lin, L.; Tan, X. Clinical study on 37 cases of COVID-19 treated with integrated traditional Chinese and Western Medicine. Ther. Adv. Vaccines Immunother. 2020, 31, 600–604. [Google Scholar] [CrossRef]
- Wu, C.; Wong, A.R.; Chen, Q.; Yang, S.; Chen, M.; Sun, X.; Zhou, L.; Liu, Y.; Yang, A.W.H.; Bi, J.; et al. Identification of inhibitors from a functional food-based plant Perillae Folium against hyperuricemia via metabolomics profiling, network pharmacology and all-atom molecular dynamics simulations. Front. Endocrinol. 2024, 15, 1320092. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Alheety, M.A.; Srivastava, V. Molecular docking and ADMET study of spice-derived potential phytochemicals against human DNA Topoisomerase III Alpha. Macromol. Symp. 2023, 407, 2200108. [Google Scholar] [CrossRef]
- Hatamipour, M.; Ramezani, M.; Tabassi, S.A.S.; Johnston, T.P.; Sahebkar, A. Demethoxycurcumin: A naturally occurring curcumin analogue for treating non-cancerous diseases. J. Cell. Physiol. 2019, 234, 19320–19330. [Google Scholar] [CrossRef]
- Yalcin-Ozkat, G. Molecular modeling strategies of cancer multidrug resistance. Drug Resist. Updat. 2021, 59, 100789. [Google Scholar] [CrossRef]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y.; et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminform. 2014, 6, 13. [Google Scholar] [CrossRef]
- Li, H.; Hung, A.; Yang, A.W.H. Herb-target virtual screening and network pharmacology for prediction of molecular mechanism of Danggui Beimu Kushen Wan for prostate cancer. Sci. Rep. 2021, 11, 6656. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Marincola, F.M. Combination therapy: The next opportunity and challenge of medicine. J. Transl. Med. 2011, 9, 115. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Makhoba, X.H.; Viegas, C., Jr.; Mosa, R.A.; Viegas, F.P.D.; Pooe, O.J. Potential impact of the multi-target drug approach in the treatment of some complex diseases. Drug Des. Devel. Ther. 2020, 14, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, evaluation, and treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Xu, J.; Yang, Y. Traditional Chinese medicine in the Chinese health care system. Health Policy 2009, 90, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Bi, Y.M.; Sun, L.; Huang, J.Z.; Zhao, J.; Yao, J.; Li, A.X.; Wang, X.Z.; Fan, G.J. Efficacy of integrated traditional Chinese and Western medicine for treating COVID-19: A systematic review and meta-analysis of RCTs. Front. Public Health 2021, 9, 622707. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The development of Coronavirus 3C-Like protease (3CL(pro)) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Hu, H.; Gao, M.; et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzyme. Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Duchon, A.; Nikolaitchik, O.A.; Pathak, V.K.; Hu, W.S. Development of a cell-based luciferase complementation assay for identification of SARS-CoV-2 3CL(pro) inhibitors. Viruses 2021, 13, 173. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Pitsillou, E.; Karagiannis, C.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: Molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. [Google Scholar] [CrossRef]
- Liang, J.; Pitsillou, E.; Ververis, K.; Guallar, V.; Hung, A.; Karagiannis, T.C. Small molecule interactions with the SARS-CoV-2 main protease: In silico all-atom microsecond MD simulations, PELE Monte Carlo simulations, and determination of in vitro activity inhibition. J. Mol. Graph. Model. 2022, 110, 108050. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, X.; Chen, X.; Li, M.; Komori, A.; Sun, X.; Liu, Y.; Wei Hong Yang, A.; Hung, A.; Zhao, X.; et al. Computational biomedical modeling and screening for prediction of molecular mechanisms of Simiao Pill against hyperuricemia. J. Mol. Liq. 2023, 381, 121827. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.C.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- da Sousa Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Roberts, E.; Eargle, J.; Wright, D.; Luthey-Schulten, Z. MultiSeq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinform. 2006, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Konecny, R.; Baker, N.A.; McCammon, J.A. iAPBS: A programming interface to Adaptive Poisson-Boltzmann Solver (APBS). Comput. Sci. Discov. 2012, 5, 015005. [Google Scholar] [CrossRef]
- Baidya, S.K.; Banerjee, S.; Ghosh, B.; Jha, T.; Adhikari, N. Assessing structural insights into in-house arylsulfonyl L-(+) glutamine MMP-2 inhibitors as promising anticancer agents through structure-based computational modelling approaches. SAR QSAR Environ. Res. 2023, 34, 805–830. [Google Scholar] [CrossRef]
- Banerjee, S.; Baidya, S.K.; Ghosh, B.; Jha, T.; Adhikari, N. Exploring the key structural attributes and chemico-biological interactions of pyridinone-based SARS-CoV-2 3CL(pro) inhibitors through validated structure-based drug design strategies. Heliyon 2024, 10, e40404. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, E.; Cai, C.; Zhang, M.; Li, D.; Cai, S.; Zeng, G.; Yin, Z.; Wang, B.; Li, P.; et al. Combined treatment of tanshinone I and epirubicin revealed enhanced inhibition of hepatocellular carcinoma by targeting PI3K/AKT/HIF-1α. Drug Des. Devel. Ther. 2022, 16, 3197–3213. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Gautam, P.; Kononov, A.; Potdar, S.; Saarela, J.; Wennerberg, K.; Aittokallio, T. Prediction of drug combination effects with a minimal set of experiments. Nat. Mach. Intell. 2019, 1, 568–577. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, J.; Shen, F.; Ye, L.; Cheng, C.; Li, Y.; Zhang, Q.; Niu, L.; Hou, Y.; Bai, G. A novel mode of action for COX-2 inhibition: Targeting ATPase domain of HSP90 induces ubiquitin degradation of new client protein COX-2. Clin. Transl. Med. 2022, 12, e705. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug A | Drug B | Drug A Dosage (IC50) | Drug B Dosage (IC50) | Drug A Dosage (μM) | Drug B Dosage (μM) | Effects | Chou-Talalay CI | Bliss Synergy Score | Ratio (IC50) | Ratio (μM) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arbidol | Baicalein | 10 | 10 | 160 | 31.2 | 0.072 | 0.60062 | 2.000854 | 1:1 | 200:39 | ≈ | 5:1 |
| Arbidol | Baicalein | 10 | 20 | 160 | 62.4 | 0.093 | 0.56821 | 1.810456 | 1:2 | 100:39 | ≈ | 3:1 |
| Arbidol | Baicalein | 10 | 40 | 160 | 124.8 | 0.114 | 0.72318 | 0.359348 | 1:4 | 50:39 | ≈ | 1:1 |
| Arbidol | Baicalein | 10 | 60 | 160 | 187.2 | 0.128 | 0.8929 | −0.43471 | 1:6 | 100:117 | ≈ | 1:1 |
| Arbidol | Baicalein | 10 | 80 | 160 | 249.6 | 0.138 | 1.06439 | −0.329 | 1:8 | 25:39 | ≈ | 1:1 |
| Arbidol | Baicalein | 10 | 100 | 160 | 312 | 0.153 | 1.16796 | 0.159993 | 1:10 | 20:39 | ≈ | 1:1 |
| Arbidol | Baicalein | 20 | 10 | 320 | 31.2 | 0.076 | 0.86929 | 1.928658 | 2:1 | 400:39 | ≈ | 10:1 |
| Arbidol | Baicalein | 20 | 20 | 320 | 62.4 | 0.101 | 0.67028 | 2.203209 | 1:1 | 200:39 | ≈ | 5:1 |
| Arbidol | Baicalein | 20 | 40 | 320 | 124.8 | 0.128 | 0.72071 | 1.388058 | 1:2 | 100:39 | ≈ | 3:1 |
| Arbidol | Baicalein | 20 | 60 | 320 | 187.2 | 0.139 | 0.88779 | 0.29493 | 1:3 | 200:117 | ≈ | 2:1 |
| Arbidol | Baicalein | 20 | 80 | 320 | 249.6 | 0.148 | 1.04871 | 0.322774 | 1:4 | 50:39 | ≈ | 1:1 |
| Arbidol | Baicalein | 20 | 100 | 320 | 312 | 0.174 | 1.05344 | 1.89968 | 1:5 | 40:39 | ≈ | 1:1 |
| Arbidol | Baicalein | 40 | 10 | 640 | 31.2 | 0.082 | 1.29173 | 0.608507 | 4:1 | 800:39 | ≈ | 21:1 |
| Arbidol | Baicalein | 40 | 20 | 640 | 62.4 | 0.113 | 0.81097 | 1.548198 | 2:1 | 400:39 | ≈ | 10:1 |
| Arbidol | Baicalein | 40 | 40 | 640 | 124.8 | 0.143 | 0.76168 | 1.113669 | 1:1 | 200:39 | ≈ | 5:1 |
| Arbidol | Baicalein | 40 | 60 | 640 | 187.2 | 0.157 | 0.87332 | 0.445113 | 2:3 | 400:117 | ≈ | 3:1 |
| Arbidol | Baicalein | 40 | 80 | 640 | 249.6 | 0.165 | 1.01788 | 0.265352 | 1:2 | 100:39 | ≈ | 3:1 |
| Arbidol | Baicalein | 40 | 100 | 640 | 312 | 0.19 | 1.01695 | 1.862945 | 2:5 | 80:39 | ≈ | 2:1 |
| Arbidol | Baicalein | 50 | 10 | 800 | 31.2 | 0.098 | 1.06719 | 0.773526 | 5:1 | 1000:39 | ≈ | 26:1 |
| Arbidol | Baicalein | 50 | 20 | 800 | 62.4 | 0.126 | 0.75942 | 1.438732 | 5:2 | 500:39 | ≈ | 13:1 |
| Arbidol | Baicalein | 50 | 40 | 800 | 124.8 | 0.148 | 0.7872 | 0.284372 | 5:4 | 250:39 | ≈ | 6:1 |
| Arbidol | Baicalein | 50 | 60 | 800 | 187.2 | 0.163 | 0.87914 | −0.34147 | 5:6 | 500:117 | ≈ | 4:1 |
| Arbidol | Baicalein | 50 | 80 | 800 | 249.6 | 0.174 | 0.99087 | −0.12508 | 5:8 | 125:39 | ≈ | 3:1 |
| Arbidol | Baicalein | 50 | 100 | 800 | 312 | 0.203 | 0.9618 | 1.859403 | 1:2 | 100:39 | ≈ | 3:1 |
| Arbidol | Baicalein | 60 | 10 | 960 | 31.2 | 0.101 | 1.16692 | 0.691017 | 6:1 | 400:13 | ≈ | 31:1 |
| Arbidol | Baicalein | 60 | 20 | 960 | 62.4 | 0.139 | 0.7056 | 2.2956 | 3:1 | 200:13 | ≈ | 15:1 |
| Arbidol | Baicalein | 60 | 40 | 960 | 124.8 | 0.166 | 0.70144 | 1.612178 | 3:2 | 100:13 | ≈ | 8:1 |
| Arbidol | Baicalein | 60 | 60 | 960 | 187.2 | 0.174 | 0.84117 | 0.388165 | 1:1 | 200:39 | ≈ | 5:1 |
| Arbidol | Baicalein | 60 | 80 | 960 | 249.6 | 0.191 | 0.90367 | 1.217708 | 3:4 | 50:13 | ≈ | 4:1 |
| Arbidol | Baicalein | 60 | 100 | 960 | 312 | 0.214 | 0.92087 | 2.598663 | 3:5 | 40:13 | ≈ | 3:1 |
| Arbidol | Baicalein | 80 | 10 | 1280 | 31.2 | 0.126 | 0.91745 | 0.917917 | 8:1 | 1600:39 | ≈ | 41:1 |
| Arbidol | Baicalein | 80 | 20 | 1280 | 62.4 | 0.146 | 0.77646 | 0.814347 | 4:1 | 800:39 | ≈ | 21:1 |
| Arbidol | Baicalein | 80 | 40 | 1280 | 124.8 | 0.171 | 0.75828 | −0.01067 | 2:1 | 400:39 | ≈ | 10:1 |
| Arbidol | Baicalein | 80 | 60 | 1280 | 187.2 | 0.181 | 0.87029 | −0.96663 | 4:3 | 800:117 | ≈ | 7:1 |
| Arbidol | Baicalein | 80 | 80 | 1280 | 249.6 | 0.202 | 0.89239 | 0.277942 | 1:1 | 200:39 | ≈ | 5:1 |
| Arbidol | Baicalein | 80 | 100 | 1280 | 312 | 0.233 | 0.85803 | 2.457203 | 4:5 | 160:39 | ≈ | 4:1 |
| Arbidol | Baicalein | 100 | 10 | 1600 | 31.2 | 0.163 | 0.61863 | 2.07305 | 10:1 | 2000:39 | ≈ | 51:1 |
| Arbidol | Baicalein | 100 | 20 | 1600 | 62.4 | 0.177 | 0.60655 | 1.415308 | 5:1 | 1000:39 | ≈ | 26:1 |
| Arbidol | Baicalein | 100 | 40 | 1600 | 124.8 | 0.193 | 0.67776 | −0.23855 | 5:2 | 500:39 | ≈ | 13:1 |
| Arbidol | Baicalein | 100 | 60 | 1600 | 187.2 | 0.199 | 0.80732 | −1.58835 | 5:3 | 1000:117 | ≈ | 9:1 |
| Arbidol | Baicalein | 100 | 80 | 1600 | 249.6 | 0.214 | 0.86781 | −0.93566 | 5:4 | 250:39 | ≈ | 6:1 |
| Arbidol | Baicalein | 100 | 100 | 1600 | 312 | 0.251 | 0.80203 | 1.891295 | 1:1 | 200:39 | ≈ | 5:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Su, H.; Komori, A.; Yang, S.; Luo, H.; Yang, A.W.H.; Sun, X.; Li, H.; Hung, A.; Zhao, X. Supercomputing Multi-Ligand Modeling, Simulation, Wavelet Analysis and Surface Plasmon Resonance to Develop Novel Combination Drugs: A Case Study of Arbidol and Baicalein Against Main Protease of SARS-CoV-2. Pharmaceuticals 2025, 18, 1054. https://doi.org/10.3390/ph18071054
Li H, Su H, Komori A, Yang S, Luo H, Yang AWH, Sun X, Li H, Hung A, Zhao X. Supercomputing Multi-Ligand Modeling, Simulation, Wavelet Analysis and Surface Plasmon Resonance to Develop Novel Combination Drugs: A Case Study of Arbidol and Baicalein Against Main Protease of SARS-CoV-2. Pharmaceuticals. 2025; 18(7):1054. https://doi.org/10.3390/ph18071054
Chicago/Turabian StyleLi, Hong, Hailong Su, Akari Komori, Shuxuan Yang, Hailang Luo, Angela Wei Hong Yang, Xiaomin Sun, Hongwei Li, Andrew Hung, and Xiaoshan Zhao. 2025. "Supercomputing Multi-Ligand Modeling, Simulation, Wavelet Analysis and Surface Plasmon Resonance to Develop Novel Combination Drugs: A Case Study of Arbidol and Baicalein Against Main Protease of SARS-CoV-2" Pharmaceuticals 18, no. 7: 1054. https://doi.org/10.3390/ph18071054
APA StyleLi, H., Su, H., Komori, A., Yang, S., Luo, H., Yang, A. W. H., Sun, X., Li, H., Hung, A., & Zhao, X. (2025). Supercomputing Multi-Ligand Modeling, Simulation, Wavelet Analysis and Surface Plasmon Resonance to Develop Novel Combination Drugs: A Case Study of Arbidol and Baicalein Against Main Protease of SARS-CoV-2. Pharmaceuticals, 18(7), 1054. https://doi.org/10.3390/ph18071054