
M. avium Complex Pulmonary Infections: Therapeutic Obstacles and Progress in Drug Development

 ,
,  ,
, 
Abstract

1. Introduction
2. Overview of MAC Infection
2.1. Subspecies of MAC
2.2. Pathogenesis
2.2.1. Pathophysiology
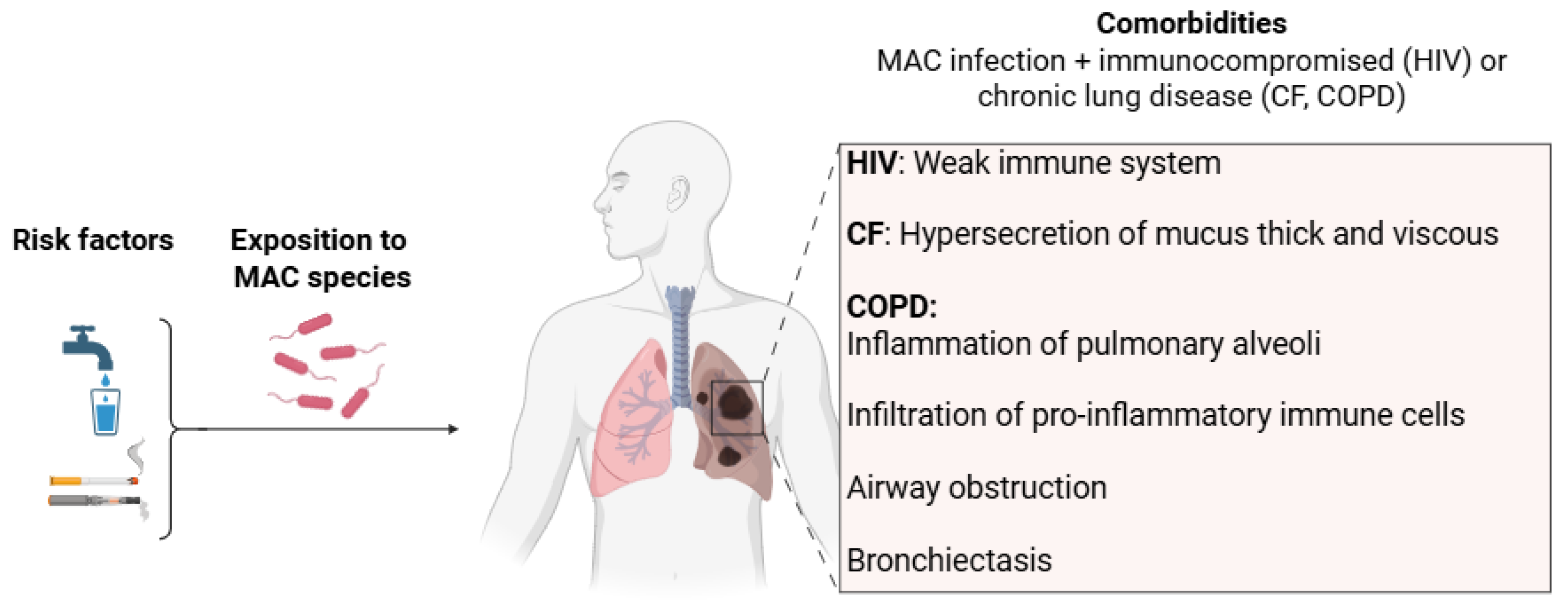
2.2.2. Risk Factors and Comorbidities
2.3. Diagnosis Criteria
3. Available Drugs for MAC Infections and Challenges
3.1. Available Medications for Treating MAC Infections
3.2. Resistance Mechanisms
3.2.1. Unique Features of the NTM Cell Envelope as Sources of Intrinsic Antibiotic Resistance
3.2.2. Other Resistance Mechanisms
3.3. Challenges
4. Current and Future Drug Targets for the Treatment of MAC Lung Infections
5. Therapeutic Approaches for the Treatment of MAC Lung Disease
5.1. Optimized Combination Therapy
5.2. New Formulations: Liposomal and Inhaled Therapies
- Aminosides: AMK

- Phenazines: CFZ

5.3. Drug Repurposing


5.4. New Anti-MAC Compounds in (Pre)Clinical Development
5.4.1. 5-Phenylpyridine: From BQ to Sudapyridine

5.4.2. Benzimidazole Ureas: From Compound 1 to Fobrepodacin (Or SPR720)
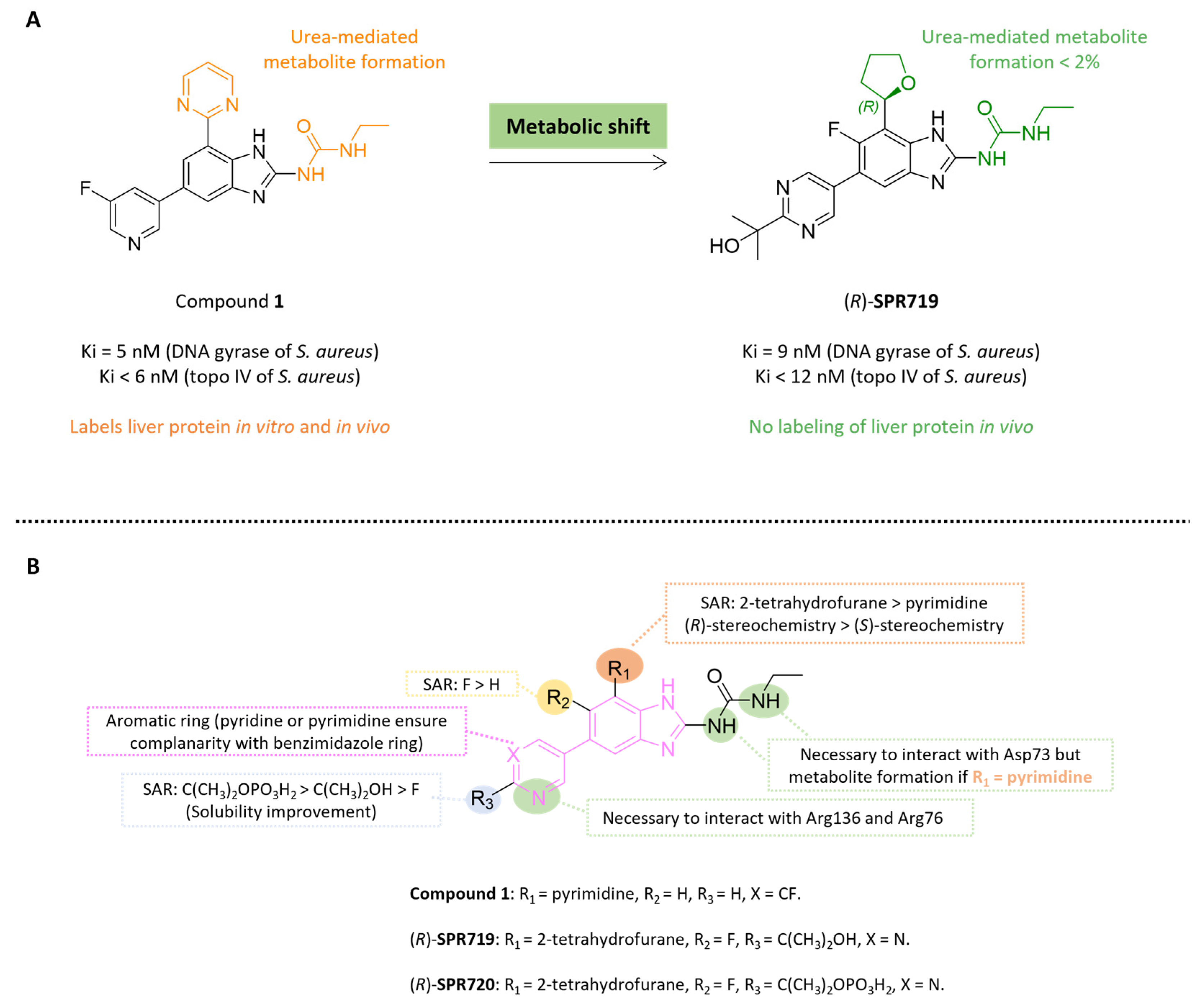
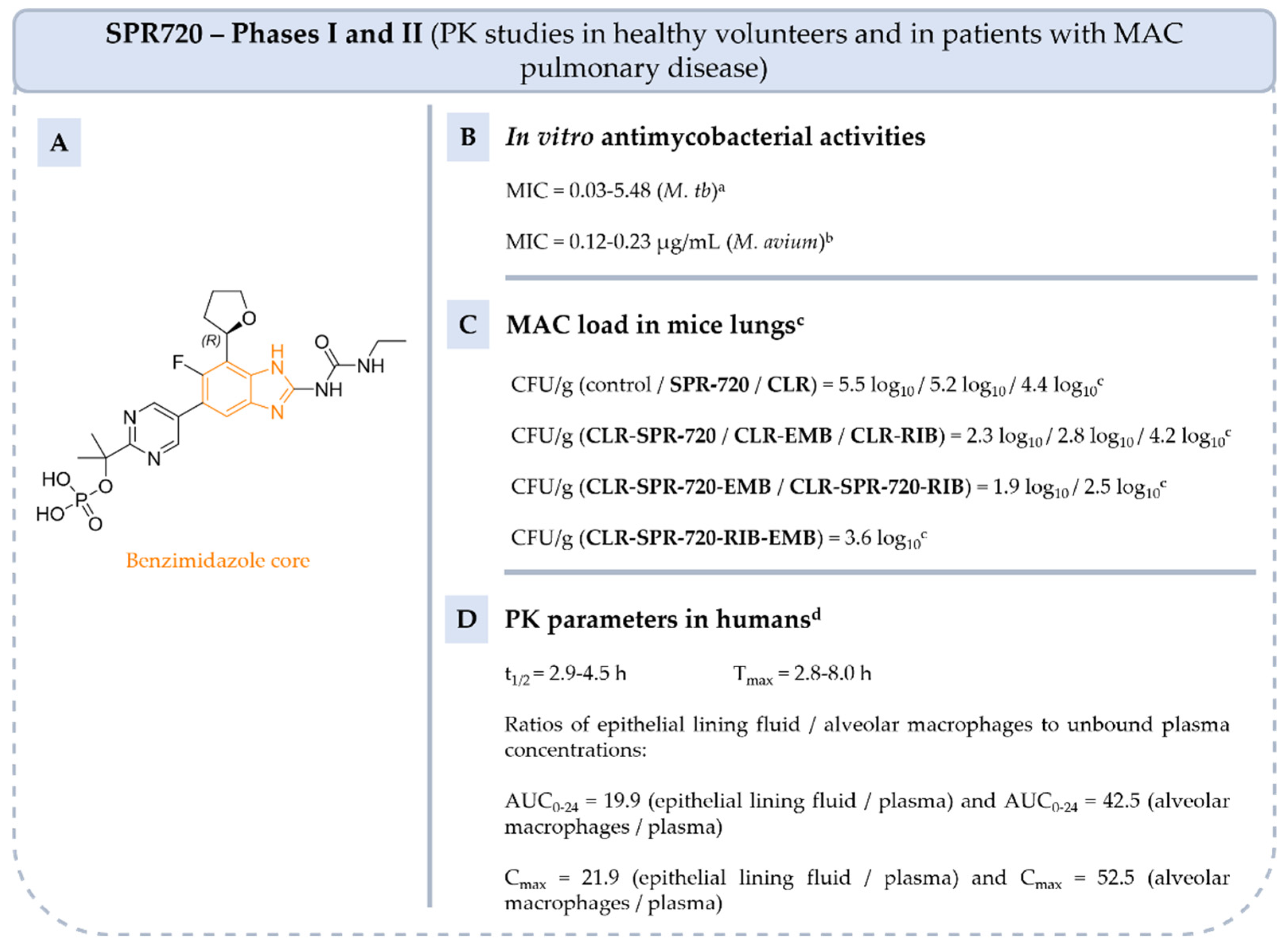
5.4.3. Benzoxaboroles: From Tavaborole to Epetraborole (AN3365)

5.4.4. Thiosemicarbazones: From Thiacetazone to SRI-286

5.4.5. Mefloquine

5.4.6. Mavintramycin A
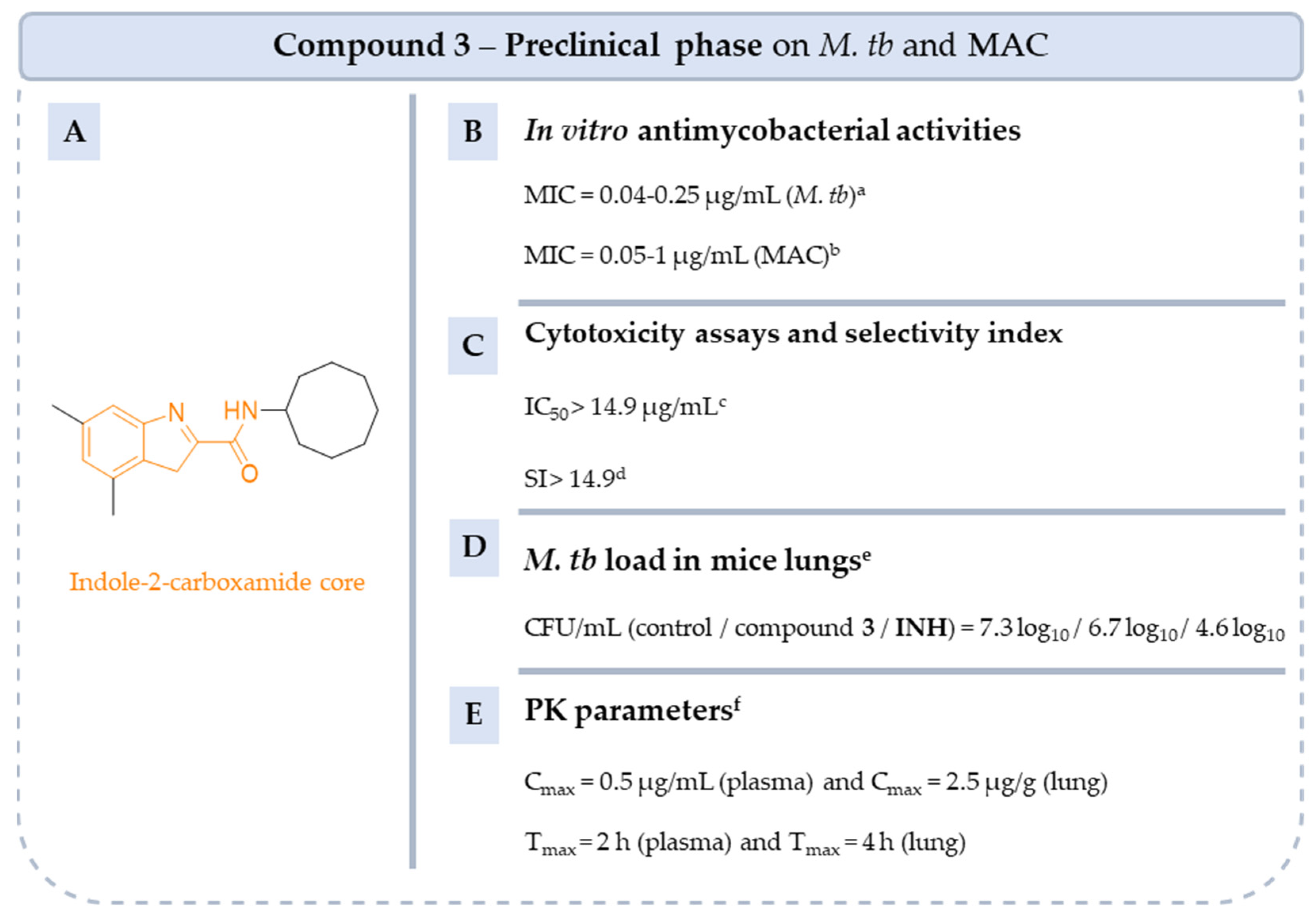
5.4.7. Indole-2-Carboxamides
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALIS | amikacin liposome inhalation suspension |
| AMK | amikacin |
| ATS | American thoracic society |
| AUC | area under the curve |
| AZI | azithromycin |
| BQ | bedaquiline |
| CD | cavitary disease |
| CF | cystic fibrosis |
| CFU | colony-forming unit |
| CFZ | clofazimine |
| CLR | clarithromycin |
| COPD | chronic obstructive pulmonary disease |
| EMB | ethambutol |
| FDA | Food and Drug Administration |
| GBT | guidelines-based therapy |
| HDT | host-directed therapies |
| IC50 | median inhibitory concentration |
| IDSA | Infectious Diseases Society of America |
| INH | isoniazid |
| Ki | inhibition constant |
| MAC | Mycobacterium avium complex |
| MIC | minimal inhibitory concentration |
| MOX | moxifloxacin |
| M. tb | Mycobacterium tuberculosis |
| MQ | mefloquine |
| ND | nodular bronchiectasis disease |
| NTM | non-tuberculous mycobacteria |
| PC | physicochemical |
| PD | pharmacodynamical |
| PDB ID | protein data bank identifiers |
| PK | pharmacokinetic |
| PSA | polar surface area |
| RD | refractory disease |
| RIB | rifabutin |
| RIF | rifampicin |
| RGM | rapidly growing mycobacteria |
| SAR | structure-activity relationships |
| SGM | slowly growing mycobacteria |
| SI | selectivity index |
| WHO | World Health Organization |
References
- Global Tuberculosis Report 2024. World Health Organization 2024. Featured Publication 29 October 2024. Available online: https://iris.who.int/bitstream/handle/10665/379339/9789240101531-eng.pdf (accessed on 29 October 2024).
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An Official ATS/IDSA Statement: Diagnosis, Treatment, and Prevention of Nontuberculous Mycobacterial Diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.M.; Gomes, M.S.; Silva, T. Looking beyond Typical Treatments for Atypical Mycobacteria. Antibiotics 2020, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Busatto, C.; Vianna, J.S.; da Silva, L.V.; Ramis, I.B.; da Silva, P.E.A. Mycobacterium avium: An Overview. Tuberculosis 2019, 114, 127–134. [Google Scholar] [CrossRef]
- Daley, C.L.; Iaccarino, J.M.; Lange, C.; Cambau, E.; Wallace, R.J.; Andrejak, C.; Böttger, E.C.; Brozek, J.; Griffith, D.E.; Guglielmetti, L.; et al. Treatment of Nontuberculous Mycobacterial Pulmonary Disease: An Official ATS/ERS/ESCMID/IDSA Clinical Practice Guideline. Eur. Respir. J. 2020, 56, 2000535. [Google Scholar] [CrossRef]
- Diel, R.; Nienhaus, A.; Ringshausen, F.C.; Richter, E.; Welte, T.; Rabe, K.F.; Loddenkemper, R. Microbiologic Outcome of Interventions Against Mycobacterium avium Complex Pulmonary Disease: A Systematic Review. Chest 2018, 153, 888–921. [Google Scholar] [CrossRef]
- Cuttino, J.T.; McCABE, A.M. Pure Granulomatous Nocardiosis, a New Fungus Disease Distinguished by Intracellular Parasitism; a Description of a New Disease in Man Due to a Hitherto Undescribed Organism, Nocardia Intracellularis, n. Sp., Including a Study of the Biologic and Pathogenic Properties of This Species. Am. J. Pathol. 1949, 25, 1–47. [Google Scholar] [PubMed]
- Turenne, C.Y.; Wallace, R., Jr.; Behr, M.A. Mycobacterium avium in the Postgenomic Era. Clin. Microbiol. Rev. 2007, 20, 205–229. [Google Scholar] [CrossRef]
- Leão, C.; Canto, A.; Machado, D.; Sanches, I.S.; Couto, I.; Viveiros, M.; Inácio, J.; Botelho, A. Relatedness of Mycobacterium avium Subspecies Hominissuis Clinical Isolates of Human and Porcine Origins Assessed by MLVA. Vet. Microbiol. 2014, 173, 92–100. [Google Scholar] [CrossRef]
- Thomson, R.M.; Armstrong, J.G.; Looke, D.F. Gastroesophageal Reflux Disease, Acid Suppression, and Mycobacterium avium Complex Pulmonary Disease. Chest 2007, 131, 1166–1172. [Google Scholar] [CrossRef]
- To, K.; Cao, R.; Yegiazaryan, A.; Owens, J.; Venketaraman, V. General Overview of Nontuberculous Mycobacteria Opportunistic Pathogens: Mycobacterium avium and Mycobacterium abscessus. JCM 2020, 9, 2541. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Wagner, D.; Sosnowska, D. Mechanisms of Mycobacterium avium Pathogenesis. In Inflammation; Górski, A., Krotkiewski, H., Zimecki, M., Eds.; Springer: Dordrecht, The Netherlands, 2001; pp. 153–166. ISBN 978-90-481-5852-2. [Google Scholar]
- Mullis, S.N.; Falkinham, J.O. Adherence and Biofilm Formation of Mycobacterium avium, Mycobacterium intracellulare and Mycobacterium abscessus to Household Plumbing Materials. J. Appl. Microbiol. 2013, 115, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.H.; Falkinham, J.O.; Norton, C.D.; LeChevallier, M.W. Chlorine, Chloramine, Chlorine Dioxide, and Ozone Susceptibility of Mycobacterium avium. Appl. Environ. Microbiol. 2000, 66, 1702–1705. [Google Scholar] [CrossRef]
- Lewis, A.H.; Falkinham, J.O. Microaerobic Growth and Anaerobic Survival of Mycobacterium avium, Mycobacterium intracellulare and Mycobacterium scrofulaceum. Int. J. Mycobacteriol. 2015, 4, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Gao, J. Progress in Non-Viral Localized Delivery of siRNA Therapeutics for Pulmonary Diseases. Acta Pharm. Sin. B 2023, 13, 1400–1428. [Google Scholar] [CrossRef] [PubMed]
- Andréjak, C.; Nielsen, R.; Thomsen, V.Ø.; Duhaut, P.; Sørensen, H.T.; Thomsen, R.W. Chronic Respiratory Disease, Inhaled Corticosteroids and Risk of Non-Tuberculous Mycobacteriosis. Thorax 2013, 68, 256–262. [Google Scholar] [CrossRef]
- Canan, A.; Batra, K.; Saboo, S.S.; Landay, M.; Kandathil, A. Radiological Approach to Cavitary Lung Lesions. Postgrad. Med. J. 2021, 97, 521–531. [Google Scholar] [CrossRef]
- Parkar, A.P.; Kandiah, P. Differential Diagnosis of Cavitary Lung Lesions. J. Belg. Soc. Radiol. 2016, 100, 100. [Google Scholar] [CrossRef]
- Kwon, Y.-S.; Koh, W.-J.; Daley, C.L. Treatment of Mycobacterium avium Complex Pulmonary Disease. Tuberc. Respir. Dis. 2019, 82, 15. [Google Scholar] [CrossRef]
- Clofazimine Side Effects: Common, Severe, Long Term. Available online: https://www.drugs.com/sfx/clofazimine-side-effects.html (accessed on 14 May 2025).
- Svensson, E.M.; Murray, S.; Karlsson, M.O.; Dooley, K.E. Rifampicin and Rifapentine Significantly Reduce Concentrations of Bedaquiline, a New Anti-TB Drug. J. Antimicrob. Chemother. 2015, 70, 1106–1114. [Google Scholar] [CrossRef]
- Chatterjee, D.; Khoo, K.-H. The Surface Glycopeptidolipids of Mycobacteria: Structures and Biological Properties. CMLS Cell. Mol. Life Sci. 2001, 58, 2018–2042. [Google Scholar] [CrossRef]
- Brennan, P.J.; Nikaido, H. The Envelope of Mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Mdluli, K.; Swanson, J.; Fischer, E.; Lee, R.E.; Barry Iii, C.E. Mechanisms Involved in the Intrinsic Isoniazid Resistance of Mycobacterium avium. Mol. Microbiol. 1998, 27, 1223–1233. [Google Scholar] [CrossRef]
- Raynaud, C.; Lanéelle, M.-A.; Senaratne, R.H.; Draper, P.; Lanéelle, G.; Daffé, M. Mechanisms of Pyrazinamide Resistance in Mycobacteria: Importance of Lack of Uptake in Addition to Lack of Pyrazinamidase Activity. Microbiology 1999, 145, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Van Ingen, J.; Boeree, M.J.; van Soolingen, D.; Mouton, J.W. Resistance Mechanisms and Drug Susceptibility Testing of Nontuberculous Mycobacteria. Drug Resist. Updates 2012, 15, 149–161. [Google Scholar] [CrossRef]
- Zhu, J.-H.; Wang, B.-W.; Pan, M.; Zeng, Y.-N.; Rego, H.; Javid, B. Rifampicin Can Induce Antibiotic Tolerance in Mycobacteria via Paradoxical Changes in rpoB Transcription. Nat. Commun. 2018, 9, 4218. [Google Scholar] [CrossRef]
- Bastian, S.; Veziris, N.; Roux, A.-L.; Brossier, F.; Gaillard, J.-L.; Jarlier, V.; Cambau, E. Assessment of Clarithromycin Susceptibility in Strains Belonging to the Mycobacterium abscessus Group by Erm (41) and Rrl Sequencing. Antimicrob. Agents Chemother. 2011, 55, 775–781. [Google Scholar] [CrossRef]
- Philalay, J.S.; Palermo, C.O.; Hauge, K.A.; Rustad, T.R.; Cangelosi, G.A. Genes Required for Intrinsic Multidrug Resistance in Mycobacterium avium. Antimicrob. Agents Chemother. 2004, 48, 3412–3418. [Google Scholar] [CrossRef]
- Schorey, J.S.; Sweet, L. The Mycobacterial Glycopeptidolipids: Structure, Function, and Their Role in Pathogenesis. Glycobiology 2008, 18, 832–841. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Danelishvili, L.; Wu, M.; Hidaka, E.; Katsuyama, T.; Stang, B.; Petrofsky, M.; Bildfell, R.; Bermudez, L.E. The Ability to Form Biofilm Influences Mycobacterium avium Invasion and Translocation of Bronchial Epithelial Cells. Cell. Microbiol. 2006, 8, 806–814. [Google Scholar] [CrossRef]
- Nakajima, Y. Mode of Action and Resistance Mechanisms of Antimicrobial Macrolides. In Macrolide Antibiotics; Elsevier: Amsterdam, The Netherlands, 2003; pp. 453–499. ISBN 978-0-12-526451-8. [Google Scholar]
- Mikusová, K.; Slayden, R.A.; Besra, G.S.; Brennan, P.J. Biogenesis of the Mycobacterial Cell Wall and the Site of Action of Ethambutol. Antimicrob. Agents Chemother. 1995, 39, 2484–2489. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.-W. RifamycinMode of Action, Resistance, and Biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, Y.; Gao, Y.; Wu, L.; Gao, R.; Zhang, Q.; Wang, Y.; Wu, C.; Wu, F.; Gurcha, S.S.; et al. Structures of Cell Wall Arabinosyltransferases with the Anti-Tuberculosis Drug Ethambutol. Science 2020, 368, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The Arabinosyltransferase EmbC Is Inhibited by Ethambutol in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Iakhiaeva, E.; Griffith, D.E.; Woods, G.L.; Stout, J.E.; Wolfe, C.R.; Turenne, C.Y.; Wallace, R.J. In Vitro Activity of Amikacin against Isolates of Mycobacterium avium Complex with Proposed MIC Breakpoints and Finding of a 16S rRNA Gene Mutation in Treated Isolates. J. Clin. Microbiol. 2013, 51, 3389–3394. [Google Scholar] [CrossRef]
- Van Rensburg, C.E.; Jooné, G.K.; O’Sullivan, J.F.; Anderson, R. Antimicrobial Activities of Clofazimine and B669 Are Mediated by Lysophospholipids. Antimicrob. Agents Chemother. 1992, 36, 2729–2735. [Google Scholar] [CrossRef]
- Yao, R.; Wang, B.; Fu, L.; Li, L.; You, K.; Li, Y.-G.; Lu, Y. Sudapyridine (WX-081), a Novel Compound against Mycobacterium tuberculosis. Microbiol. Spectr. 2022, 10, e02477-21. [Google Scholar] [CrossRef]
- Matassova, N.B.; Rodnina, M.V.; Endermann, R.; Kroll, H.-P.; Pleiss, U.; Wild, H.; Wintermeyer, W. Ribosomal RNA Is the Target for Oxazolidinones, a Novel Class of Translational Inhibitors. RNA 1999, 5, 939–946. [Google Scholar] [CrossRef]
- Charifson, P.S.; Grillot, A.-L.; Grossman, T.H.; Parsons, J.D.; Badia, M.; Bellon, S.; Deininger, D.D.; Drumm, J.E.; Gross, C.H.; LeTiran, A.; et al. Novel Dual-Targeting Benzimidazole Urea Inhibitors of DNA Gyrase and Topoisomerase IV Possessing Potent Antibacterial Activity: Intelligent Design and Evolution through the Judicious Use of Structure-Guided Design and Stucture−Activity Relationships. J. Med. Chem. 2008, 51, 5243–5263. [Google Scholar] [CrossRef]
- Martín-Galiano, A.J.; Gorgojo, B.; Kunin, C.M. Mefloquine and New Related Compounds Target the F0 Complex of the F0F1 H+-ATPase of Streptococcus Pneumoniae. Antimicrob. Agent. Chemother. 2002, 46, 1680–1687. [Google Scholar] [CrossRef]
- Dos Santos, M.C.; Scaini, J.L.R.; Lopes, M.V.C.; Rodrigues, B.G.; Silva, N.O.; Borges, C.R.L.; dos Santos, S.C.; dos Santos Machado, K.; Werhli, A.V.; da Silva, P.E.A.; et al. Mefloquine Synergism with Anti-Tuberculosis Drugs and Correlation to Membrane Effects: Biologic, Spectroscopic and Molecular Dynamics Simulations Studies. Bioorg. Chem. 2021, 110, 104786. [Google Scholar] [CrossRef]
- Liang, X.; Liu, Z.; Wang, Y.; Zhang, Y.; Deng, W.; Liu, Q.; Lu, Z.; Li, K.; Chang, Y.; Wei, L. Progress in the Study of Mefloquine as an Antibiotic Adjuvant for Combination Bacterial Inhibition Treatment. Front. Cell. Infect. Microbiol. 2024, 14, 1470891. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, N.; Jayanthan, H.S.; Port, J.; Rossatto, F.C.; Mylonakis, E. Mefloquine Reduces the Bacterial Membrane Fluidity of Acinetobacter Baumannii and Distorts the Bacterial Membrane When Combined with Polymyxin B. Antimicrob. Chemother. 2025, 16, e04016-24. [Google Scholar] [CrossRef] [PubMed]
- Crilly, N.P.; Ayeh, S.K.; Karakousis, P.C. The New Frontier of Host-Directed Therapies for Mycobacterium avium Complex. Front. Immunol. 2021, 11, 623119. [Google Scholar] [CrossRef]
- Kilinç, G.; Ottenhoff, T.H.M.; Saris, A. Phenothiazines Boost Host Control of Mycobacterium avium Infection in Primary Human Macrophages. Biomed. Pharmacother. 2025, 185, 117941. [Google Scholar] [CrossRef]
- Lanoix, J.-P.; Joseph, C.; Peltier, F.; Castelain, S.; Andréjak, C. Synergistic Activity of Clofazimine and Clarithromycin in an Aerosol Mouse Model of Mycobacterium avium Infection. Antimicrob. Agents Chemother. 2020, 64, e02349-19. [Google Scholar] [CrossRef]
- Zhang, J.; Leifer, F.; Rose, S.; Chun, D.Y.; Thaisz, J.; Herr, T.; Nashed, M.; Joseph, J.; Perkins, W.R.; DiPetrillo, K. Amikacin Liposome Inhalation Suspension (ALIS) Penetrates Non-Tuberculous Mycobacterial Biofilms and Enhances Amikacin Uptake Into Macrophages. Front. Microbiol. 2018, 9, 915. [Google Scholar] [CrossRef]
- Chalmers, J.D.; van Ingen, J.; van der Laan, R.; Herrmann, J.-L. Liposomal Drug Delivery to Manage Nontuberculous Mycobacterial Pulmonary Disease and Other Chronic Lung Infections. Eur. Respir. Rev. 2021, 30, 210010. [Google Scholar] [CrossRef]
- Zhang, Y.; Hill, A.T. Amikacin Liposome Inhalation Suspension as a Treatment for Patients with Refractory Mycobacterium avium Complex Lung Infection. Expert Rev. Respir. Med. 2021, 15, 737–744. [Google Scholar] [CrossRef]
- Fielding, R.M.; Moon-Mcdermott, L.; Lewis, R.O.; Horner, M.J. Pharmacokinetics and Urinary Excretion of Amikacin in Low-Clearance Unilamellar Liposomes after a Single or Repeated Intravenous Administration in the Rhesus Monkey. Antimicrob. Agents Chemother. 1999, 43, 503–509. [Google Scholar] [CrossRef]
- Fielding, R.M.; Ramilla, O.; Lewis, L.M.M. Altered Tissue Distribution and Elimination of Amikacin Encapsulated in Unilamellar, Low Clearance Liposomes (MiKasome). Pharm. Res. 1998, 15, 177–1781. [Google Scholar] [CrossRef]
- Barry, V.C.; Conalty, M.L. The Antimycobacterial Activity of B663. Lepr. Rev. 1965, 36, 3–7. [Google Scholar] [PubMed]
- Lamprene (R). Novartis. 2002; Revised: 2019. Available online: https://www.accessdata.fda.gov/Drugsatfda_docs/Label/2016/019500s013lbl.Pdf (accessed on 7 July 2002).
- Cholo, M.C.; Steel, H.C.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current Status and Future Prospects. J. Antimicrob. Chemother. 2012, 67, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Levy, L. Pharmacologic Studies of Clofazimine. Am. J. Trop. Med. Hyg. 1974, 23, 1097–1109. [Google Scholar] [CrossRef]
- Banaschewski, B.; Verma, D.; Pennings, L.J.; Zimmerman, M.; Ye, Q.; Gadawa, J.; Dartois, V.; Ordway, D.; Van Ingen, J.; Ufer, S.; et al. Clofazimine Inhalation Suspension for the Aerosol Treatment of Pulmonary Nontuberculous Mycobacterial Infections. J. Cyst. Fibros. 2019, 18, 714–720. [Google Scholar] [CrossRef]
- Kunkel, M.; Doyle-Eisele, M.; Kuehl, P.; Rotermund, K.; Hittinger, M.; Ufer, S.; Reed, M.; Grant, M.; Hofmann, T. Clofazimine Inhalation Suspension Demonstrates Promising Toxicokinetics in Canines for Treating Pulmonary Nontuberculous Mycobacteria Infection. Antimicrob. Agents Chemother. 2023, 67, e01144-22. [Google Scholar] [CrossRef] [PubMed]
- Van Ingen, J. Why Do We Use 100 Mg of Clofazimine in TB and NTM Treatment? J. Antimicrob. Chemother. 2024, 79, 697–702. [Google Scholar] [CrossRef]
- Murashov, M.D.; LaLone, V.; Rzeczycki, P.M.; Keswani, R.K.; Yoon, G.S.; Sud, S.; Rajeswaran, W.; Larsen, S.; Stringer, K.A.; Rosania, G.R. The Physicochemical Basis of Clofazimine-Induced Skin Pigmentation. J. Investig. Dermatol. 2018, 138, 697–703. [Google Scholar] [CrossRef]
- Gangadharal, P.R.; Candler, E.R. Activity of Some Antileprosy Compounds against Mycobacterium intracellulare in Vitro. Am. Rev. Respir. Dis. 1977, 115, 705–708. [Google Scholar] [CrossRef]
- Saivin, S.; Houin, G. Clinical Pharmacokinetics of Doxycycline and Minocycline. Clin. Pharmacokinet. 1988, 15, 355–366. [Google Scholar] [CrossRef]
- Ruth, M.M.; Magombedze, G.; Gumbo, T.; Bendet, P.; Sangen, J.J.N.; Zweijpfenning, S.; Hoefsloot, W.; Pennings, L.; Koeken, V.A.C.M.; Wertheim, H.F.L.; et al. Minocycline Treatment for Pulmonary Mycobacterium avium Complex Disease Based on Pharmacokinetics/Pharmacodynamics and Bayesian Framework Mathematical Models. J. Antimicrob. Chemother. 2019, 74, 1952–1961. [Google Scholar] [CrossRef]
- Naline, E.; Sanceaume, M.; Toty, L.; Bakdach, H.; Pays, M.; Advenier, C. Penetration of Minocycline into Lung Tissues. Br. J. Clin. Pharmacol. 1991, 32, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Agwuh, K.N. Pharmacokinetics and Pharmacodynamics of the Tetracyclines Including Glycylcyclines. J. Antimicrob. Chemother. 2006, 58, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Luo, W.; Xu, D.; Guo, F.; Yang, M.; Zhu, Y.; Shen, L.; Chen, S.; Tang, D.; Li, L.; et al. Discovery and Preclinical Profile of Sudapyridine (WX-081), a Novel Anti-Tuberculosis Agent. Bioorg. Med. Chem. Lett. 2022, 71, 128824. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, H.; Qi, X.; Zhang, W.; Wang, B.; Fu, L.; Chen, X.; Chen, X.; Lu, Y. Sudapyridine (WX-081) Antibacterial Activity against Mycobacterium avium, Mycobacterium abscessus, and Mycobacterium chelonae in Vitro and in Vivo. mSphere 2024, 9, e00518-23. [Google Scholar] [CrossRef]
- Villellas, C.; Stevenaert, F.; Remmerie, B.; Andries, K. Sub-MIC Levels of Bedaquiline and Clofazimine Can Select Mycobacterium tuberculosis Mutants with Increased MIC. Antimicrob. Agents Chemother. 2024, 68, e01275-23. [Google Scholar] [CrossRef]
- He, C.; Preiss, L.; Wang, B.; Fu, L.; Wen, H.; Zhang, X.; Cui, H.; Meir, T.; Yin, D. Structural Simplification of Bedaquiline: The Discovery of 3-(4((N,N-Dimethylaminomethyl)Phenyl)Quinoline-Derived Antitubercular Lead Compounds. ChemMeChem 2017, 12, 106–119. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Dahiya, N. Bedaquiline for the Treatment of Resistant Tuberculosis: Promises and Pitfalls. Tuberculosis 2014, 94, 357–362. [Google Scholar] [CrossRef]
- Patel, H.; Pawara, R.; Pawara, K.; Ahmed, F.; Shirkhedkar, A.; Surana, S. A Structural Insight of Bedaquiline for the Cardiotoxicity and Hepatotoxicity. Tuberculosis 2019, 117, 79–84. [Google Scholar] [CrossRef]
- Rouan, M.-C.; Lounis, N.; Gevers, T.; Dillen, L.; Gilissen, R.; Raoof, A.; Andries, K. Pharmacokinetics and Pharmacodynamics of TMC207 and Its N-Desmethyl Metabolite in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1444–1451. [Google Scholar] [CrossRef]
- Grillot, A.-L.; Tiran, A.L.; Shannon, D.; Krueger, E.; Liao, Y.; O’Dowd, H.; Tang, Q.; Ronkin, S.; Wang, T.; Waal, N.; et al. Second-Generation Antibacterial Benzimidazole Ureas: Discovery of a Preclinical Candidate with Reduced Metabolic Liability. J. Med. Chem. 2014, 57, 8792–8816. [Google Scholar] [CrossRef]
- Locher, C.P.; Jones, S.M.; Hanzelka, B.L.; Perola, E.; Shoen, C.M.; Cynamon, M.H.; Ngwane, A.H.; Wiid, I.J.; Van Helden, P.D.; Betoudji, F.; et al. A Novel Inhibitor of Gyrase B Is a Potent Drug Candidate for Treatment of Tuberculosis and Nontuberculosis Mycobacterial Infections. Antimicrob. Agents Chemother. 2015, 59, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Rubio, A.; Wallace, R.J. In Vitro Susceptibility Testing of a Novel Benzimidazole, SPR719, against Nontuberculous Mycobacteria. Antimicrob. Agents Chemother. 2018, 62, e01503-18. [Google Scholar] [CrossRef]
- Winthrop, K.L.; Flume, P.; Hamed, K.A. Nontuberculous Mycobacterial Pulmonary Disease and the Potential Role of SPR720. Expert Rev. Anti-Infect. Ther. 2023, 21, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Cotroneo, N.; Stokes, S.S.; Pucci, M.J.; Rubio, A.; Hamed, K.A.; Critchley, I.A. Efficacy of SPR720 in Murine Models of Non-Tuberculous Mycobacterial Pulmonary Infection. J. Antimicrob. Chemother. 2024, 79, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Talley, A.K.; Thurston, A.; Moore, G.; Gupta, V.K.; Satterfield, M.; Manyak, E.; Stockes, S.; Dane, A.; Melnick, D. First-in-Human Evaluation of the Safety, Tolerability, and Pharmacokinetics of SPR720, a Novel Oral Bacterial DNA Gyrase (GyrB) Inhibitor for Mycobacterial Infections. Antimicrob. Agents Chemother. 2021, 65, e01208-21. [Google Scholar] [CrossRef]
- Rodvold, K.A.; Gotfried, M.H.; Ussery, X.T.; Wong, S.L.; Hamed, K.A. Intrapulmonary Pharmacokinetics of SPR719 Following Oral Administration of SPR720 to Healthy Volunteers. Antimicrob. Agents Chemother. 2024, 68, e01103-24. [Google Scholar] [CrossRef]
- Ciaravino, V.; Plattner, J.; Chanda, S. An Assessment of the Genetic Toxicology of Novel Boron-containing Therapeutic Agents. Environ. Mol. Mutagen. 2013, 54, 338–346. [Google Scholar] [CrossRef]
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in Drug Design: Recent Advances in the Development of New Therapeutic Agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef]
- Pang, Y.L.J.; Poruri, K.; Martinis, S.A. TRNA Synthetase: TRNA Aminoacylation and Beyond. WIREs RNA 2014, 5, 461–480. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crépin, T.; Zhou, H.; Zhang, Y.-K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An Antifungal Agent Inhibits an Aminoacyl-tRNA Synthetase by Trapping tRNA in the Editing Site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef]
- Ganapathy, U.S.; Gengenbacher, M.; Dick, T. Epetraborole Is Active against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2021, 65, e01156-21. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, M.S.; Shoen, C.M.; Alley, M.R.K.; Cynamon, M.H. 1713. In Vitro Activities of Epetraborole, a Novel Bacterial Leucyl-tRNA Synthetase Inhibitor, Against Mycobacterium avium Complex Isolates. Open Forum Infect. Dis. 2022, 9, ofac492.1343. [Google Scholar] [CrossRef]
- Hernandez, V.; Crépin, T.; Palencia, A.; Cusack, S.; Akama, T.; Baker, S.J.; Bu, W.; Feng, L.; Freund, Y.R.; Liu, L.; et al. Discovery of a Novel Class of Boron-Based Antibacterials with Activity against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2013, 57, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- De, K.; DeStefano, M.S.; Shoen, C.; Cynamon, M.H.; Alley, M.R.K. 1704. Epetraborole, a Novel Bacterial Leucyl-tRNA Synthetase Inhibitor, Demonstrates Potent Efficacy and Improves Efficacy of Standard of Care Regimen Against Mycobacterium avium Complex in a Chronic Mouse Lung Infection Model. Open Forum Infect. Dis. 2022, 9, ofac492.1334. [Google Scholar] [CrossRef]
- AN2 Therapeutics. AN2 Therapeutics Reports Phase 1b Data for Oral Epetraborole; AN2 Therapeutics: Menlo Park, CA, USA, 2022. [Google Scholar]
- Eckburg, P.B.; Clarke, D.; Long, J.; Chanda, S.; Krause, K.M.; Easom, E.; Talbot, G.; Rubino, C.M.; Molga, A. 1727. Phase 1b Dose-Ranging Study Demonstrates Tolerability and Pharmacokinetics (PK) of Oral Epetraborole at the Predicted Therapeutic Dosage for Mycobacterium avium Complex (MAC) Lung Disease. Open Forum Infect. Dis. 2022, 9, ofac492.1357. [Google Scholar] [CrossRef]
- Keshavjee, S.; Farmer, P.E. Tuberculosis, Drug Resistance, and the History of Modern Medicine. N. Engl. J. Med. 2012, 367, 931–936. [Google Scholar] [CrossRef]
- Qian, L.; Ortiz De Montellano, P.R. Oxidative Activation of Thiacetazone by the Mycobacterium tuberculosis Flavin Monooxygenase EtaA and Human FMO1 and FMO3. Chem. Res. Toxicol. 2006, 19, 443–449. [Google Scholar] [CrossRef]
- Alahari, A.; Trivelli, X.; Guérardel, Y.; Dover, L.G.; Besra, G.S.; Sacchettini, J.C.; Reynolds, R.C.; Coxon, G.D.; Kremer, L. Thiacetazone, an Antitubercular Drug That Inhibits Cyclopropanation of Cell Wall Mycolic Acids in Mycobacteria. PLoS ONE 2007, 2, e1343. [Google Scholar] [CrossRef]
- Grzegorzewicz, A.E.; Korduláková, J.; Jones, V.; Born, S.E.M.; Belardinelli, J.M.; Vaquié, A.; Gundi, V.A.K.B.; Madacki, J.; Slama, N.; Laval, F.; et al. A Common Mechanism of Inhibition of the Mycobacterium tuberculosis Mycolic Acid Biosynthetic Pathway by Isoxyl and Thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef]
- Sriram, D.; Yogeeswari, P.; Thirumurugan, R.; Pavana, R.K. Discovery of New Antitubercular Oxazolyl Thiosemicarbazones. J. Med. Chem. 2006, 49, 3448–3450. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Reynolds, R.; Kolonoski, P.; Aralar, P.; Inderlied, C.B.; Young, L.S. Thiosemicarbazole (Thiacetazone-Like) Compound with Activity against Mycobacterium avium in Mice. Antimicrob. Agents Chemother. 2003, 47, 2685–2687. [Google Scholar] [CrossRef] [PubMed]
- Heifets, L.B.; Lindholm-Levy, P.J.; Flory, M. Thiacetazone: In Vitro Activity against Mycobacterium avium and m. Tuberculosis. Tubercle 1990, 71, 287–291. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Meek, L. Mefloquine and Its Enantiomers Are Active against Mycobacterium tuberculosis In Vitro and in Macrophages. Tuberc. Res. Treat. 2014, 2014, 530815. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Kolonoski, P.; Wu, M.; Aralar, P.A.; Inderlied, C.B.; Young, L.S. Mefloquine Is Active In Vitro and In Vivo against Mycobacterium avium Complex. Antimicrob. Agents Chemother. 1999, 43, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Froment, A.; Delomez, J.; Da Nascimento, S.; Dassonville-Klimpt, A.; Andréjak, C.; Peltier, F.; Joseph, C.; Sonnet, P.; Lanoix, J.-P. Efficacy of Mefloquine and Its Enantiomers in a Murine Model of Mycobacterium avium Infection. PLoS ONE 2024, 19, e0311167. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, L.E.; Inderlied, C.B.; Kolonoski, P.; Chee, C.B.; Aralar, P.; Petrofsky, M.; Parman, T.; Green, C.E.; Lewin, A.H.; Ellis, W.Y.; et al. Identification of (+)-Erythro-Mefloquine as an Active Enantiomer with Greater Efficacy than Mefloquine against Mycobacterium avium Infection in Mice. Antimicrob. Agents Chemother. 2012, 56, 4202–4206. [Google Scholar] [CrossRef]
- Martin, C.; Gimenez, F.; Bangchang, K.N.; Karbwang, J.; Farinotti, R.; Wainer, I.W. Whole Blood Concentrations of Mefloquine Enantiomers in Healthy Thai Volunteers. Eur. J. Clin. Pharmacol. 1994, 47, 85–87. [Google Scholar] [CrossRef]
- Hosoda, K.; Koyama, N.; Shigeno, S.; Nishimura, T.; Hasegawa, N.; Kanamoto, A.; Ohshiro, T.; Tomoda, H. Mavintramycin A Is a Promising Antibiotic for Treating Mycobacterium avium Complex Infectious Disease. Antimicrob. Agents Chemother. 2024, 68, e00917-23. [Google Scholar] [CrossRef]
- Lun, S.; Guo, H.; Onajole, O.K.; Pieroni, M.; Gunosewoyo, H.; Chen, G.; Tipparaju, S.K.; Ammerman, N.C.; Kozikowski, A.P.; Bishai, W.R. Indoleamides Are Active against Drug-Resistant Mycobacterium tuberculosis. Nat. Commun. 2013, 4, 2907. [Google Scholar] [CrossRef]
- Stec, J.; Onajole, O.K.; Lun, S.; Guo, H.; Merenbloom, B.; Vistoli, G.; Bishai, W.R.; Kozikowski, A.P. Indole-2-Carboxamide-Based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem. 2016, 59, 6232–6247. [Google Scholar] [CrossRef]
- Franz, N.D.; Belardinelli, J.M.; Kaminski, M.A.; Dunn, L.C.; Calado Nogueira de Moura, V.; Blaha, M.A.; Truong, D.D.; Li, W.; Jackson, M.; North, E.J. Design, Synthesis and Evaluation of Indole-2-Carboxamides with Pan Anti-Mycobacterial Activity. Bioorg. Med. Chem. 2017, 25, 3746–3755. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Disease | Regimen | Recommendations | |
|---|---|---|---|
| European | North American | ||
| ND | GBT: CLR/AZI RIF EMB | 3 times weekly or daily | |
| 1000 mg/250 mg 10 mg/kg 15–20 mg/kg | 1000 mg/500 mg 600 mg 25 mg/kg | ||
| CD | GBT: CLR/AZI RIF EMB And AMK IV | 3 times weekly or daily | |
| 1000 mg/250 mg 10 mg/kg Or RIB: 150–300 mg 15 mg/kg And 10 mg/kg | 1000 mg/250–500 mg 450–600 mg 15 mg/kg And 15 mg/kg | ||
| RD | (daily) GBT and ALIS or AMK IV (or streptomycin) | ||
| Molecules | Mode of Action | Develop Specifically Against MAC |
|---|---|---|
| Fobrepodacin | Inhibition of DNA replication | - |
| Rifampicin | Inhibition of transcription | - |
| Rifamycin | - | |
| Clarithromycin | Inhibition of protein synthesis | - |
| Azithromycin | - | |
| Streptomycin | - | |
| Linezolid | - | |
| Amikacin | - | |
| Minocycline | - | |
| Epetraborole | x | |
| Mavintramycin A | x | |
| SRI-286 | Inhibition of mycolic acids biosynthesis | - |
| Ethambutol | Inhibition of arabinogalactan biosynthesis | - |
| Indole-2-carboxamides | Inhibition of MmpL3 transporter | - |
| Clofazimine | Inhibition of the electron transport chain (NADH-quinone oxidoreductase II) | - |
| Bedaquiline | Inhibition of the electron transport chain (ATP synthase) | - |
| Sudapyridine | - | |
| Mefloquine | Inhibition of ATP synthase, efflux pumps and disruption of cell membrane integrity | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charrier, E.S.A.; Dassonville-Klimpt, A.; Andréjak, C.; Sonnet, P. M. avium Complex Pulmonary Infections: Therapeutic Obstacles and Progress in Drug Development. Pharmaceuticals 2025, 18, 891. https://doi.org/10.3390/ph18060891
Charrier ESA, Dassonville-Klimpt A, Andréjak C, Sonnet P. M. avium Complex Pulmonary Infections: Therapeutic Obstacles and Progress in Drug Development. Pharmaceuticals. 2025; 18(6):891. https://doi.org/10.3390/ph18060891
Chicago/Turabian StyleCharrier, Elise Si Ahmed, Alexandra Dassonville-Klimpt, Claire Andréjak, and Pascal Sonnet. 2025. "M. avium Complex Pulmonary Infections: Therapeutic Obstacles and Progress in Drug Development" Pharmaceuticals 18, no. 6: 891. https://doi.org/10.3390/ph18060891
APA StyleCharrier, E. S. A., Dassonville-Klimpt, A., Andréjak, C., & Sonnet, P. (2025). M. avium Complex Pulmonary Infections: Therapeutic Obstacles and Progress in Drug Development. Pharmaceuticals, 18(6), 891. https://doi.org/10.3390/ph18060891