
In Vitro Activity of the Triazinyl Diazepine Compound FTSD2 Against Drug-Resistant Mycobacterium tuberculosis Strains

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Susceptibility Testing
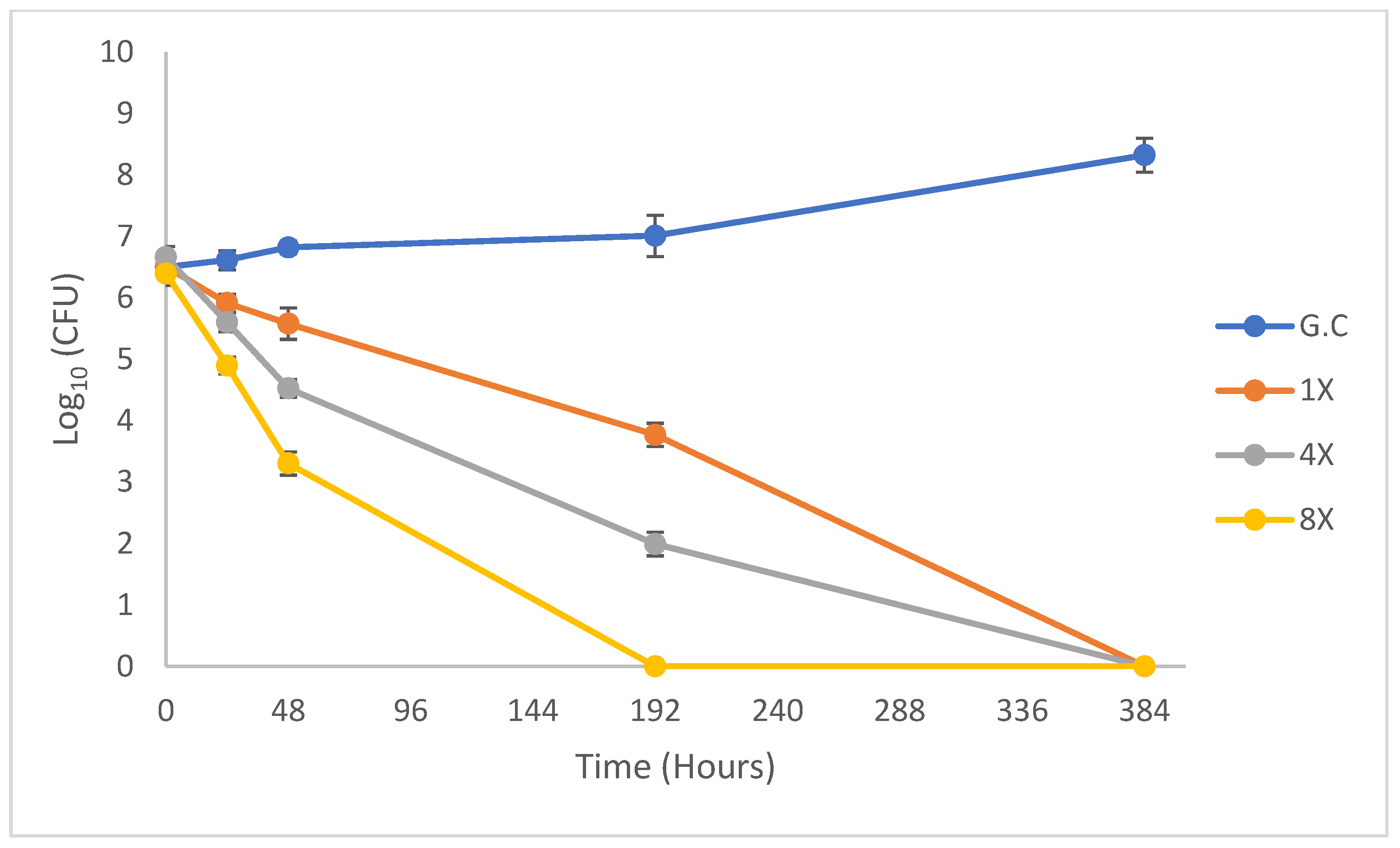
2.2. Dose−Response of M. tuberculosis
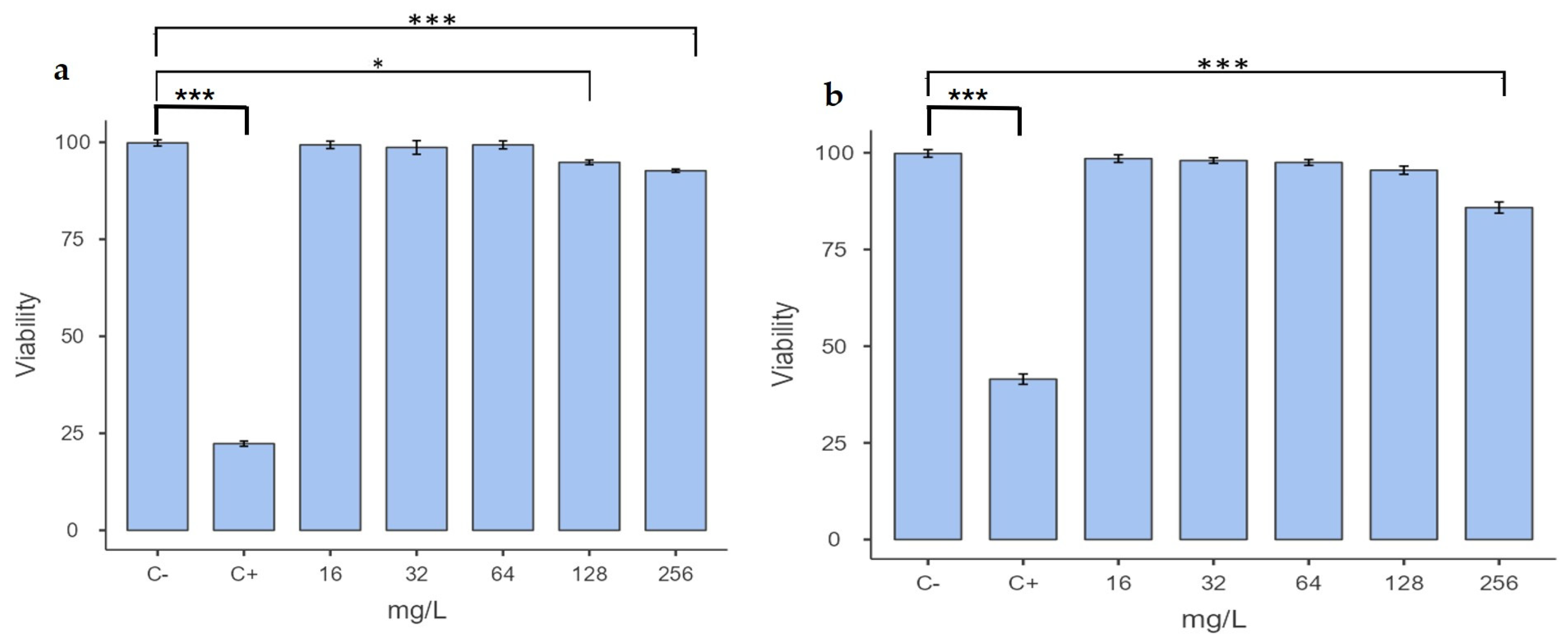
2.3. Determination of Cytotoxicity
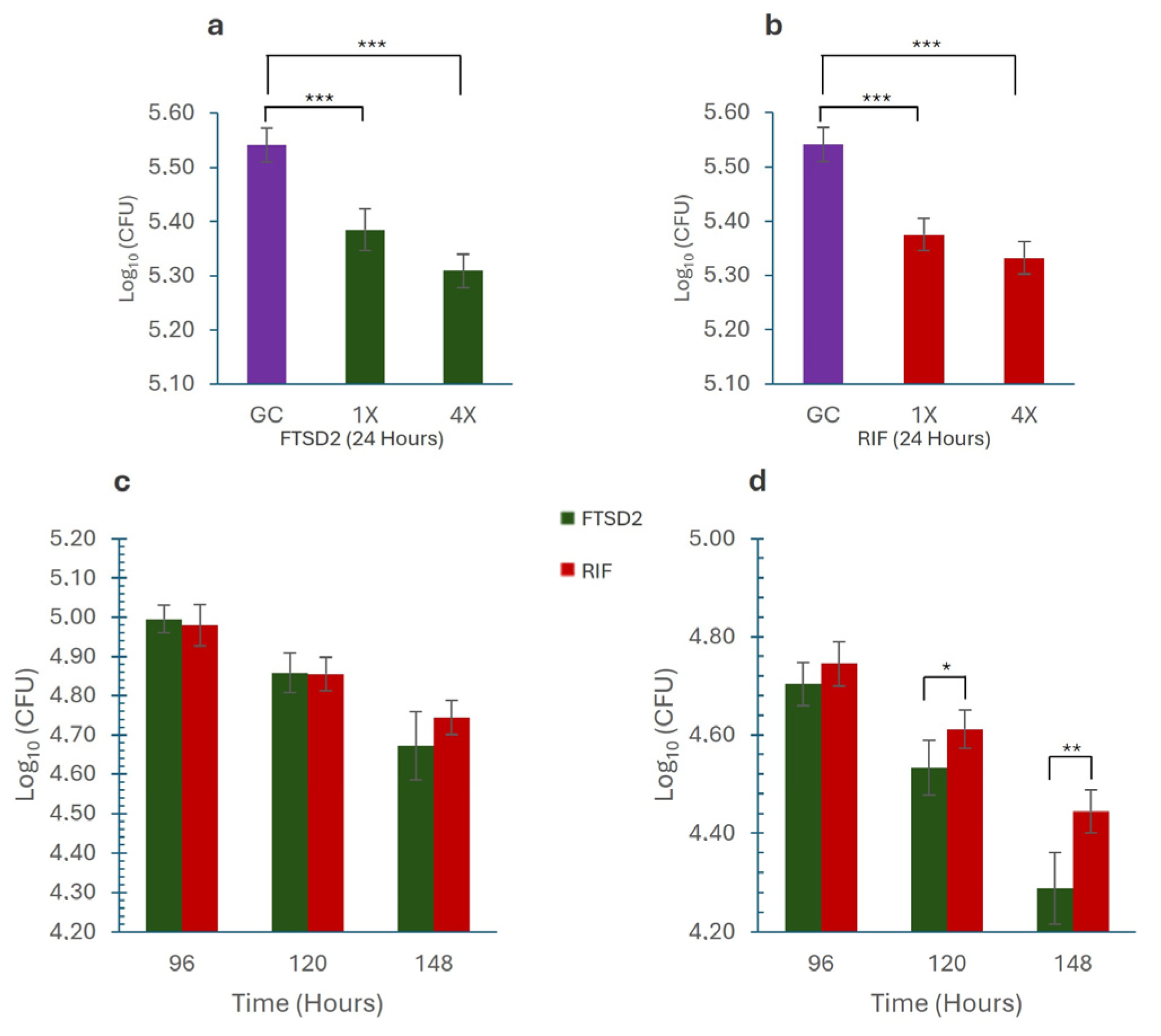
2.4. Macrophage Assay
3. Discussion
4. Materials and Methods
4.1. Strains
4.2. Triaminopyrimidine-Diazepane Compounds
4.3. Preparation of Stock Solutions
4.4. Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
4.5. Kill Rate Determination
4.6. Cytotoxicity Assay in Macrophages
4.6.1. MMT Assay
4.6.2. MTT Assay
4.7. Macrophage Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MIC | Minimum inhibitory concentration |
| MBC | Minimum bactericidal concentration |
| TB | Tuberculosis |
| MDR-TB | Multidrug-resistant tuberculosis |
| hMDMs | Human monocyte-derived macrophages |
| MMT | Macrophage-to-myofibroblast transition |
| DPBS | Dulbecco’s phosphate buffered saline |
| DMSO | Dimethyl sulfoxide |
References
- World Health Organization. Global Tuberculosis Report 2024; WHO: Geneva, Switzerland, 2024.
- Cohen, J. Approval of Novel TB Drug Celebrated—With Restraint. Science (1979) 2013, 339, 130. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J.; Lo, J.H. Delamanid: First Global Approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef]
- Thakare, R.; Dasgupta, A.; Chopra, S. Pretomanid for the Treatment of Pulmonary Tuberculosis. Drugs Today 2020, 56, 655. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Susceptible Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2022.
- World Health Organization Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023; ISBN 9789240083851.
- Moe, S.; Rekart, M.L.; Hernandez, D.; Sholpan, A.; Ismailov, A.; Oluya, M.; Bayniyazova, A.; Zinaida, T.; Nargiza, P.; Gomez-Restrepo, C.; et al. Primary Bedaquiline Resistance in Karakalpakstan, Uzbekistan. Int. J. Tuberc. Lung Dis. 2023, 27, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Huitric, E.; Verhasselt, P.; Koul, A.; Andries, K.; Hoffner, S.; Andersson, D.I. Rates and Mechanisms of Resistance Development in Mycobacterium Tuberculosis to a Novel Diarylquinoline ATP Synthase Inhibitor. Antimicrob. Agents. Chemother. 2010, 54, 1022–1028. [Google Scholar] [CrossRef]
- Kadura, S.; King, N.; Nakhoul, M.; Zhu, H.; Theron, G.; Köser, C.U.; Farhat, M. Systematic Review of Mutations Associated with Resistance to the New and Repurposed Mycobacterium Tuberculosis Drugs Bedaquiline, Clofazimine, Linezolid, Delamanid and Pretomanid. J. Antimicrob. Chemother. 2020, 75, 2031–2043. [Google Scholar] [CrossRef]
- Andries, K.; Villellas, C.; Coeck, N.; Thys, K.; Gevers, T.; Vranckx, L.; Lounis, N.; de Jong, B.C.; Koul, A. Acquired Resistance of Mycobacterium Tuberculosis to Bedaquiline. PLoS ONE 2014, 9, e102135. [Google Scholar] [CrossRef]
- Millard, J.; Rimmer, S.; Nimmo, C.; O’Donnell, M. Therapeutic Failure and Acquired Bedaquiline and Delamanid Resistance in Treatment of Drug-Resistant TB. Emerg. Infect. Dis. 2023, 29, 1081. [Google Scholar] [CrossRef]
- Shang, Y.; Chen, S.; Shi, W.; Nie, W.; Jing, W.; Huo, F.; Xue, Y.; Dong, L.; Jiang, G.; Huang, H.; et al. Bedaquiline Resistance Pattern in Clofazimine-Resistant Clinical Isolates of Tuberculosis Patients. J. Glob. Antimicrob. Resist. 2023, 33, 294–300. [Google Scholar] [CrossRef]
- Mallick, J.S.; Nair, P.; Abbew, E.T.; Van Deun, A.; Decroo, T. Acquired Bedaquiline Resistance during the Treatment of Drug-Resistant Tuberculosis: A Systematic Review. JAC Antimicrob. Resist. 2022, 4, dlac029. [Google Scholar] [CrossRef]
- Diriba, G.; Alemu, A.; Yenew, B.; Tola, H.H.; Gamtesa, D.F.; Mollalign, H.; Eshetu, K.; Moga, S.; Abdella, S.; Tollera, G.; et al. Epidemiology of Extensively Drug-Resistant Tuberculosis among Patients with Multidrug-Resistant Tuberculosis: A Systematic Review and Meta-Analysis. Int. J. Infect. Dis. 2023, 132, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.M.; Quiroga, J.; Abonia, R.; Crespo, M.D.; Aranaga, C.; Martínez-Martínez, L.; Sortino, M.; Barreto, M.; Burbano, M.E.; Insuasty, B. Synthesis of Novel Triazine-Based Chalcones and 8,9-Dihydro-7H-Pyrimido[4,5-b][1,4]Diazepines as Potential Leads in the Search of Anticancer, Antibacterial and Antifungal Agents. Int. J. Mol. Sci. 2024, 25, 3623. [Google Scholar] [CrossRef]
- Grieco, I.; Bassani, D.; Trevisan, L.; Salmaso, V.; Cescon, E.; Prencipe, F.; Da Ros, T.; Martinez-Gonzalez, L.; Martinez, A.; Spalluto, G.; et al. 7-Amino-[1,2,4]Triazolo[1,5-a][1,3,5]Triazines as CK1δ Inhibitors: Exploring Substitutions at the 2 and 5-Positions. Bioorg. Chem. 2024, 151, 107659. [Google Scholar] [CrossRef]
- Liu, B. A Systematic Review on Antitumor Agents with 1, 3, 5-Triazines. Med. Chem. 2015, 5, 131–148. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Liu, Y.; Harshbarger, W.; Huang, L.; Wang, J.; Deng, X.; Capuzzi, S.J.; Muratov, E.N.; Tropsha, A.; Muthuswamy, S.; et al. Synthesis and Structure-Activity Relationships of DCLK1 Kinase Inhibitors Based on a 5,11-Dihydro-6 H-Benzo[ e]Pyrimido[5,4- b][1,4]Diazepin-6-One Scaffold. J. Med. Chem. 2020, 63, 7817–7826. [Google Scholar] [CrossRef]
- Zeng, J.; Platig, J.; Cheng, T.Y.; Ahmed, S.; Skaf, Y.; Potluri, L.P.; Schwartz, D.; Steen, H.; Branch Moody, D.; Husson, R.N. Protein Kinases PknA and PknB Independently and Coordinately Regulate Essential Mycobacterium Tuberculosis Physiologies and Antimicrobial Susceptibility. PLoS Pathog. 2020, 16, e1008452. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Hurtado, M.; Meffre, P.; Szurmant, H.; Benfodda, Z. Synthesis of Histidine Kinase Inhibitors and Their Biological Properties. Med. Res. Rev. 2020, 40, 1440–1495. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. 1,3,5-Triazines: A Promising Scaffold for Anticancer Drugs Development. Eur. J. Med. Chem. 2017, 142, 523–549. [Google Scholar] [CrossRef]
- Singh, R.; Dwivedi, S.P.; Gaharwar, U.S.; Meena, R.; Rajamani, P.; Prasad, T. Recent Updates on Drug Resistance in Mycobacterium Tuberculosis. J. Appl. Microbiol. 2020, 128, 1547–1567. [Google Scholar] [CrossRef]
- Coll, P. Active Drugs against Mycobacterium Tuberculosis. Enferm. Infecc. Microbiol. Clin. 2009, 27, 474–480. [Google Scholar] [CrossRef]
- Santos, N.C.D.S.; Scodro, R.B.D.L.; Leal, D.C.B.; Do Prado, S.M.T.; Micheletti, D.F.; Sampiron, E.G.; Costacurta, G.F.; De Almeida, A.L.; Da Silva, L.A.F.; Ieque, A.L.; et al. Determination of Minimum Bactericidal Concentration, in Single or Combination Drugs, against Mycobacterium Tuberculosis. Future Microbiol. 2020, 15, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, R.; Tornheim, J.A.; Diricks, M.; De Bruyne, K.; Sadani, M.; Shetty, A.; Rodrigues, C. Linezolid Resistance in Mycobacterium Tuberculosis Isolates at a Tertiary Care Centre in Mumbai, India. Indian J. Med. Res. Suppl. 2021, 154, 85–89. [Google Scholar] [CrossRef]
- Bax, H.I.; Bakker-Woudenberg, I.A.J.M.; de Vogel, C.P.; van der Meijden, A.; Verbon, A.; de Steenwinkel, J.E.M. The Role of the Time-Kill Kinetics Assay as Part of a Preclinical Modeling Framework for Assessing the Activity of Anti-Tuberculosis Drugs. Tuberculosis 2017, 105, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Gumbo, T.; Louie, A.; Liu, W.; Ambrose, P.G.; Bhavnani, S.M.; Brown, D.; Drusano, G.L. Isoniazid’s Bactericidal Activity Ceases Because of the Emergence of Resistance, Not Depletion of Mycobacterium Tuberculosis in the Log Phase of Growth. J. Infect. Dis. 2007, 195, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Gurumurthy, M.; Chaik, B.; Yeo, M.; Lu, Q.; Naftalin, C.M.; Paton, N.I. Effects of Increasing Concentrations of Rifampicin on Different Mycobacterium Tuberculosis Lineages in a Whole-Blood Bactericidal Activity Assay. Antimicrob. Agents Chemother. 2022, 66, e01699-21. [Google Scholar] [CrossRef]
- Schwan, W.R. SK-03-92 Drug Kills Intracellular Mycobacterium Tuberculosis. Antibiotics 2023, 12, 1385. [Google Scholar] [CrossRef]
- Schaaf, K.; Hayley, V.; Speer, A.; Wolschendorf, F.; Niederweis, M.; Kutsch, O.; Sun, J. A Macrophage Infection Model to Predict Drug Efficacy Against Mycobacterium Tuberculosis. Assay Drug Dev. Technol. 2016, 14, 345–354. [Google Scholar] [CrossRef]
- Schön, T.; Werngren, J.; Machado, D.; Borroni, E.; Wijkander, M.; Lina, G.; Mouton, J.; Matuschek, E.; Kahlmeter, G.; Giske, C.; et al. Antimicrobial Susceptibility Testing of Mycobacterium Tuberculosis Complex Isolates—the EUCAST Broth Microdilution Reference Method for MIC Determination. Clin. Microbiol. Infect. 2020, 26, 1488–1492. [Google Scholar] [CrossRef]
- Ochoa, R.; Rocha-Roa, C.; Marín-Villa, M.; Robledo, S.M.; Varela-M, R.E. Search of Allosteric Inhibitors and Associated Proteins of an AKT-like Kinase from Trypanosoma Cruzi. Int. J. Mol. Sci. 2018, 19, 3951. [Google Scholar] [CrossRef]
- Khara, J.S.; Priestman, M.; Uhía, I.; Hamilton, M.S.; Krishnan, N.; Wang, Y.; Yang, Y.Y.; Langford, P.R.; Newton, S.M.; Robertson, B.D.; et al. Unnatural Amino Acid Analogues of Membrane-Active Helical Peptides with Anti-Mycobacterial Activity and Improved Stability. J. Antimicrob. Chemother. 2016, 71, 2181–2191. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. tuberculosis Strains | Number of Strains | MIC Range (mg/L) | MBC/MIC Ratio |
|---|---|---|---|
| H37Rv | 1 | 1 | 1 |
| H37Ra | 1 | 1 | 1 |
| ATCC 35822 | 1 | 0.5 | 1 |
| ATCC 35820 | 1 | 1 | 1 |
| ATCC 35837 | 1 | 1 | 1 |
| Clinical Isolates | |||
| Isoniazid-Resistant | 3 | 0.5–1 | 1 |
| Isoniazid and Rifampicin-Resistant | 2 | 0.25–0.5 | 1 |
| Isoniazid, Rifampicin, and Ethionamide-Resistant | 2 | 0.5–1 | 1 |
| Nontuberculous Mycobacteria | |||
| M. avium complex | 2 | >50 | - |
| M. abscessus | 3 | >50 | - |
| M. chelonae | 2 | >50 | - |
| M. fortuitum | 5 | >50 | - |
| M. smegmatis mc2155 | 1 | >50 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aranaga, C.; Varela, R.; Falco, A.; Villa, J.; Moreno, L.M.; Causse, M.; Martínez-Martínez, L. In Vitro Activity of the Triazinyl Diazepine Compound FTSD2 Against Drug-Resistant Mycobacterium tuberculosis Strains. Pharmaceuticals 2025, 18, 360. https://doi.org/10.3390/ph18030360
Aranaga C, Varela R, Falco A, Villa J, Moreno LM, Causse M, Martínez-Martínez L. In Vitro Activity of the Triazinyl Diazepine Compound FTSD2 Against Drug-Resistant Mycobacterium tuberculosis Strains. Pharmaceuticals. 2025; 18(3):360. https://doi.org/10.3390/ph18030360
Chicago/Turabian StyleAranaga, Carlos, Ruben Varela, Aura Falco, Janny Villa, Leydi M. Moreno, Manuel Causse, and Luis Martínez-Martínez. 2025. "In Vitro Activity of the Triazinyl Diazepine Compound FTSD2 Against Drug-Resistant Mycobacterium tuberculosis Strains" Pharmaceuticals 18, no. 3: 360. https://doi.org/10.3390/ph18030360
APA StyleAranaga, C., Varela, R., Falco, A., Villa, J., Moreno, L. M., Causse, M., & Martínez-Martínez, L. (2025). In Vitro Activity of the Triazinyl Diazepine Compound FTSD2 Against Drug-Resistant Mycobacterium tuberculosis Strains. Pharmaceuticals, 18(3), 360. https://doi.org/10.3390/ph18030360