
Preclinical Characterization of Efficacy and Pharmacodynamic Properties of Finotonlimab, a Humanized Anti-PD-1 Monoclonal Antibody

 ,
,
Abstract

1. Introduction
2. Results
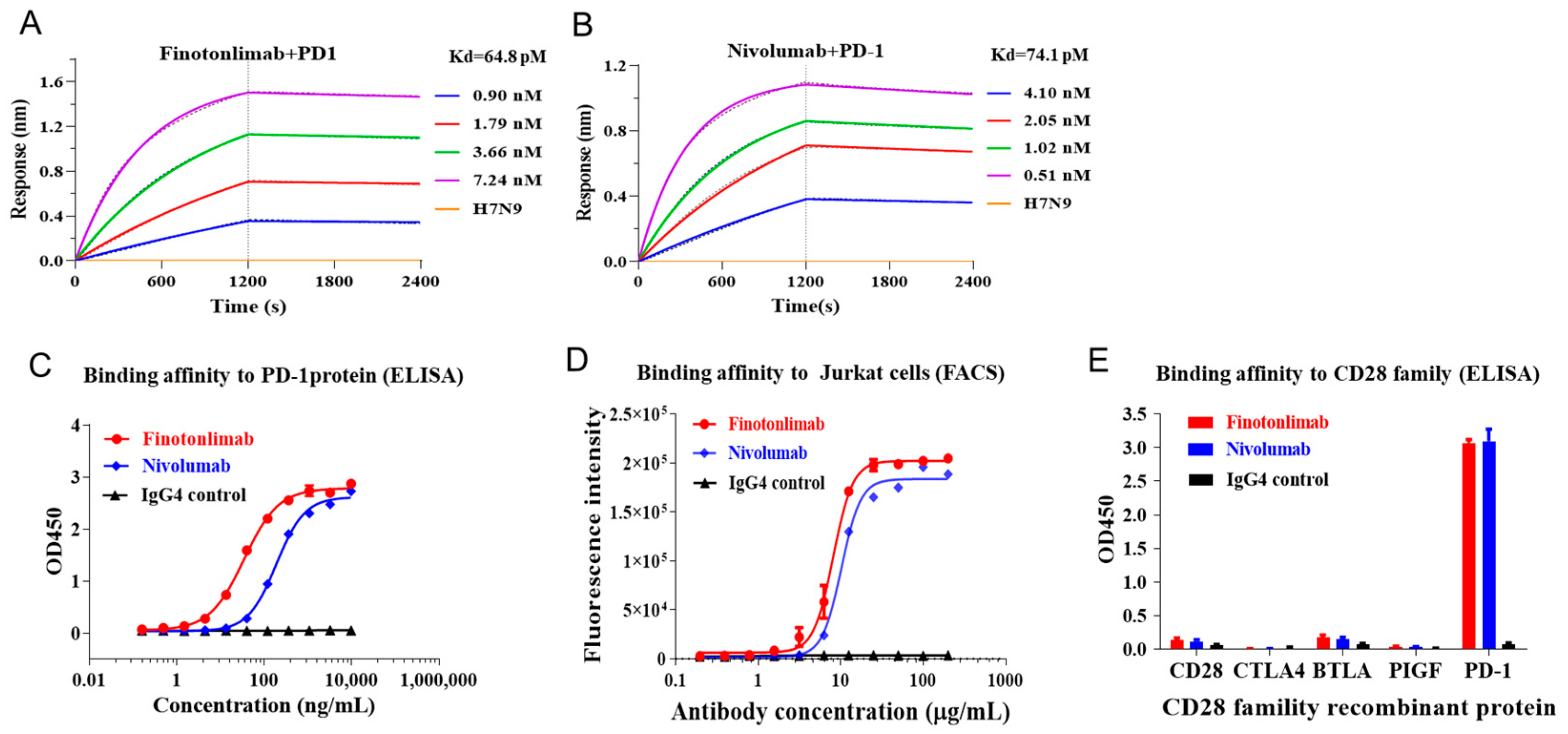
2.1. High Affinity and Binding Specificity of Finotonlimab to hPD-1
2.2. Epitope of Finotonlimab Overlaps with PD-L1/PD-L2 Binding Sites
2.3. In Vitro Effects on the Function of T Cell Activation by Finotonlimab
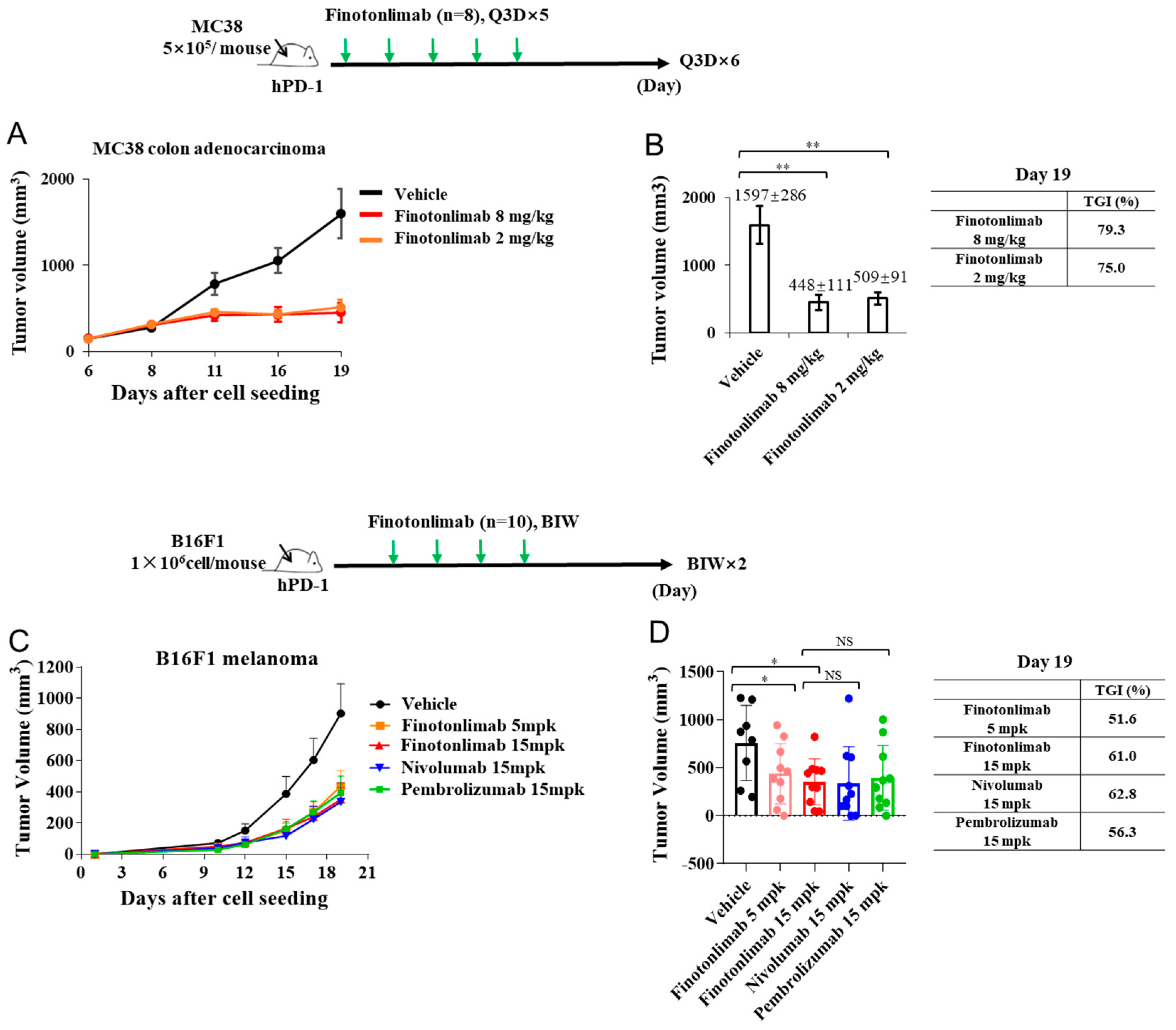
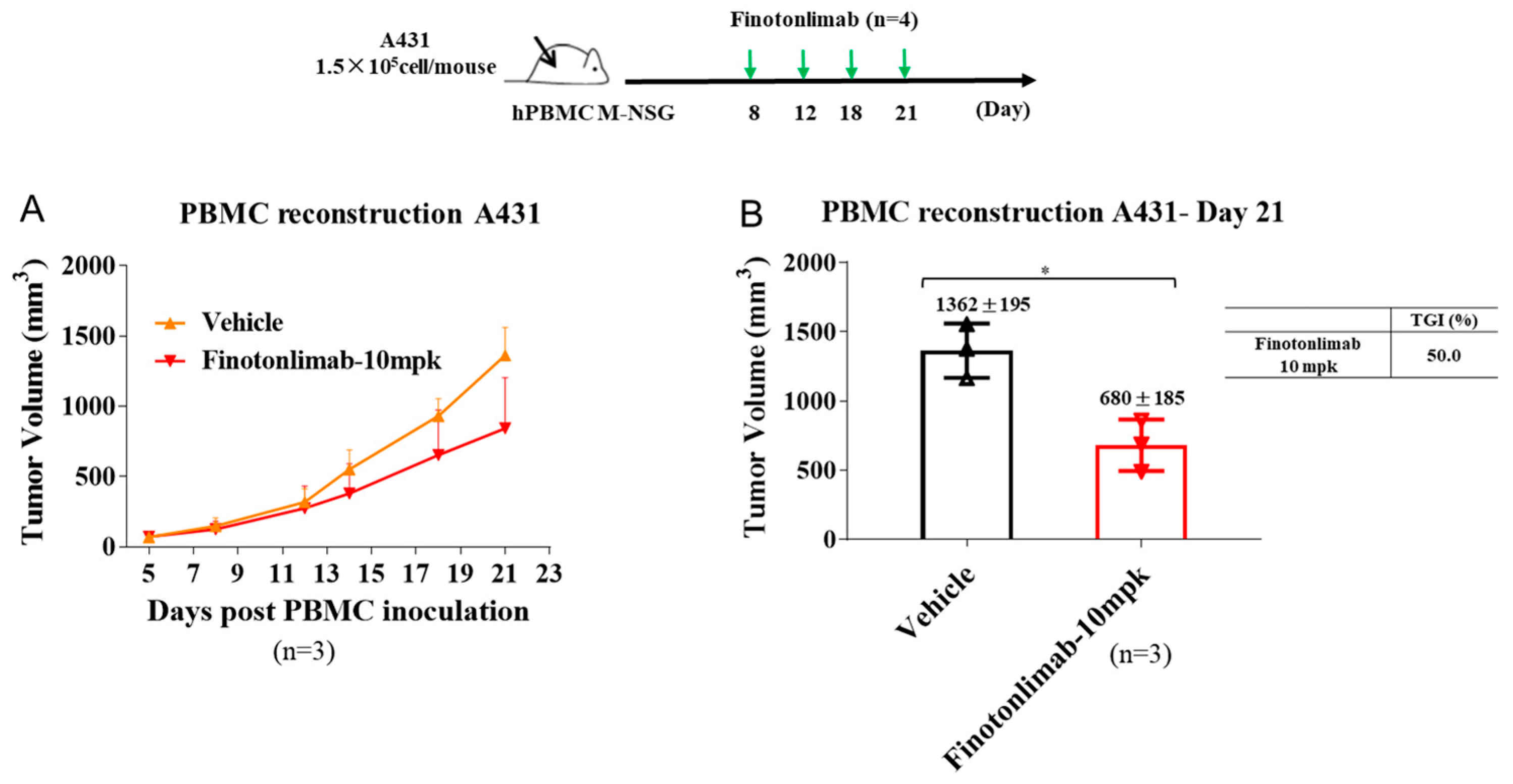
2.4. Finotonlimab Exhibited Antitumor Activity in Mouse Xenograft Models
2.5. Fc-Mediated Effector Functions of Finotonlimab
2.6. Comparative Pharmacokinetics of Finotonlimab and Anti-PD-1 Antibodies in Cynomolgus Monkeys
2.7. Pharmacokinetic and Pharmacodynamic Profiles of Finotonlimab in Cynomolgus Monkeys Following Single and Repeated Administrations
2.8. Toxicology Evaluation Following Repeated Administrations of Finotonlimab in Cynomolgus Monkeys
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Affinity Measurements by Octet
4.4. Epitope Mapping of PD-L1/PD-L2 and Finotonlimab Binding
4.5. Structural Modeling of Finotonlimab/PD-1 Complex and Epitope Analysis
4.6. Binding Affinity of Finotonlimab by ELISA
4.7. Blocking Capability of Finotonlimab by ELISA
4.8. Binding of the Finotonlimab to PD-1 Cell Surfaces by FACS
4.9. Evaluating Human PD-1/PD-L1 Blocking Capability of Finotonlimab by FACS
4.10. T Cell Activation Assay-Associated Luciferase Reporter System
4.11. In Vitro Mixed Lymphocyte Reaction
4.12. ADCC/ADCP-Activation Assays
4.13. CDC Cytotoxicity Assay
4.14. MC38 Tumor Mouse Models
4.15. B16F1 Tumor Mouse Models
4.16. hPBMC-Reconstituted A431 Mouse Models
4.17. Pharmacokinetic and Toxicity Assessments of Finotonlimab in Cynomolgus Monkeys
4.18. Anesthesia and Euthanasia
4.19. Receptor Occupancy Assay
4.20. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, J.J.; Chu, E. Recent Advances in the Clinical Development of Immune Checkpoint Blockade Therapy for Mismatch Repair Proficient (pMMR)/non-MSI-H Metastatic Colorectal Cancer. Clin. Color. Cancer 2018, 17, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Centanni, M.; Moes, D.; Trocóniz, I.; Ciccolini, J.; Van Hasselt, J.G.C. Clinical Pharmacokinetics and Pharmacodynamics of Immune Checkpoint Inhibitors. Clin. Pharmacokinet. 2019, 58, 835–857. [Google Scholar] [CrossRef]
- Parvez, A.; Choudhary, F.; Mudgal, P.; Khan, R.; Qureshi, K.A.; Farooqi, H.; Aspatwar, A. PD-1 and PD-L1: Architects of immune symphony and immunotherapy breakthroughs in cancer treatment. Front. Immunol. 2023, 14, 1296341. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Chen, L. PD-1 as an immune modulatory receptor. Cancer J. 2014, 20, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G.; et al. PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. Int. J. Mol. Sci. 2021, 22, 5123. [Google Scholar] [CrossRef]
- Yasutoshi, A.; Akemi, K.; Hiroyuki, N.; Yasumasa, I.; Takeshi, T.; Hideo, Y.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar]
- Yao, S.; Wang, S.; Zhu, Y.; Luo, L.; Chen, L. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 2009, 113, 5811–5818. [Google Scholar] [CrossRef]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Sekaly, R.P. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Chames, P.; Regenmortel, M.V.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.Y.; Kim, H.G.; Kwak, M.W.; Kang, T.H. Fc Receptor Variants and Disease: A Crucial Factor to Consider in the Antibody Therapeutics in Clinic. Int. J. Mol. Sci. 2021, 22, 9489. [Google Scholar] [CrossRef]
- Shi, Y.; Guo, W.; Wang, W.; Wu, Y.; Fang, M.; Huang, X.; Han, P.; Zhang, Q.; Dong, P.; Zhou, X.; et al. Finotonlimab with chemotherapy in recurrent or metastatic head and neck cancer: A randomized phase 3 trial. Nat. Med. 2024, 30, 2568–2575. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcγR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef]
- Cheng, Z.J.; Garvin, D.; Paguio, A.; Moravec, R.; Engel, L.; Fan, F.; Surowy, T. Development of a robust reporter-based ADCC assay with frozen, thaw-and-use cells to measure Fc effector function of therapeutic antibodies. J. Immunol. Methods 2014, 414, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Uccellini, M.B.; Aslam, S.; Liu, S.T.H.; Alam, F.; García-Sastre, A. Development of a Macrophage-Based ADCC Assay. Vaccines 2021, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Bumbaca, D.; Pastuskovas, C.V.; Boswell, C.A.; West, D.; Cowan, K.J.; Chiu, H.; McBride, J.; Johnson, C.; Xin, Y.; et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs 2016, 8, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Kurino, T.; Matsuda, R.; Terui, A.; Suzuki, H.; Kokubo, T.; Uehara, T.; Hatakeyama, H.; Hisaka, A.; Arano, Y. Poor outcome with anti-programmed death-ligand 1 (PD-L1) antibody due to poor pharmacokinetic properties in PD-1/PD-L1 blockade-sensitive mouse models. J. Immunother. Cancer 2020, 8, e000400. [Google Scholar] [CrossRef]
- Gridelli, C.; Ardizzoni, A.; Barberis, M.; Cappuzzo, F.; Casaluce, F.; Danesi, R.; Troncone, G.; De Marinis, F. Predictive biomarkers of immunotherapy for non-small cell lung cancer: Results from an Experts Panel Meeting of the Italian Association of Thoracic Oncology. Transl. Lung Cancer Res. 2017, 6, 373–386. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef] [PubMed]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef]
- Zhang, T.; Song, X.; Xu, L.; Ma, J.; Zhang, Y.; Gong, W.; Zhang, Y.; Zhou, X.; Wang, Z.; Wang, Y.; et al. The binding of an anti-PD-1 antibody to FcγRΙ has a profound impact on its biological functions. Cancer Immunol. Immunother. 2018, 67, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Buijsse, A.O.; van den Bremer, E.T.J.; Verwilligen, A.Y.W.; Bleeker, W.K.; Thorpe, S.J.; Killestein, J.; Polman, C.H.; Aalberse, R.C.; Schuurman, J.; et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat. Biotechnol. 2009, 27, 767–771. [Google Scholar] [CrossRef]
- Swisher, J.F.; Feldman, G.M. The many faces of FcRI: Implications for therapeutic antibody function. Immunol. Rev. 2015, 268, 160–174. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Publ. Group 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Delidakis, G.; Kim, J.E.; George, K.; Georgiou, G. Improving Antibody Therapeutics by Manipulating the Fc Domain: Immunological and Structural Considerations. Annu. Rev. Biomed. Eng. 2022, 24, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Shchelokov, D.; Demin, O. (Eds.) Abstract 2233: Prediction and Comparison of PD-1 Receptor Occupancy in the Tumor after Treatment with Immune Checkpoint Inhibitors. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA, 27–28 April; 22–24 June 2020. [Google Scholar]
- Wang, J.; Fei, K.; Jing, H.; Wu, Z.; Wu, W.; Zhou, S.; Ni, H.; Chen, B.; Xiong, Y.; Liu, Y.; et al. Durable blockade of PD-1 signaling links preclinical efficacy of sintilimab to its clinical benefit. Mabs 2019, 11, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, F.; Dong, L.H.; Xing, M.J.; Song, H.F. Receptor occupancy measurement of anti-PD-1 antibody drugs in support of clinical trials. Bioanalysis 2019, 11, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.; Wei, H.; Yang, F.; Wang, S.; Yang, B.; Zheng, Y.; Zhu, J.; Yan, S. Preclinical Characterization of GLS-010 (Zimberelimab), a Novel Fully Human Anti-PD-1 Therapeutic Monoclonal Antibody for Cancer. Front. Oncol. 2021, 11, 736955. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ghosh, S.; Sharma, G.; Wang, Z.; Kehry, M.; Marino, M.; Neben, T.Y.; Lu, S.; Luo, S.; Roberts, S.; et al. Preclinical characterization of dostarlimab, a therapeutic anti-PD-1 antibody with potent activity to enhance immune function in in vitro cellular assays and in vivo animal models. mAbs 2021, 13, 1954136. [Google Scholar] [CrossRef]
- Sharma, S. Pan-TGFβ inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. OncoImmunology 2020, 9, 1811605. [Google Scholar]
- Greenlee, J.D.; King, M.R. A syngeneic MC38 orthotopic mouse model of colorectal cancer metastasis. Biol. Methods Protoc. 2022, 7, bpac024. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tanaka, Y.; Takeda, R.; Nakamoto, A.; Nakamoto, M.; Yagita, H.; Sakai, T. Soy isoflavone genistein attenuates the efficacy of immune checkpoint therapy in C57BL/6 mice inoculated with B16F1 melanoma and a high PD-L1 expression level reflects tumor resistance. J. Clin. Biochem. Nutr. 2024, 74, 57–62. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, H.; Wu, M.; Wang, J.; Wu, Z.; Fu, F.; Liu, Y.; Lu, J.; Yao, Y.; Luo, N.; et al. Pharmacology, pharmacokinetics, and toxicity characterization of a novel anti-CD73 therapeutic antibody IBI325 for cancer immunotherapy. Int. J. Biol. Macromol. 2023, 229, 158–167. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KD (M) | Kon (M−1s−1) | Kdis (s−1) | |
|---|---|---|---|
| 7.13 × 10−11 | 2.95 × 105 | 2.10 × 10−5 | |
| Finotonlimab | 5.46 × 10−11 | 3.06 × 105 | 1.67 × 10−5 |
| 6.84 × 10−11 | 3.03 × 105 | 2.07 × 10−5 | |
| Mean | 6.48 × 10−11 | 3.01 × 105 | 1.95 × 10−5 |
| 6.80 × 10−11 | 6.89 × 105 | 4.68 × 10−5 | |
| Nivolumab | 7.92 × 10−11 | 7.02 × 105 | 5.56 × 10−5 |
| 7.50 × 10−11 | 6.84 × 105 | 5.13 × 10−5 | |
| Mean | 7.41 × 10−11 | 6.92 × 105 | 5.12 × 10−5 |
| Dose (mg/kg) | Gender | / | T1/2 h | Cmax μg/mL | AUClast h·mg/mL | AUCinf h·mg/mL | MRT h | Cmax Ratio | AUC Ratio |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Mean | 178.34 | 30.13 | 2.71 | 4.46 | 92.09 | 1.00 | 1.00 |
| SD | 45.79 | 2.59 | 0.37 | 1.25 | 6.06 | ||||
| F | Mean | 128.50 | 25.02 | 3.44 | 3.87 | 136.68 | 1.00 | 1.00 | |
| SD | 25.43 | 9.95 | 0.85 | 0.91 | 25.12 | ||||
| 3 | M | Mean | 169.15 | 84.58 | 10.89 | 14.40 | 132.52 | 2.81 | 4.02 |
| SD | 26.36 | 7.20 | 2.30 | 1.58 | 34.03 | ||||
| F | Mean | 202.20 | 84.28 | 12.53 | 14.23 | 194.32 | 3.37 | 3.64 | |
| SD | 41.68 | 5.71 | 1.23 | 2.47 | 6.66 | ||||
| 10 | M | Mean | 191.71 | 339.39 | 44.22 | 61.49 | 151.24 | 11.26 | 16.33 |
| SD | 44.67 | 33.15 | 10.76 | 5.38 | 62.38 | ||||
| F | Mean | 215.62 | 299.93 | 49.72 | 60.24 | 217.85 | 11.99 | 14.45 | |
| SD | 154.11 | 27.57 | 10.02 | 20.85 | 48.02 |
| Receptor Occupation | 1 mg/kg | 3 mg/kg | 10 mg/kg | |||
|---|---|---|---|---|---|---|
| Time for Blood Collection | Mean (%) | SD (%) | Mean (%) | SD (%) | Mean (%) | SD (%) |
| Before dose | 8.65 | 2.03 | 8.94 | 2.46 | 9.67 | 4.76 |
| 2 h | 89.52 | 6.60 | 93.90 | 3.11 | 95.35 | 8.51 |
| 24 h | 88.47 | 4.38 | 87.98 | 3.87 | 93.17 | 2.61 |
| 72 h | 93.33 | 2.79 | 93.52 | 9.54 | 100.36 | 5.07 |
| 168 h | 92.09 | 5.44 | 87.68 | 4.65 | 93.69 | 4.63 |
| 336 h | 59.00 | 40.16 | 76.51 | 22.26 | 89.80 | 19.64 |
| 504 h | 33.68 | 41.48 | 68.09 | 47.21 | 70.91 | 33.26 |
| 672 h | 10.43 | 4.32 | 41.26 | 39.19 | 50.00 | 43.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Y.; Yang, X.; Li, J.; Guo, E.; Wang, H.; Sun, C.; Hong, Z.; Zhang, X.; Jia, J.; Wang, R.; et al. Preclinical Characterization of Efficacy and Pharmacodynamic Properties of Finotonlimab, a Humanized Anti-PD-1 Monoclonal Antibody. Pharmaceuticals 2025, 18, 395. https://doi.org/10.3390/ph18030395
Yao Y, Yang X, Li J, Guo E, Wang H, Sun C, Hong Z, Zhang X, Jia J, Wang R, et al. Preclinical Characterization of Efficacy and Pharmacodynamic Properties of Finotonlimab, a Humanized Anti-PD-1 Monoclonal Antibody. Pharmaceuticals. 2025; 18(3):395. https://doi.org/10.3390/ph18030395
Chicago/Turabian StyleYao, Yunqi, Xiaoning Yang, Jing Li, Erhong Guo, Huiyu Wang, Chunyun Sun, Zhangyong Hong, Xiao Zhang, Jilei Jia, Rui Wang, and et al. 2025. "Preclinical Characterization of Efficacy and Pharmacodynamic Properties of Finotonlimab, a Humanized Anti-PD-1 Monoclonal Antibody" Pharmaceuticals 18, no. 3: 395. https://doi.org/10.3390/ph18030395
APA StyleYao, Y., Yang, X., Li, J., Guo, E., Wang, H., Sun, C., Hong, Z., Zhang, X., Jia, J., Wang, R., Ma, J., Dai, Y., Deng, M., Yu, C., Sun, L., & Xie, L. (2025). Preclinical Characterization of Efficacy and Pharmacodynamic Properties of Finotonlimab, a Humanized Anti-PD-1 Monoclonal Antibody. Pharmaceuticals, 18(3), 395. https://doi.org/10.3390/ph18030395