

Discovery of Novel CRK12 Inhibitors for the Treatment of Human African Trypanosomiasis: An Integrated Computational and Experimental Approach

 , ,
, ,  and
and
Abstract

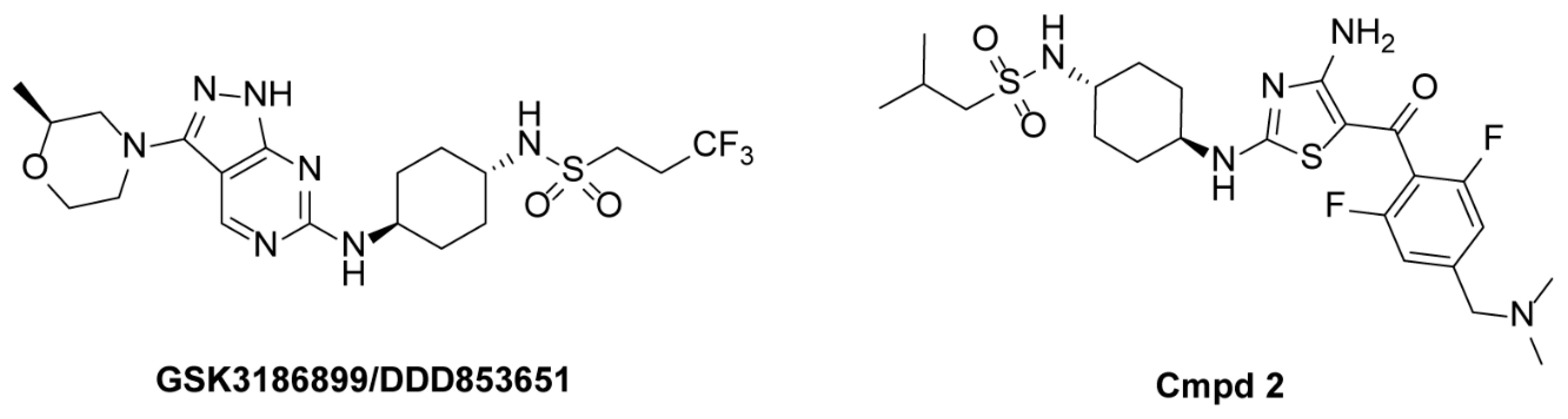
1. Introduction
2. Results and Discussion
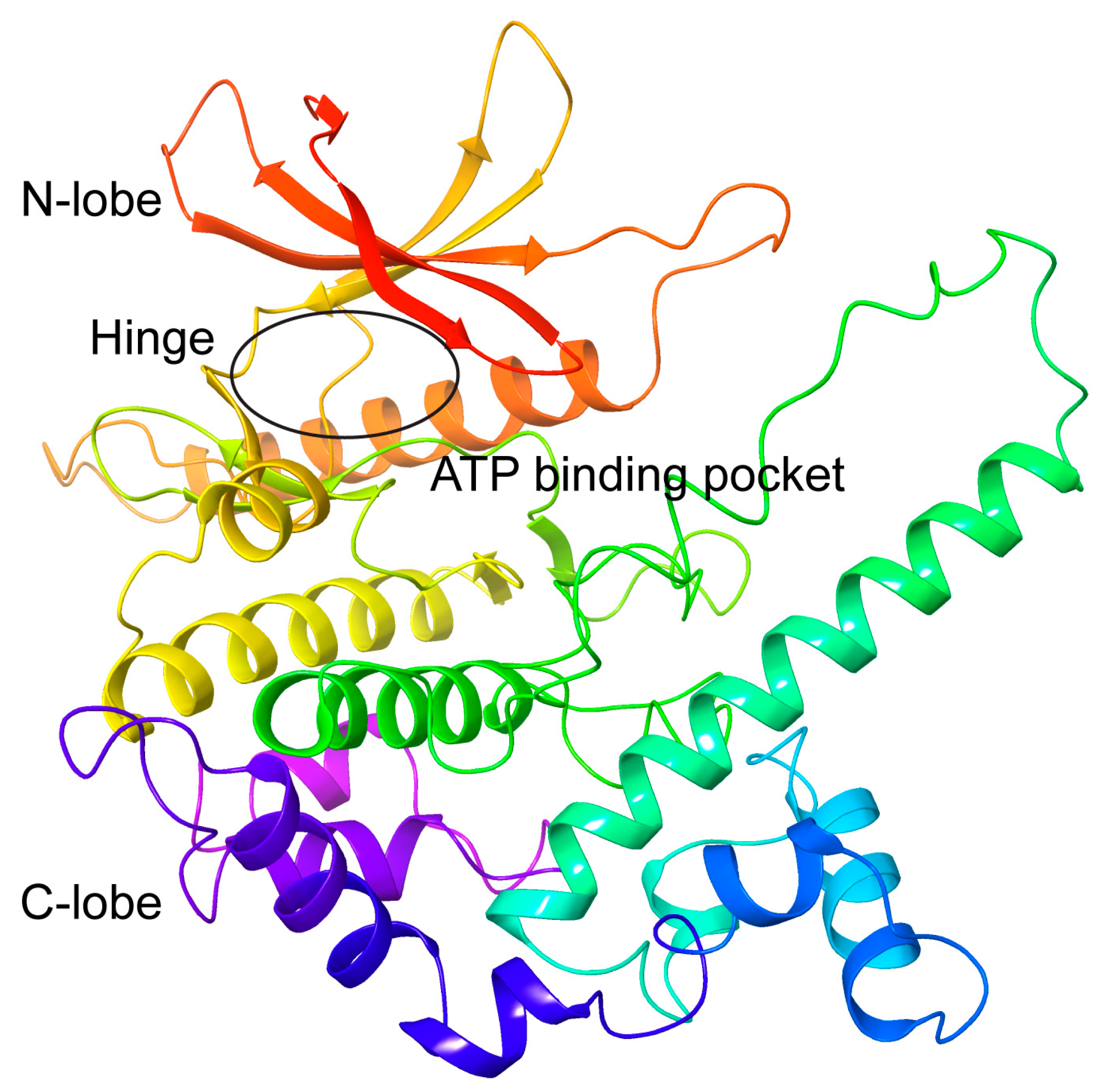
2.1. Modelling of the 3D Structure and Active Site of T. brucei CRK12
2.2. Refinement of Predicted CRK12 Structure Based on Molecular Dynamics Simulations
2.3. Assessment of Virtual Screening Performance for the AF2-Predicted CRK12 and MD-Refined CRK12
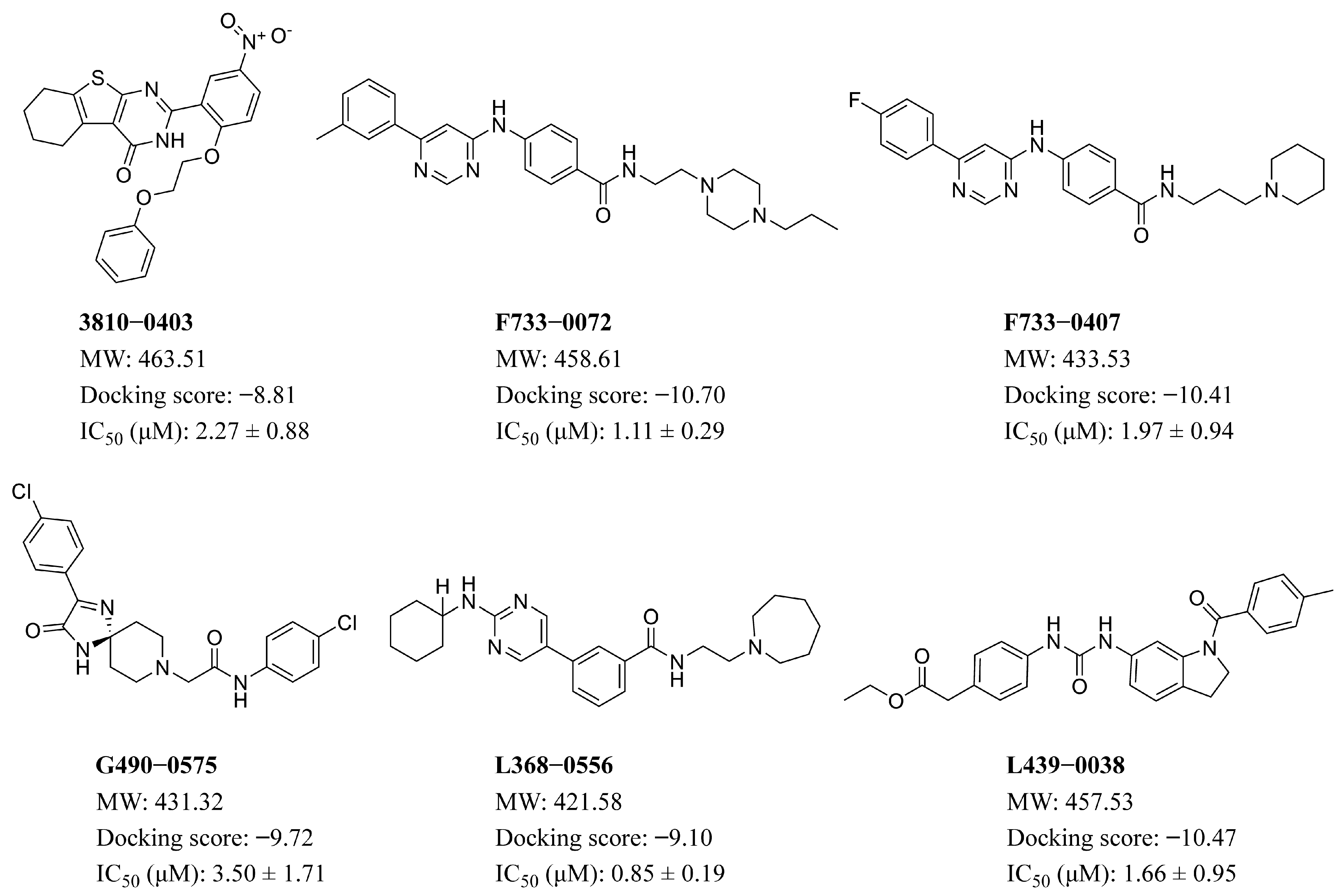
2.4. Results of Structure-Based Virtual Screening
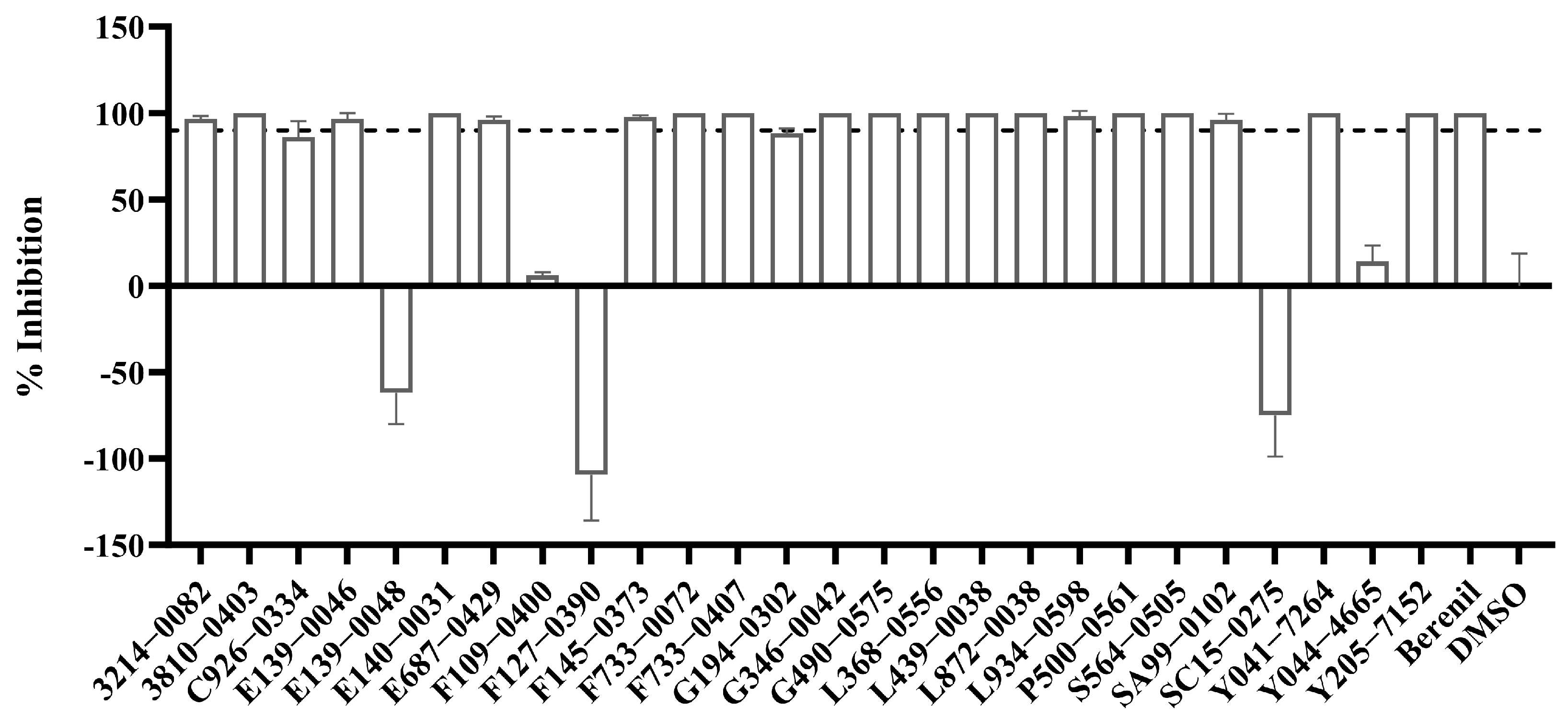
2.5. In Vitro Inhibitory Activity of the 26 Selected Compounds Against T. brucei
2.6. Molecular Dynamics Simulations
2.7. Binding Free Energy Calculation and Hot Spot Residue Identification
2.8. Hydrogen Bond Analysis Between Four Hits and CRK12
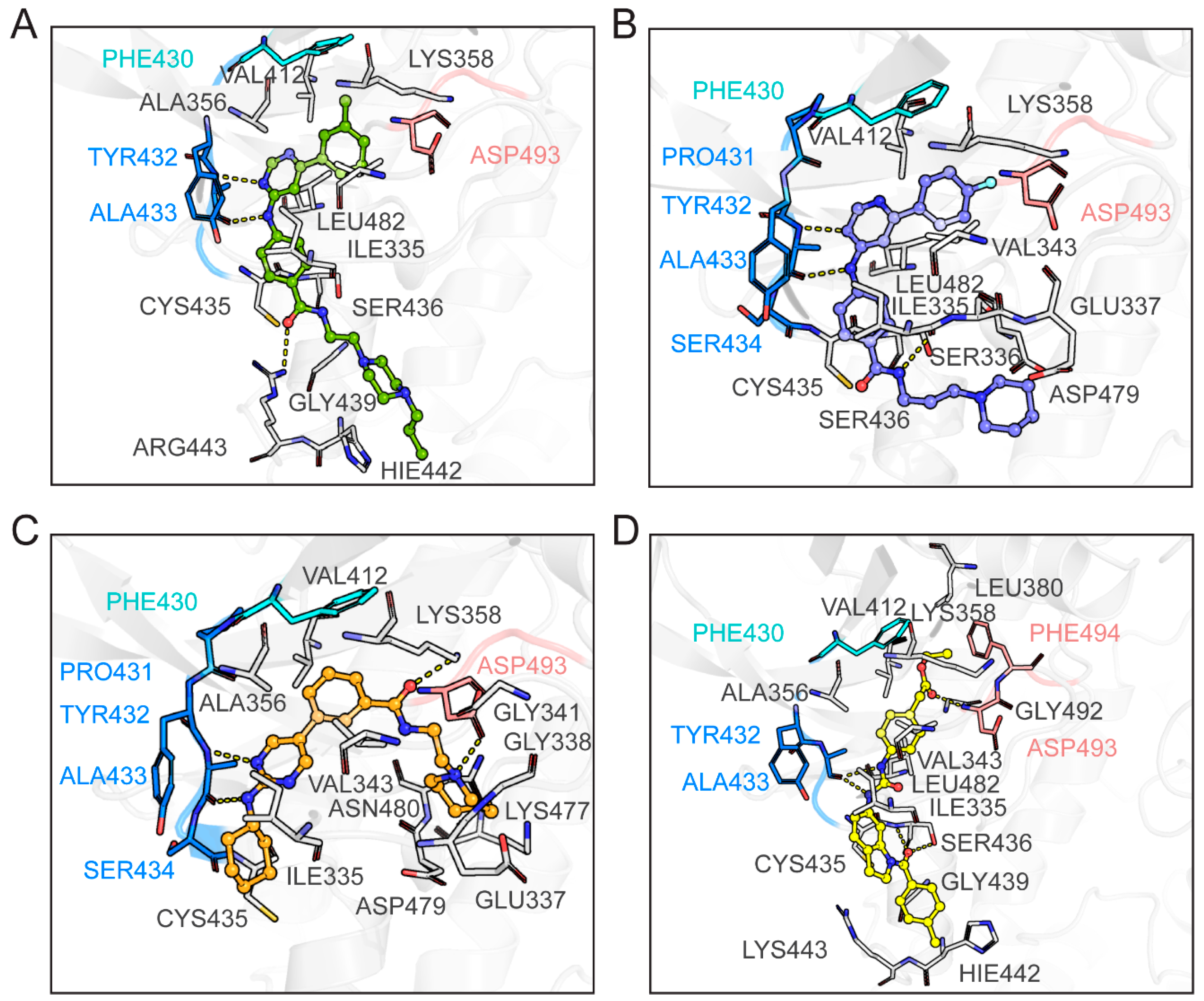
2.9. Binding Mode Analysis Between the Four Hits and CRK12
3. Materials and Methods
3.1. 3D-Structure Prediction and Binding Site Identification of Trypanosoma brucei CRK12
3.2. Molecular Docking-Based Virtual Screening
3.3. Molecular Dynamics Simulation
3.4. Molecular Mechanics Generalized-Born Surface Area Calculation
3.5. Trypanosome Cultures and Inhibitory Assays In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sutherland, C.S.; Yukich, J.; Goeree, R.; Tediosi, F. A literature review of economic evaluations for a neglected tropical disease: Human African trypanosomiasis (“sleeping sickness”). PLoS Negl. Trop. Dis. 2015, 9, e0003397. [Google Scholar] [CrossRef] [PubMed]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- WHO. Neglected Tropical Diseases; WHO: Geneva, Switzerland, 2024; Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 20 November 2024).
- Simarro, P.P.; Franco, J.R.; Cecchi, G.; Paone, M.; Diarra, A.; Ruiz Postigo, J.A.; Jannin, J.G. Human African trypanosomiasis in non-endemic countries (2000–2010). J. Travel. Med. 2012, 19, 44–53. [Google Scholar] [CrossRef]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef]
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-trypanosomatid drug discovery: Progress and challenges. Nat. Rev. Microbiol. 2023, 21, 35–50. [Google Scholar] [CrossRef]
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef]
- Isaac, C.; Ohiolei, J.A.; Ebhodaghe, F.; Igbinosa, I.B.; Eze, A.A. Animal African Trypanosomiasis in Nigeria: A long way from elimination/eradication. Acta Trop. 2017, 176, 323–331. [Google Scholar] [CrossRef]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Li, Z. Regulation of the cell division cycle in Trypanosoma brucei. Eukaryot. Cell 2012, 11, 1180–1190. [Google Scholar] [CrossRef]
- Field, M.C.; Carrington, M. The trypanosome flagellar pocket. Nat. Rev. Microbiol. 2009, 7, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.A.; Sayé, M.; Reigada, C.; Silber, A.M.; Labadie, G.R.; Miranda, M.R.; Valera-Vera, E. Computational approaches for drug discovery against trypanosomatid-caused diseases. Parasitology 2020, 147, 611–633. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, S.; Almeida Costa, C.I.; Forkert, A.C.; Benz, C.; Hamilton, A.; Tetley, L.; Burchmore, R.; Novo, C.; Mottram, J.C.; Hammarton, T.C. Identification and Functional Characterisation of CRK12:CYC9, a Novel Cyclin-Dependent Kinase (CDK)-Cyclin Complex in Trypanosoma brucei. PLoS ONE 2013, 8, e67327. [Google Scholar] [CrossRef]
- Wyllie, S.; Thomas, M.; Patterson, S.; Crouch, S.; De Rycker, M.; Lowe, R.; Gresham, S.; Urbaniak, M.D.; Otto, T.D.; Stojanovski, L.; et al. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 2018, 560, 192–197. [Google Scholar] [CrossRef]
- Smith, A.; Wall, R.J.; Patterson, S.; Rowan, T.; Rico Vidal, E.; Stojanovski, L.; Huggett, M.; Hampton, S.E.; Thomas, M.G.; Corpas Lopez, V.; et al. Repositioning of a Diaminothiazole Series Confirmed to Target the Cyclin-Dependent Kinase CRK12 for Use in the Treatment of African Animal Trypanosomiasis. J. Med. Chem. 2022, 65, 5606–5624. [Google Scholar] [CrossRef]
- Altmann, S.; Rico, E.; Carvalho, S.; Ridgway, M.; Trenaman, A.; Donnelly, H.; Tinti, M.; Wyllie, S.; Horn, D. Oligo targeting for profiling drug resistance mutations in the parasitic trypanosomatids. Nucleic Acids Res. 2022, 50, e79. [Google Scholar] [CrossRef]
- Catta-Preta, C.M.C.; Mottram, J.C. Drug candidate and target for leishmaniasis. Nature 2018, 560, 171–172. [Google Scholar] [CrossRef]
- McNae, I.W.; Kinkead, J.; Malik, D.; Yen, L.H.; Walker, M.K.; Swain, C.; Webster, S.P.; Gray, N.; Fernandes, P.M.; Myburgh, E.; et al. Fast acting allosteric phosphofructokinase inhibitors block trypanosome glycolysis and cure acute African trypanosomiasis in mice. Nat. Commun. 2021, 12, 1052. [Google Scholar] [CrossRef]
- Klug, D.M.; Mavrogiannaki, E.M.; Forbes, K.C.; Silva, L.; Diaz-Gonzalez, R.; Pérez-Moreno, G.; Ceballos-Pérez, G.; Garcia-Hernández, R.; Bosch-Navarrete, C.; Cordón-Obras, C.; et al. Lead Optimization of 3,5-Disubstituted-7-Azaindoles for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 2021, 64, 9404–9430. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput.-Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Lätti, S.; Niinivehmas, S.; Pentikäinen, O.T. Rocker: Open source, easy-to-use tool for AUC and enrichment calculations and ROC visualization. J. Cheminf. 2016, 8, 1–5. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, version C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Toukmaji, A.; Sagui, C.; Board, J.; Darden, T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000, 113, 10913–10927. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, Q.; Wang, J.; Gao, P.; Xie, G.; Liu, H.; Yao, X. Molecular Modeling Study on the Interaction Mechanism between the LRRK2 G2019S Mutant and Type I Inhibitors by Integrating Molecular Dynamics Simulation, Binding Free Energy Calculations, and Pharmacophore Modeling. ACS Chem. Neurosci. 2022, 13, 599–612. [Google Scholar] [CrossRef]
- Zhang, Q.; Tan, S.; Xiao, T.; Liu, H.; Shah, S.J.A.; Liu, H. Probing the Molecular Mechanism of Rifampin Resistance Caused by the Point Mutations S456L and D441V on Mycobacterium tuberculosis RNA Polymerase through Gaussian Accelerated Molecular Dynamics Simulation. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef]
- Gilson, M.K.; Davis, M.E.; Luty, B.A.; McCammon, J.A. Computation of electrostatic forces on solvated molecules using the Poisson-Boltzmann equation. J. Phys. Chem. 1993, 97, 3591–3600. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Delauw, M.F.; Pays, E.; Steinert, M.; Aerts, D.; Van Meirvenne, N.; Le Ray, D. Inactivation and reactivation of a variant-specific antigen gene in cyclically transmitted Trypanosoma brucei. EMBO J. 1985, 4, 989–993. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | F733-0072 | F733-0407 | L368-0556 | L439-0038 |
|---|---|---|---|---|
| −53.72 ± 0.09 | −52.87 ± 0.11 | −53.60 ± 0.11 | −54.07 ± 0.11 | |
| 103.86 ± 0.33 | 95.42 ± 0.73 | 20.58 ± 0.38 | −20.70 ± 0.17 | |
| −91.93 ± 0.32 | −80.57 ± 0.70 | −14.48 ± 0.34 | 36.07 ± 0.12 | |
| −6.59 ± 0.01 | −6.53 ± 0.01 | −7.15 ± 0.01 | −6.95 ± 0.01 | |
| 50.14 ± 0.34 | 42.55 ± 0.75 | −33.01 ± 0.40 | −74.76 ± 0.20 | |
| −98.52 ± 0.32 | −87.10 ± 0.69 | −21.63 ± 0.34 | 29.12 ± 0.12 | |
| −48.38 ± 0.12 | −44.55 ± 0.12 | −54.64 ± 0.15 | −45.64 ± 0.13 | |
| IC50 (µM) | 1.11 ± 0.29 | 1.97 ± 0.94 | 0.85 ± 0.19 | 1.66 ± 0.95 |
| Compound ID | Donor | Acceptor | Distance (Å) | Angle (°) | Occupancy (%) |
|---|---|---|---|---|---|
| F733-0072 | LIGAND@N5-H3 | ALA433@O | 2.90 | 156.69 | 99.81 |
| ALA433@N-H | LIGAND@N1 | 3.04 | 150.22 | 96.26 | |
| ARG443@NH2-HH22 | LIGAND@O1 | 2.94 | 143.20 | 30.69 | |
| LIGAND@N6-H5 | SER436@OG | 3.26 | 144.88 | 28.37 | |
| ARG443@NH1-HH12 | LIGAND@O1 | 2.99 | 144.52 | 17.41 | |
| F733-0407 | LIGAND@N4-H3 | ALA433@O | 2.93 | 158.34 | 99.74 |
| ALA433@N-H | LIGAND@N1 | 3.01 | 146.45 | 92.75 | |
| LIGAND@N5-H4 | SER436@OG | 3.23 | 143.18 | 23.41 | |
| LIGAND@N5-H4 | ILE335@O | 3.03 | 147.91 | 19.60 | |
| L368-0556 | ALA433@N-H | LIGAND@N1 | 3.13 | 156.48 | 87.48 |
| LIGAND@N3-H36 | ASP493@OD1 | 2.74 | 165.06 | 80.09 | |
| LIGAND@N4-H2 | ALA433@O | 3.04 | 149.30 | 68.75 | |
| LIGAND@N5-H5 | SER336@OG | 3.17 | 146.78 | 44.38 | |
| LYS358@NZ-HZ1 | LIGAND@O1 | 2.82 | 150.06 | 33.47 | |
| LYS358@NZ-HZ2 | LIGAND@O1 | 2.82 | 149.50 | 33.38 | |
| LYS358@NZ-HZ3 | LIGAND@O1 | 2.82 | 149.72 | 28.26 | |
| L439-0038 | LIGAND@N2-H4 | ALA433@O | 2.88 | 159.00 | 99.55 |
| LIGAND@N3-H5 | ALA433@O | 3.06 | 151.9 | 95.34 | |
| ASP493@N-H | LIGAND@O3 | 2.93 | 158.29 | 95.09 | |
| SER436@N-H | LIGAND@O1 | 2.98 | 152.47 | 91.45 | |
| SER436@OG-HG | LIGAND@O1 | 2.80 | 161.36 | 69.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Luo, J.; Fu, C.; Kang, W.; Wang, L.; Tong, H.; Lun, Z.; Zhang, Q.; Lai, D.; Liu, H. Discovery of Novel CRK12 Inhibitors for the Treatment of Human African Trypanosomiasis: An Integrated Computational and Experimental Approach. Pharmaceuticals 2025, 18, 778. https://doi.org/10.3390/ph18060778
Li Q, Luo J, Fu C, Kang W, Wang L, Tong H, Lun Z, Zhang Q, Lai D, Liu H. Discovery of Novel CRK12 Inhibitors for the Treatment of Human African Trypanosomiasis: An Integrated Computational and Experimental Approach. Pharmaceuticals. 2025; 18(6):778. https://doi.org/10.3390/ph18060778
Chicago/Turabian StyleLi, Qin, Jiayi Luo, Chenggong Fu, Wenqingqing Kang, Lingling Wang, Henry Tong, Zhaorong Lun, Qianqian Zhang, Dehua Lai, and Huanxiang Liu. 2025. "Discovery of Novel CRK12 Inhibitors for the Treatment of Human African Trypanosomiasis: An Integrated Computational and Experimental Approach" Pharmaceuticals 18, no. 6: 778. https://doi.org/10.3390/ph18060778
APA StyleLi, Q., Luo, J., Fu, C., Kang, W., Wang, L., Tong, H., Lun, Z., Zhang, Q., Lai, D., & Liu, H. (2025). Discovery of Novel CRK12 Inhibitors for the Treatment of Human African Trypanosomiasis: An Integrated Computational and Experimental Approach. Pharmaceuticals, 18(6), 778. https://doi.org/10.3390/ph18060778