
Recent Advancements in the Development of HDAC/Tubulin Dual-Targeting Inhibitors



Abstract

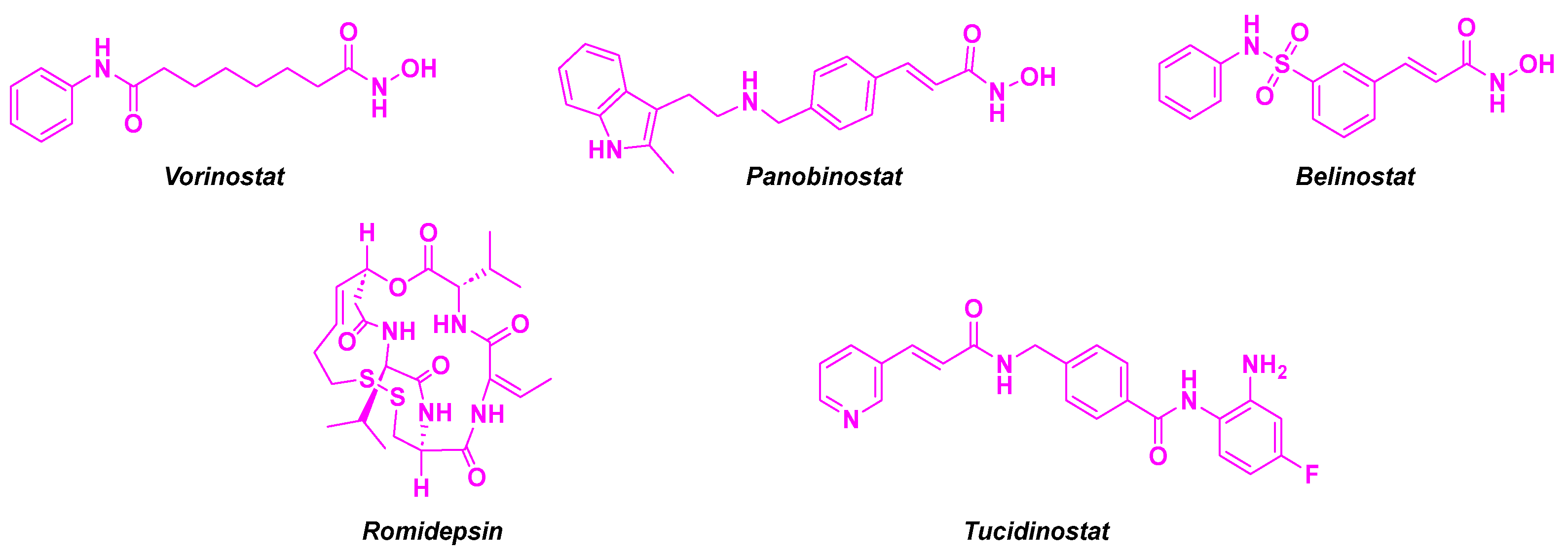
1. Introduction
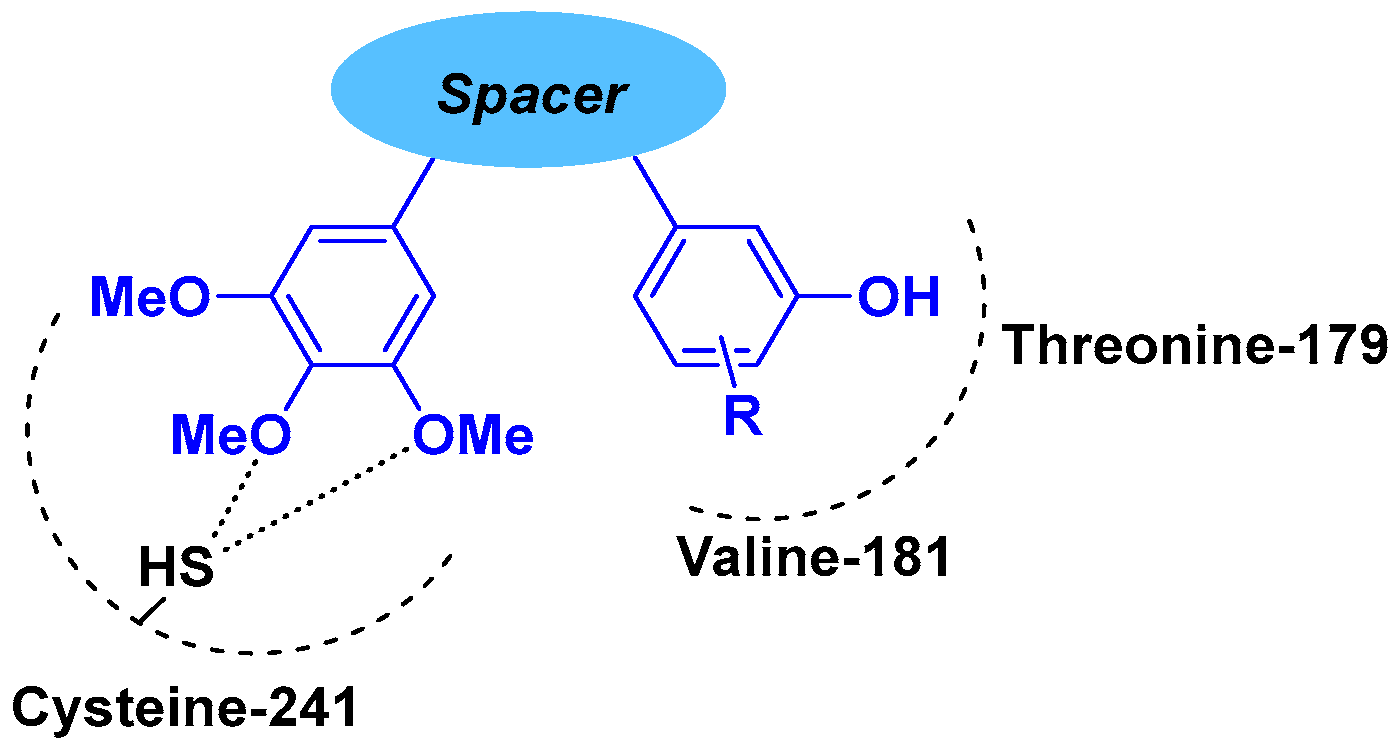
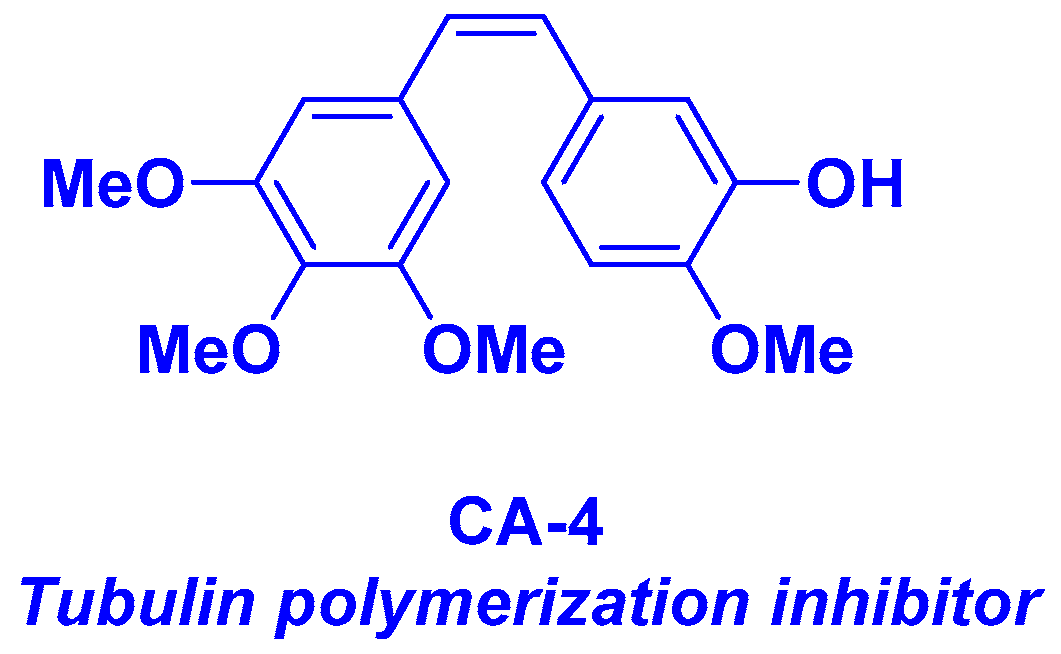
2. Combretastatin-A4 Motif and Related Structures
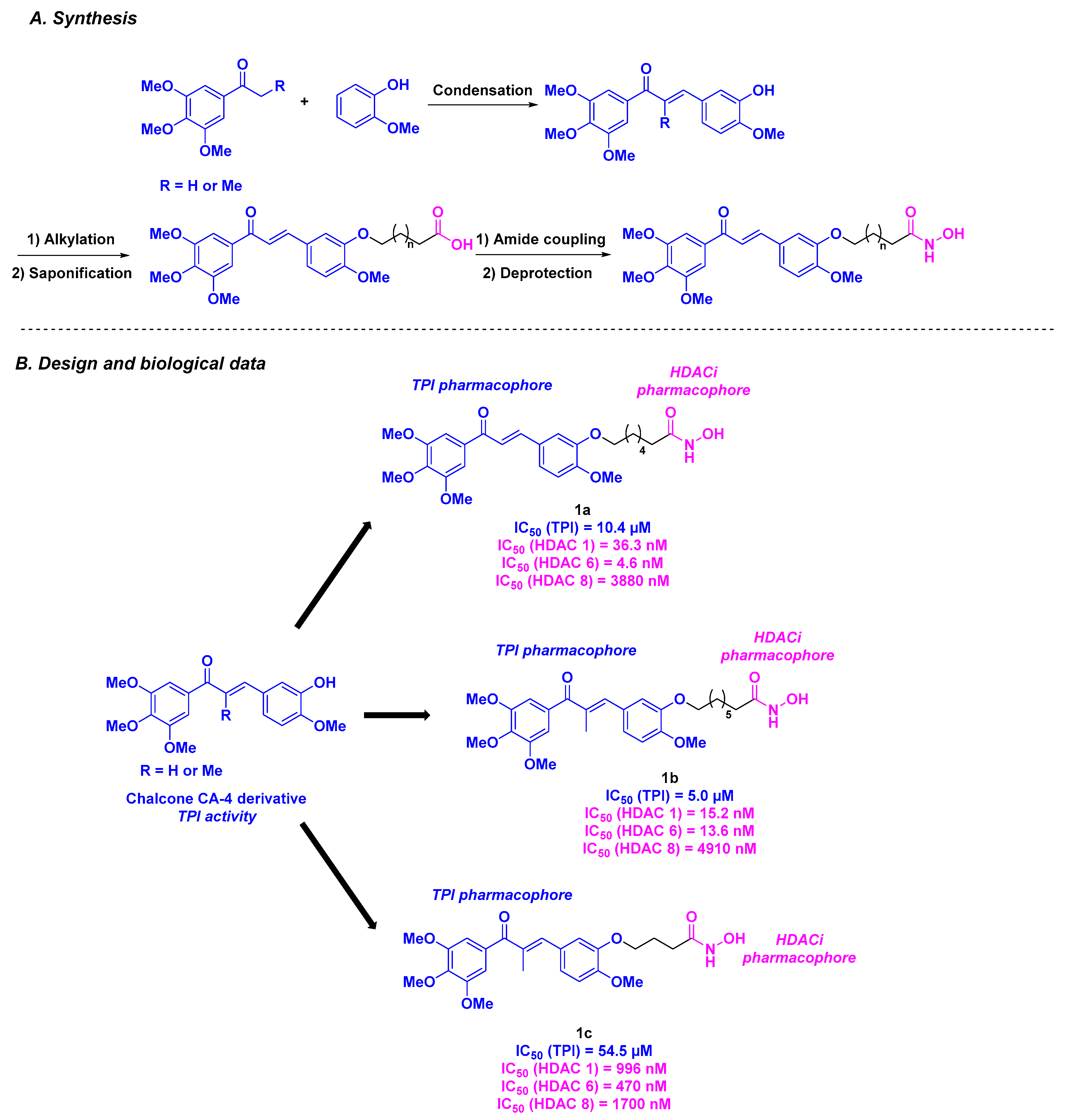
2.1. Chalcone Derivatives
2.2. Stilbene Derivatives
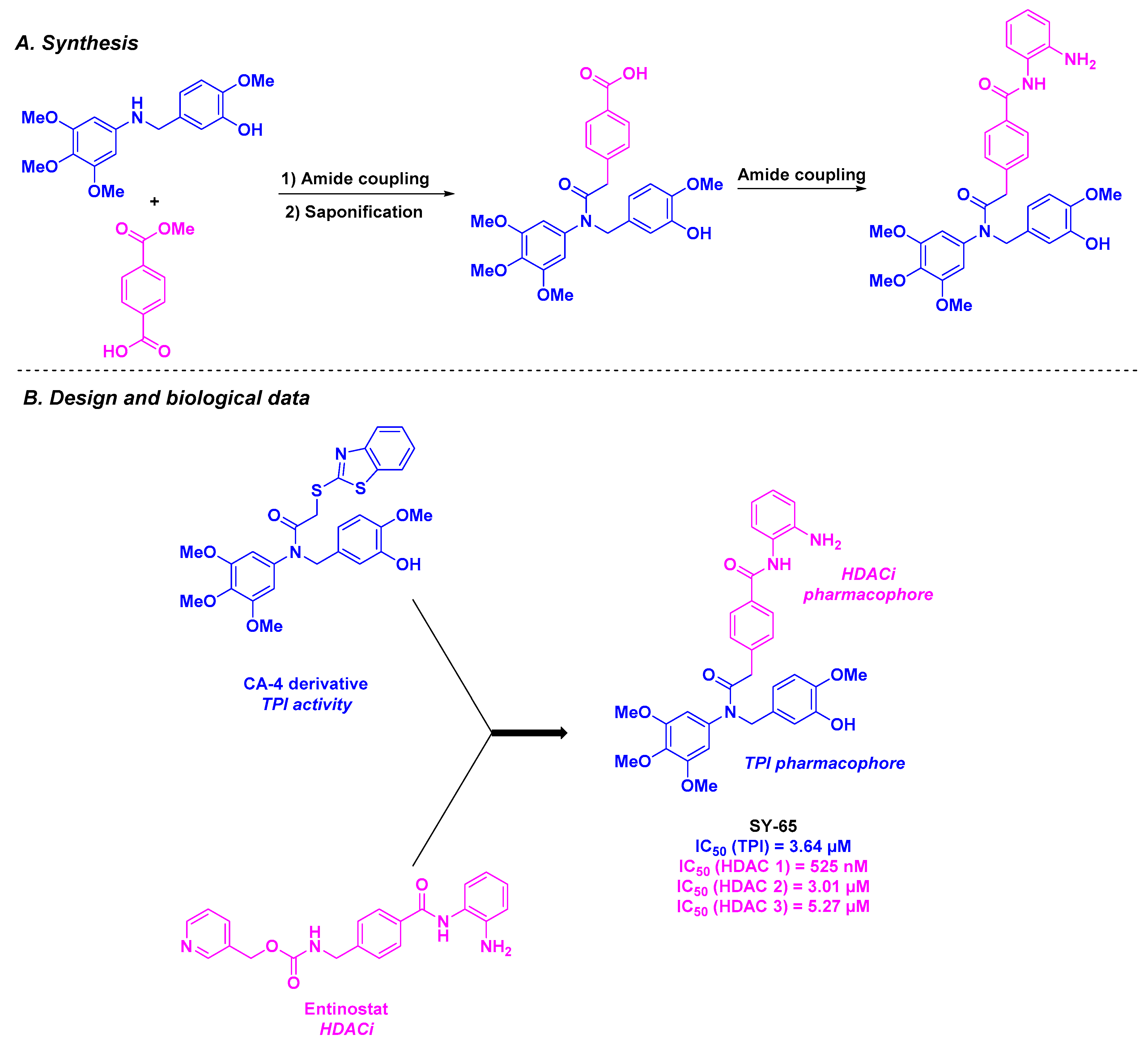
2.3. Amine Derivatives
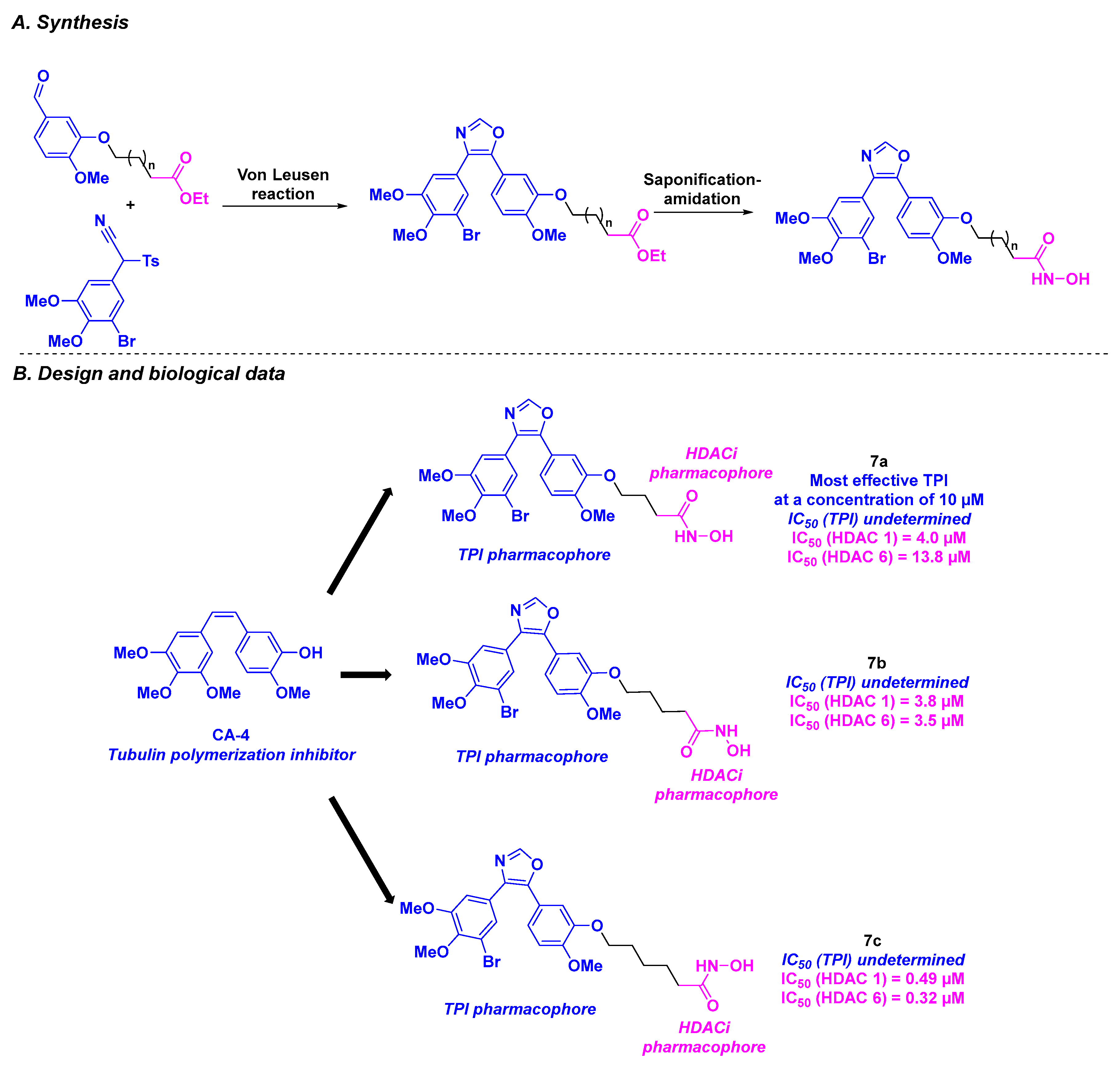
2.4. Oxazole Derivatives
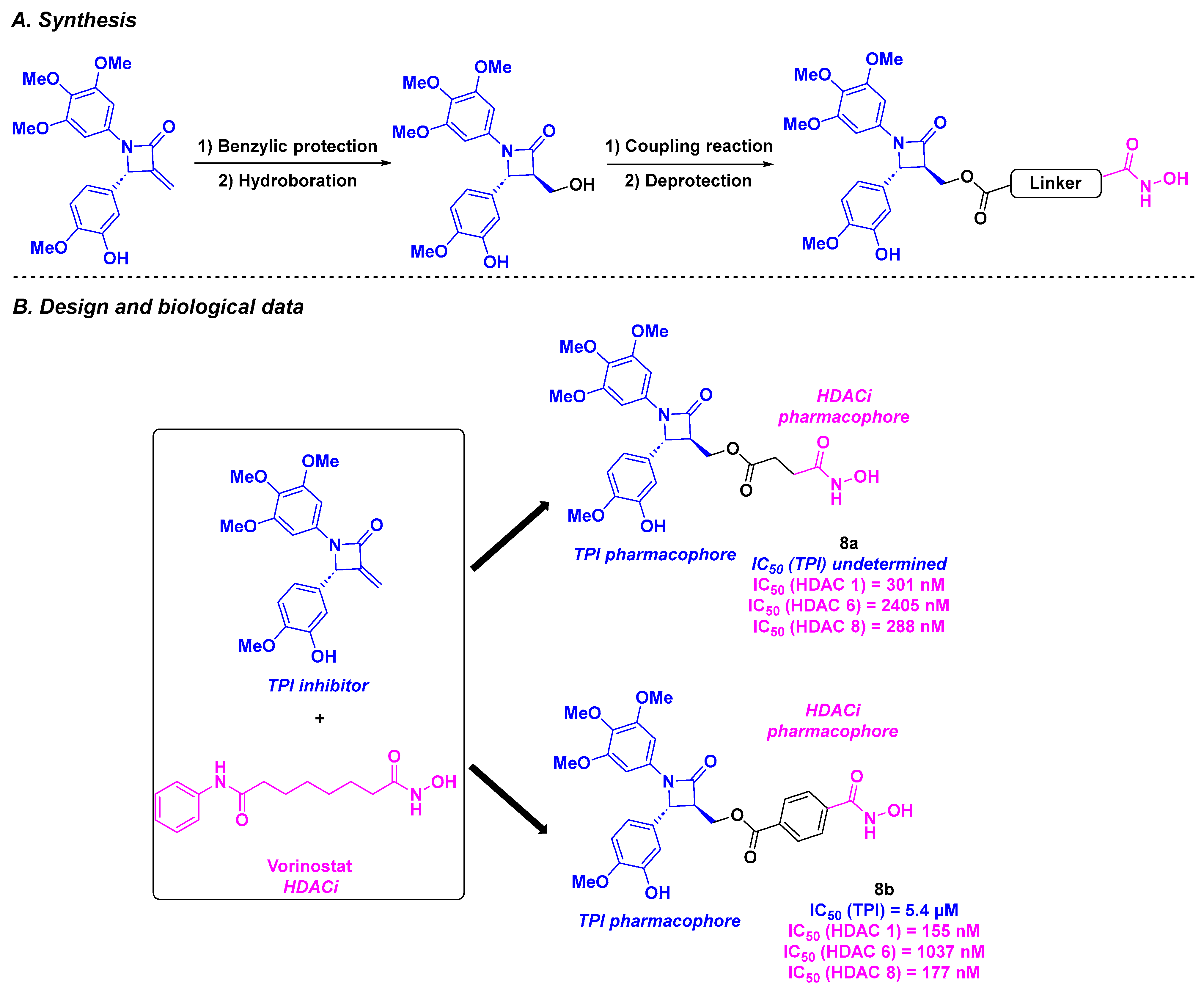
2.5. 1,4-Diarylazetidin-2-one Derivatives
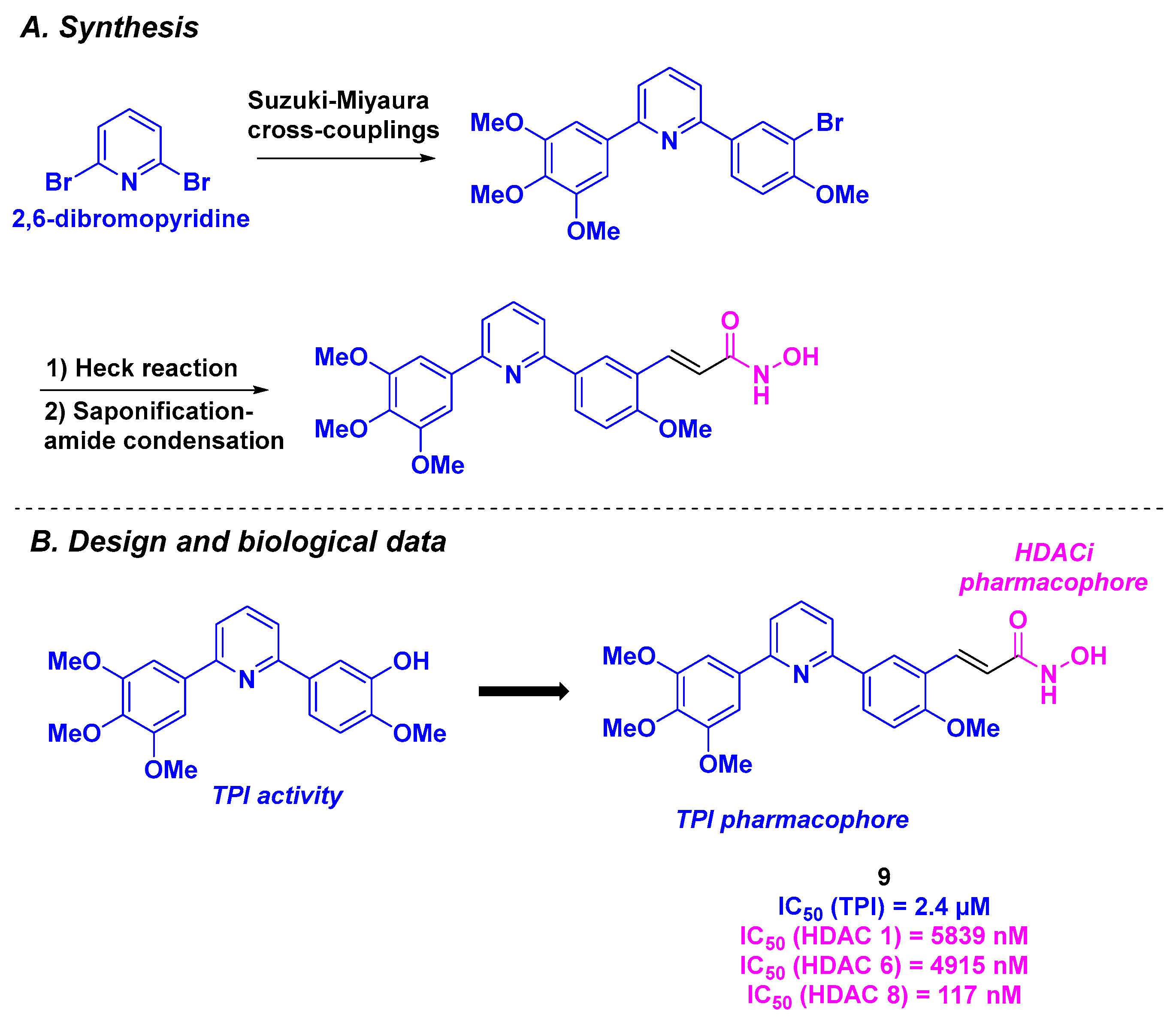
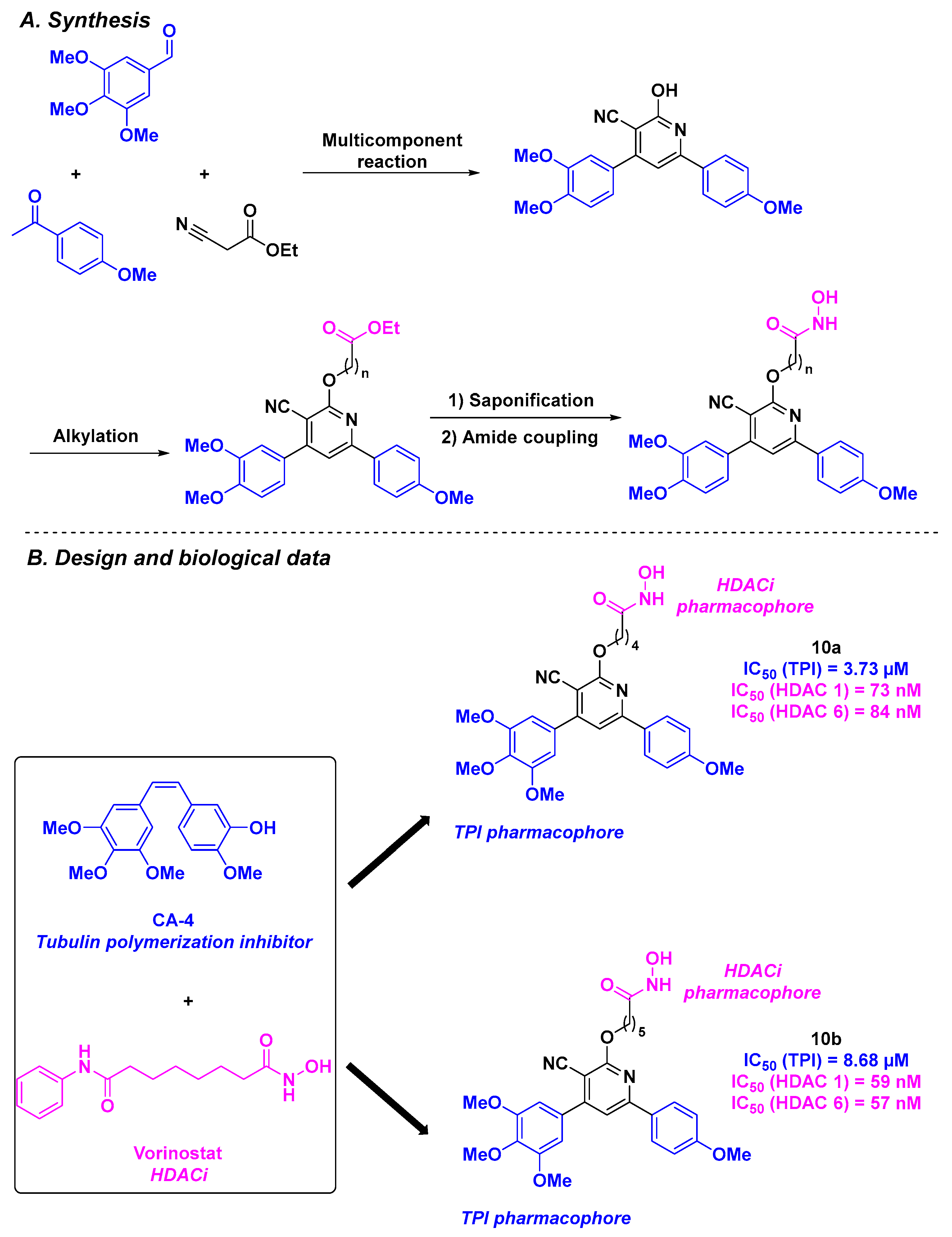
2.6. Arylpyridine Derivatives
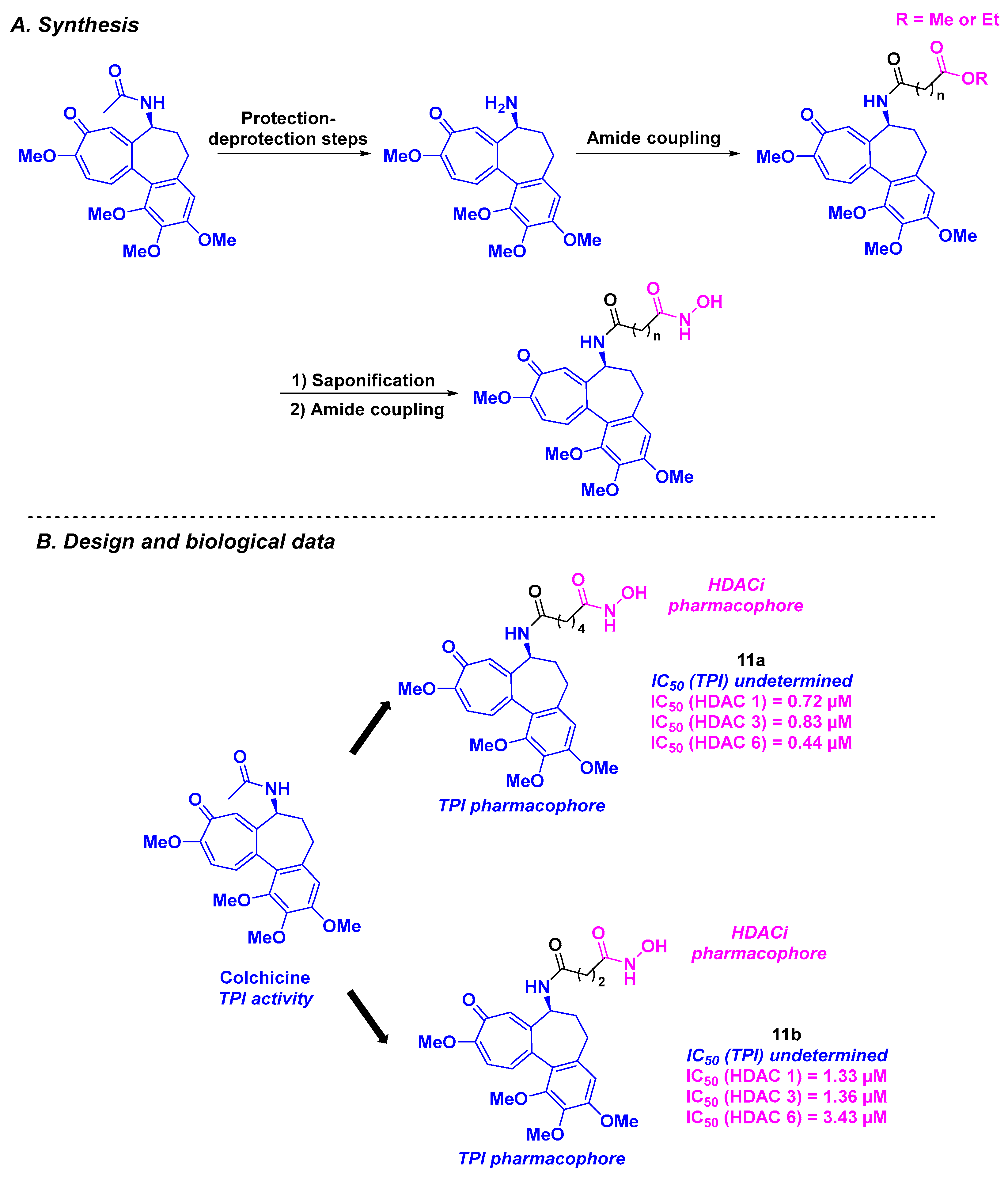
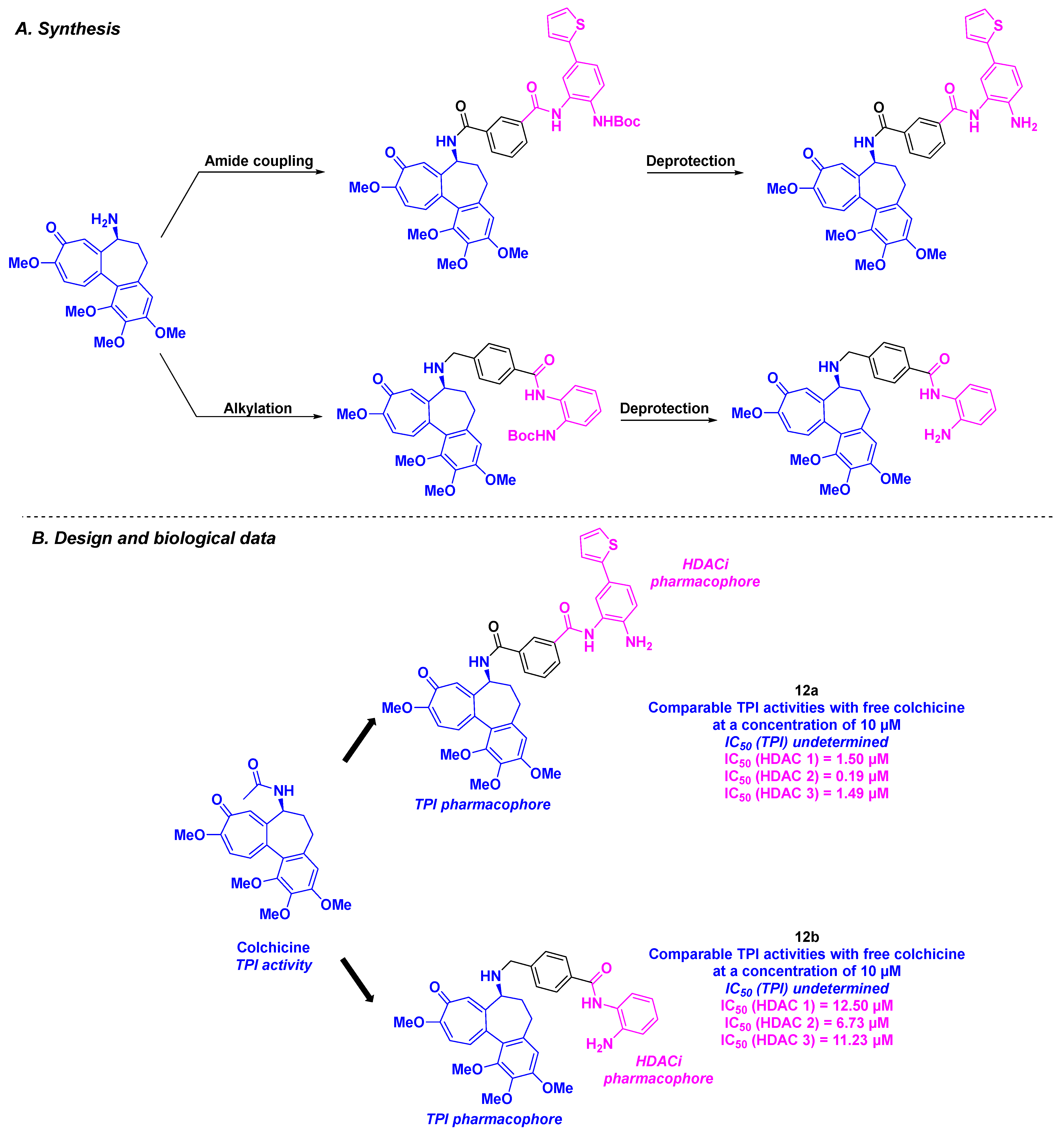
3. Colchicine Derivatives
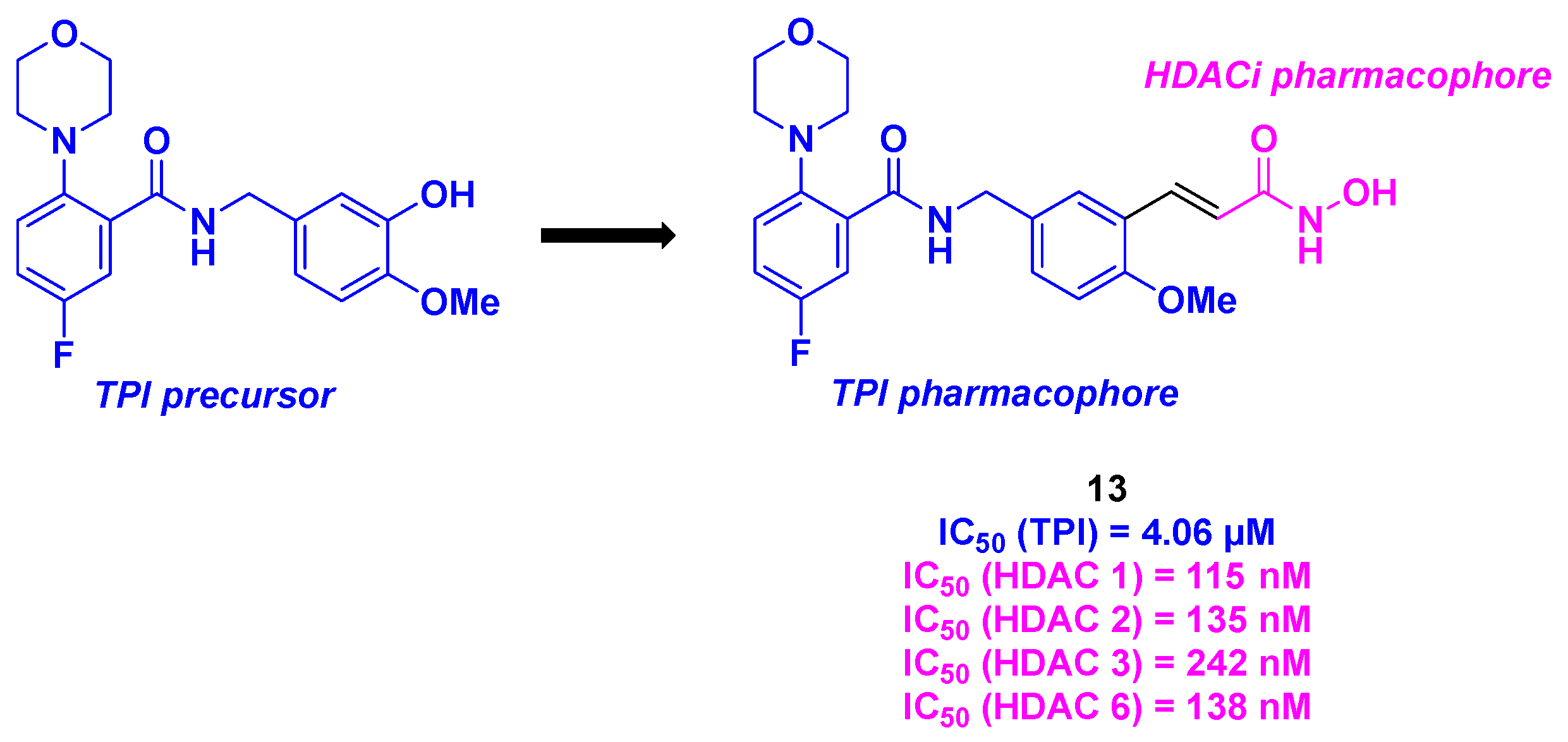
4. Aminobenzamide Core
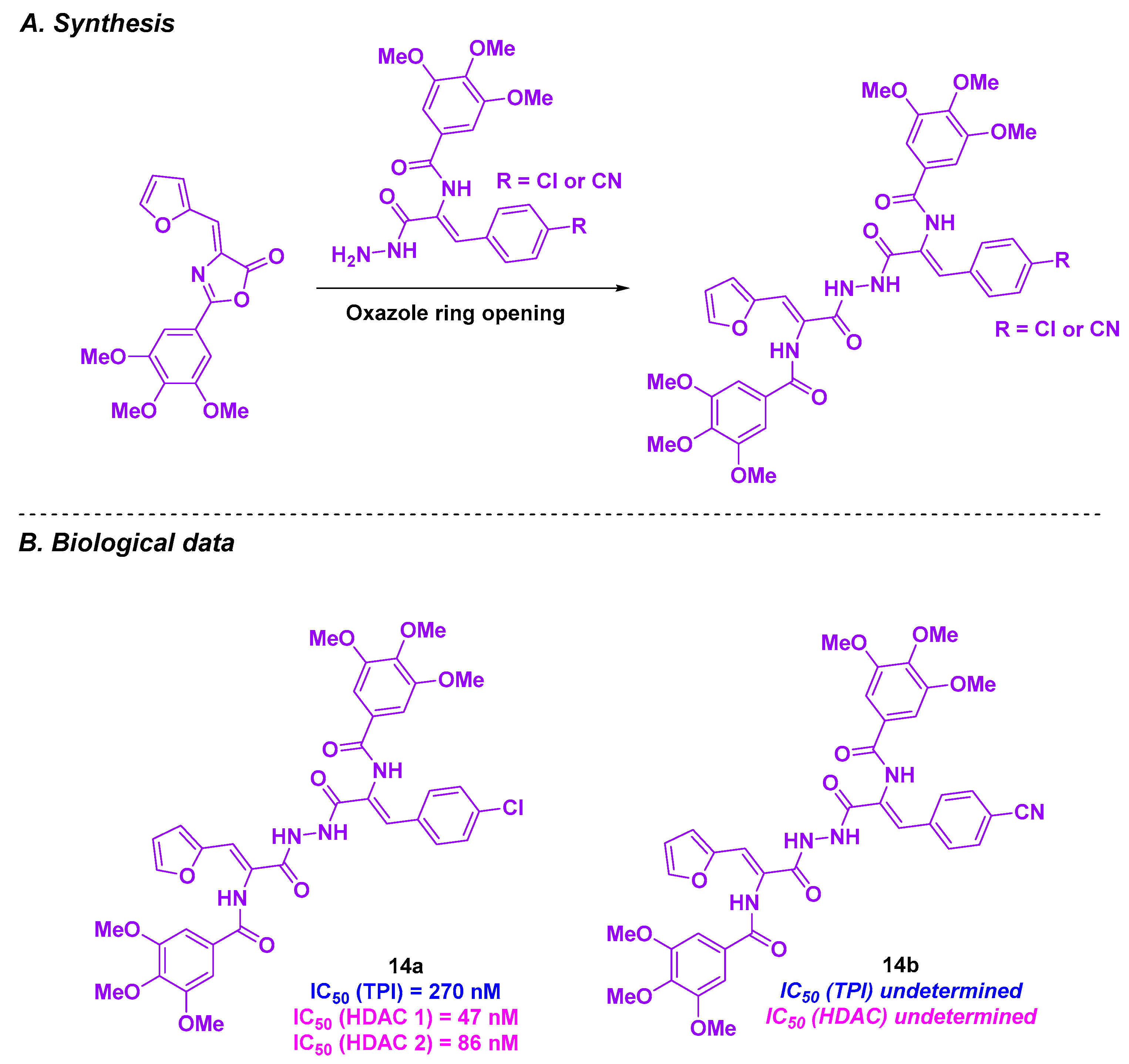
5. Amide Derivatives
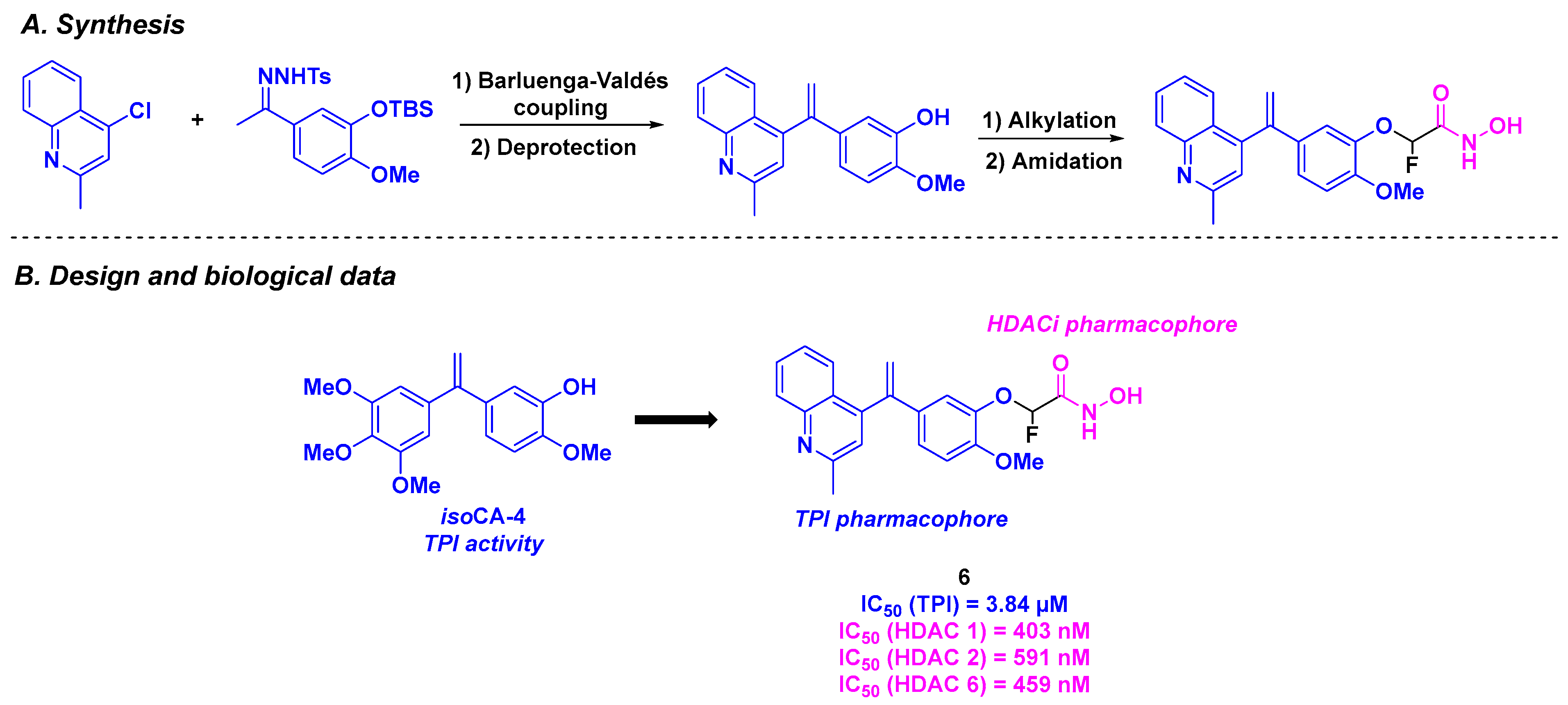
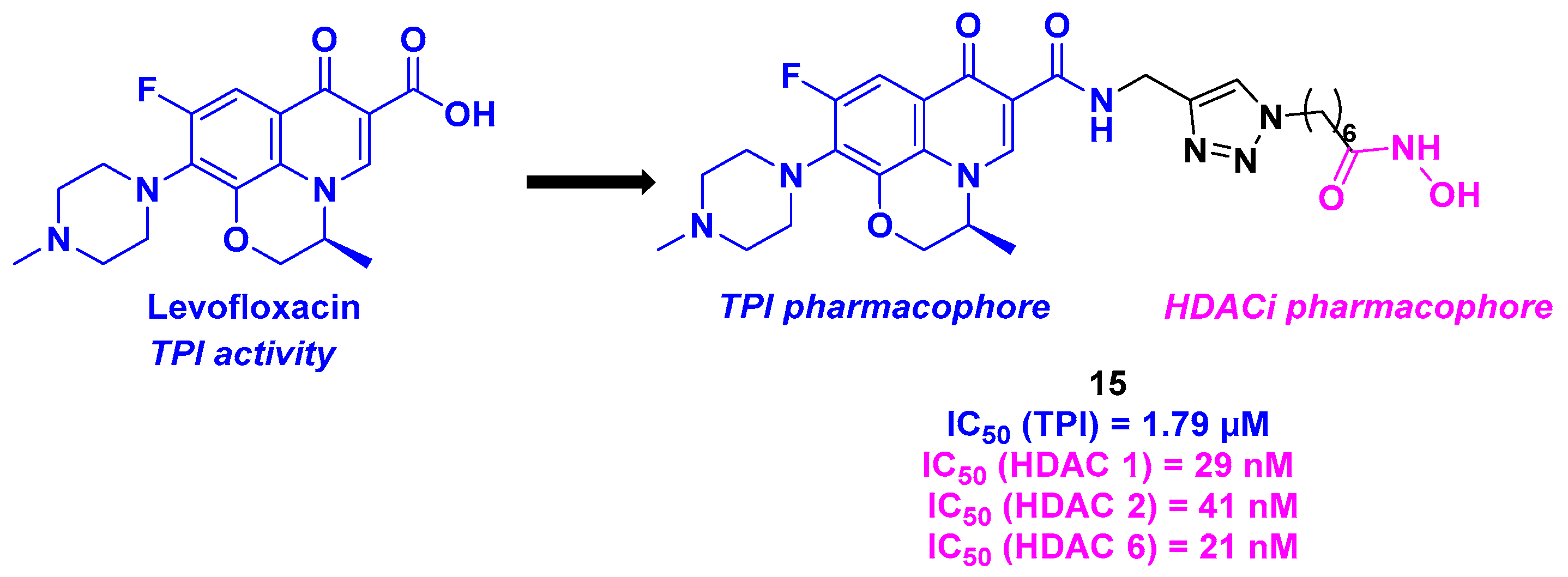
6. Quinolone Derivatives
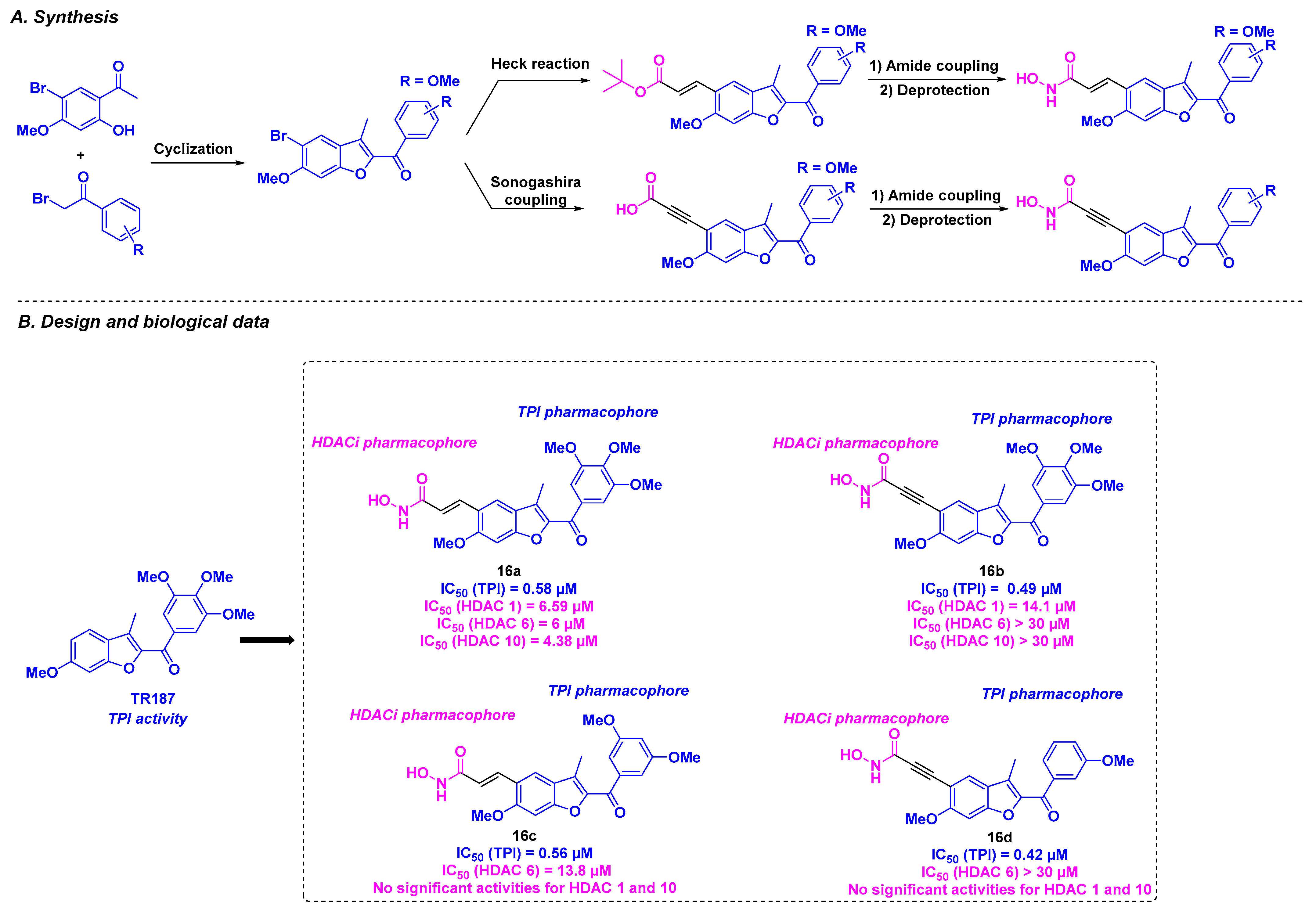
7. Benzofuran Scaffold
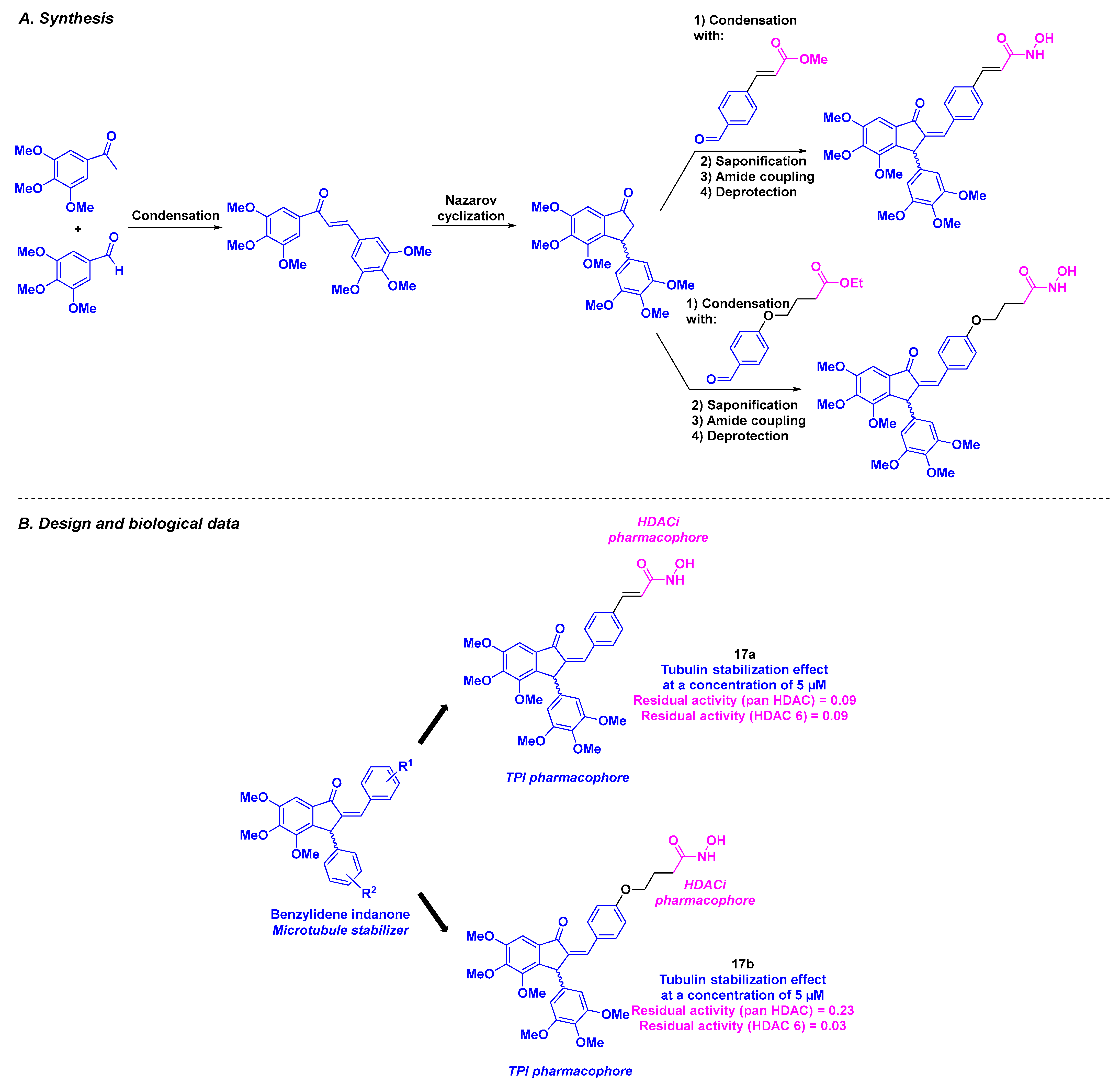
8. 2-Benzylideneindanone Scaffold
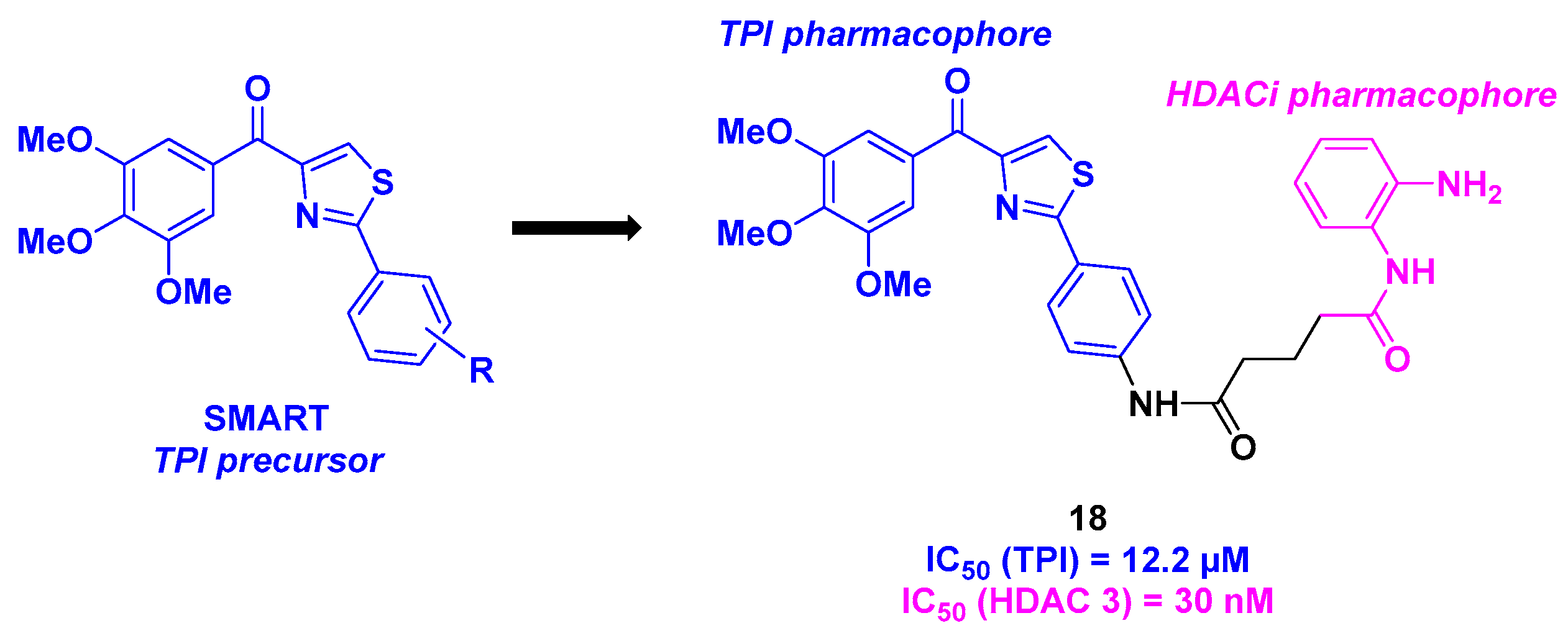
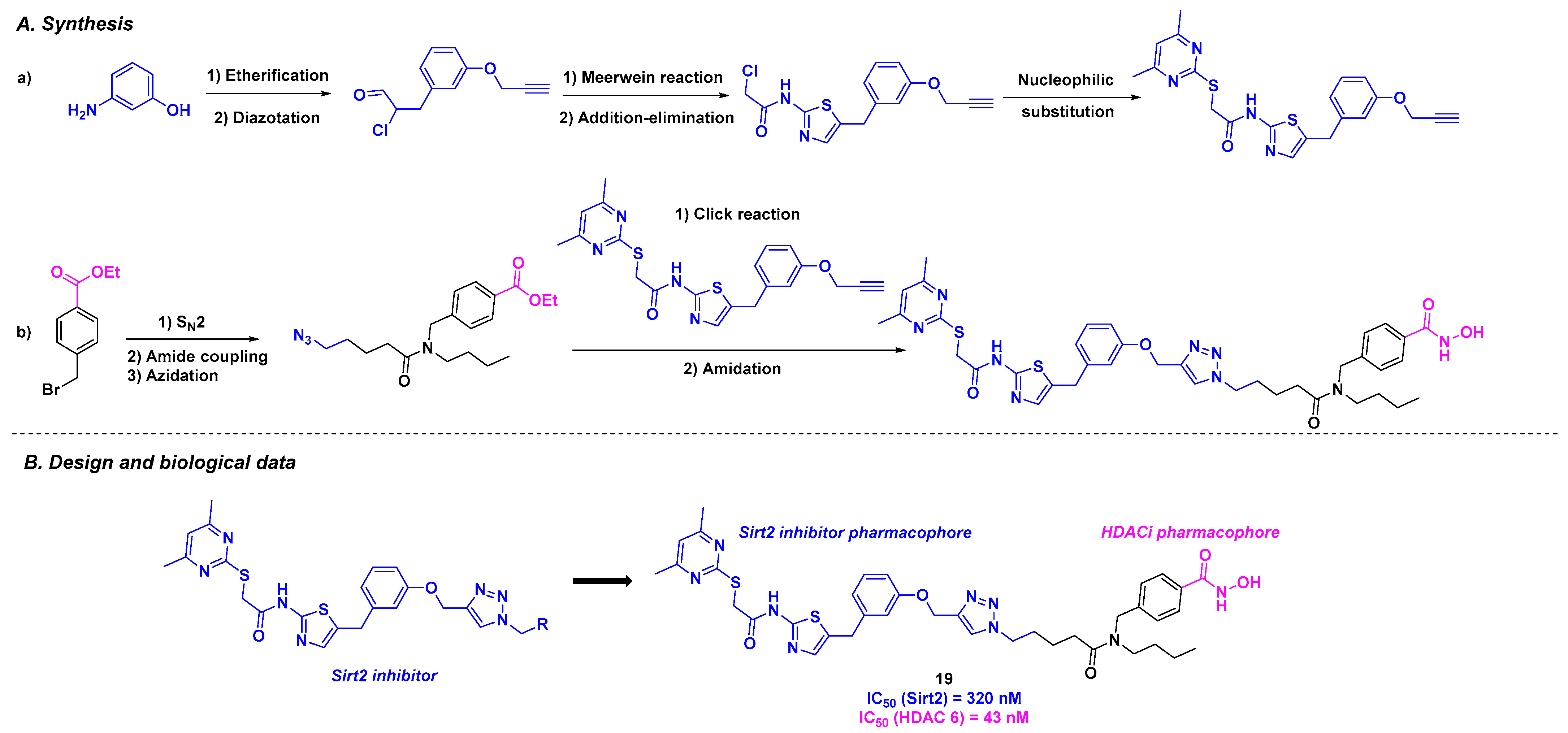
9. Aminothiazoles
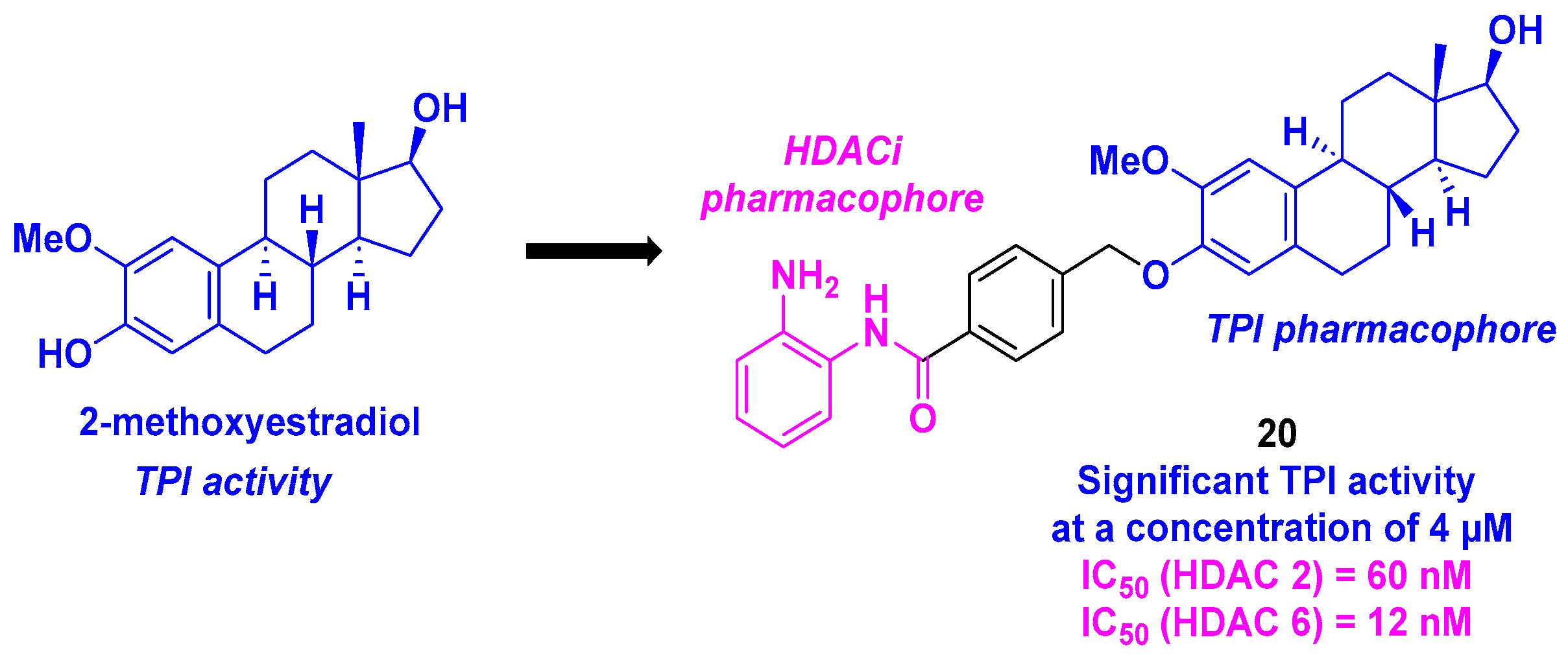
10. 2-Methoxyestradiol Core
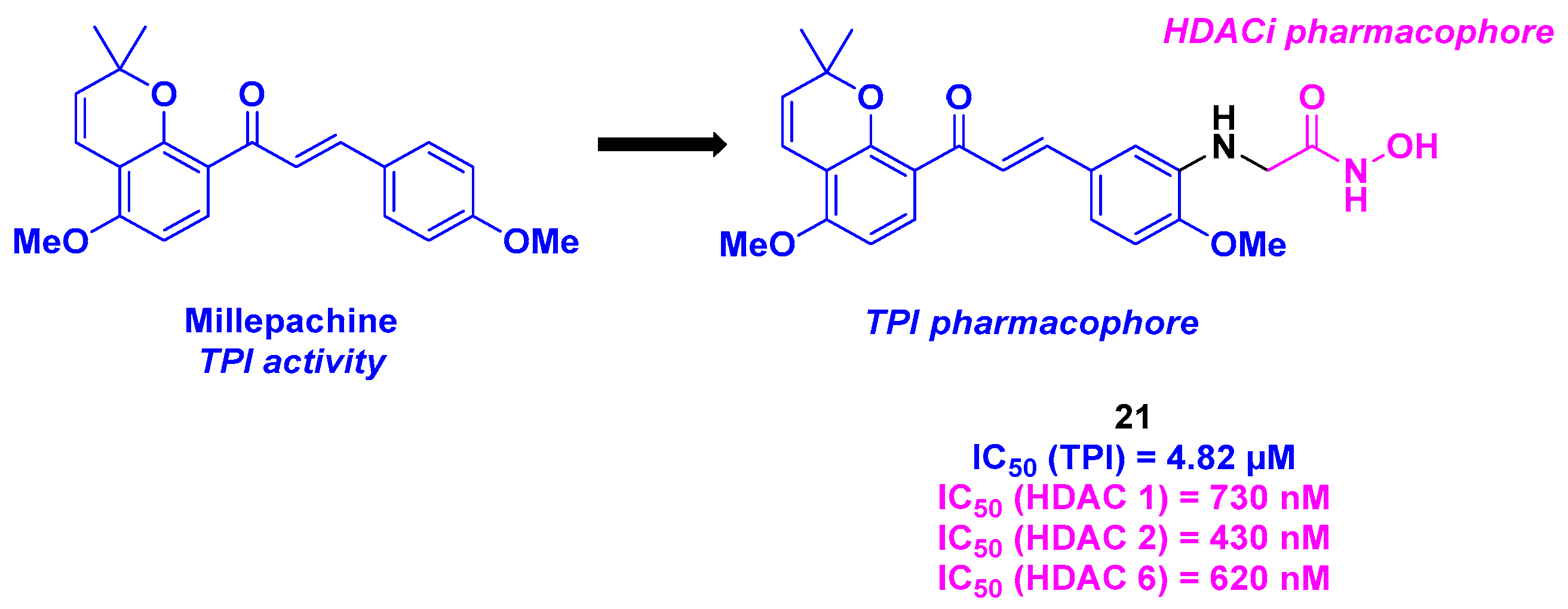
11. Millepachine Core
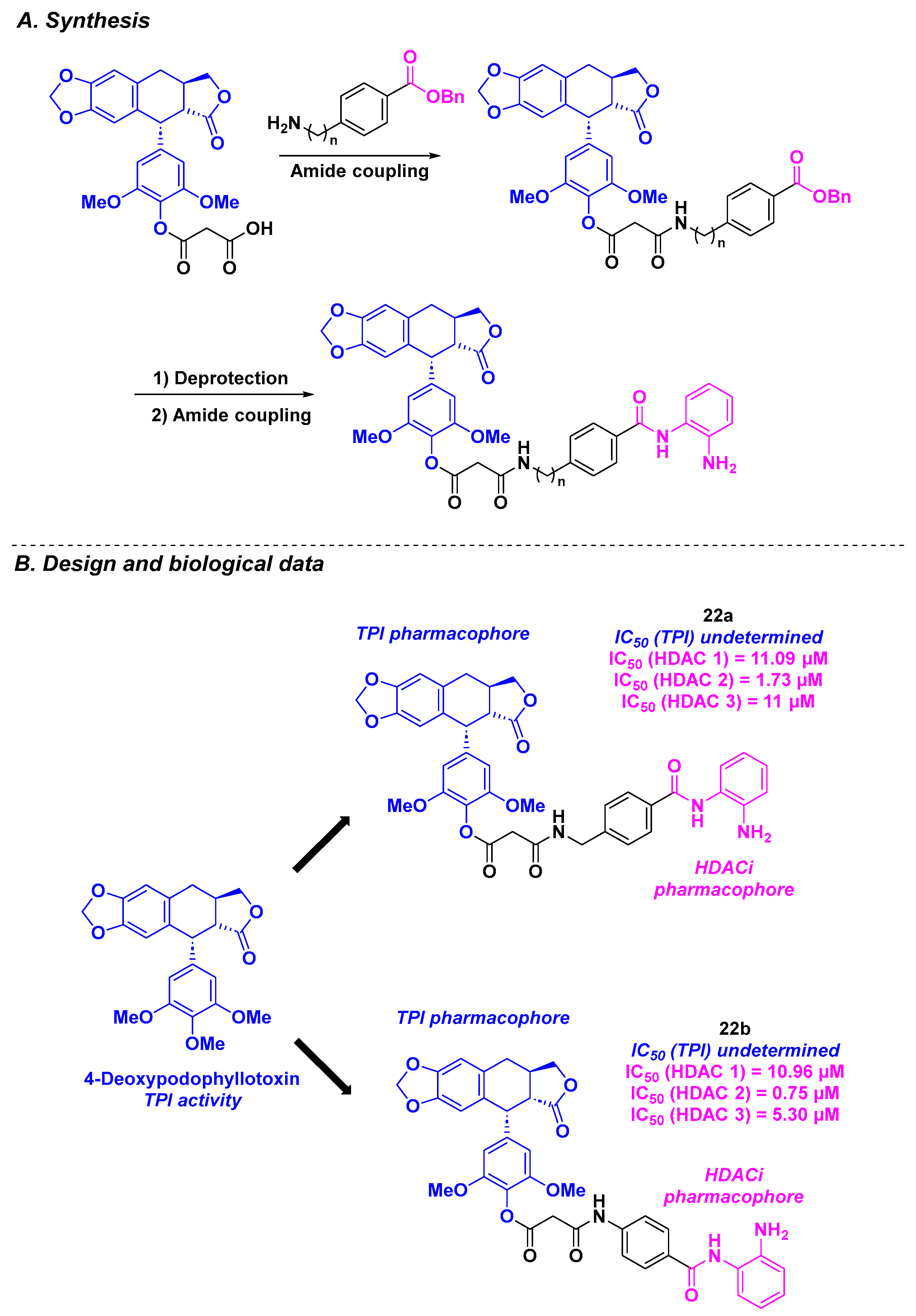
12. Deoxypodophyllotoxin Derivatives
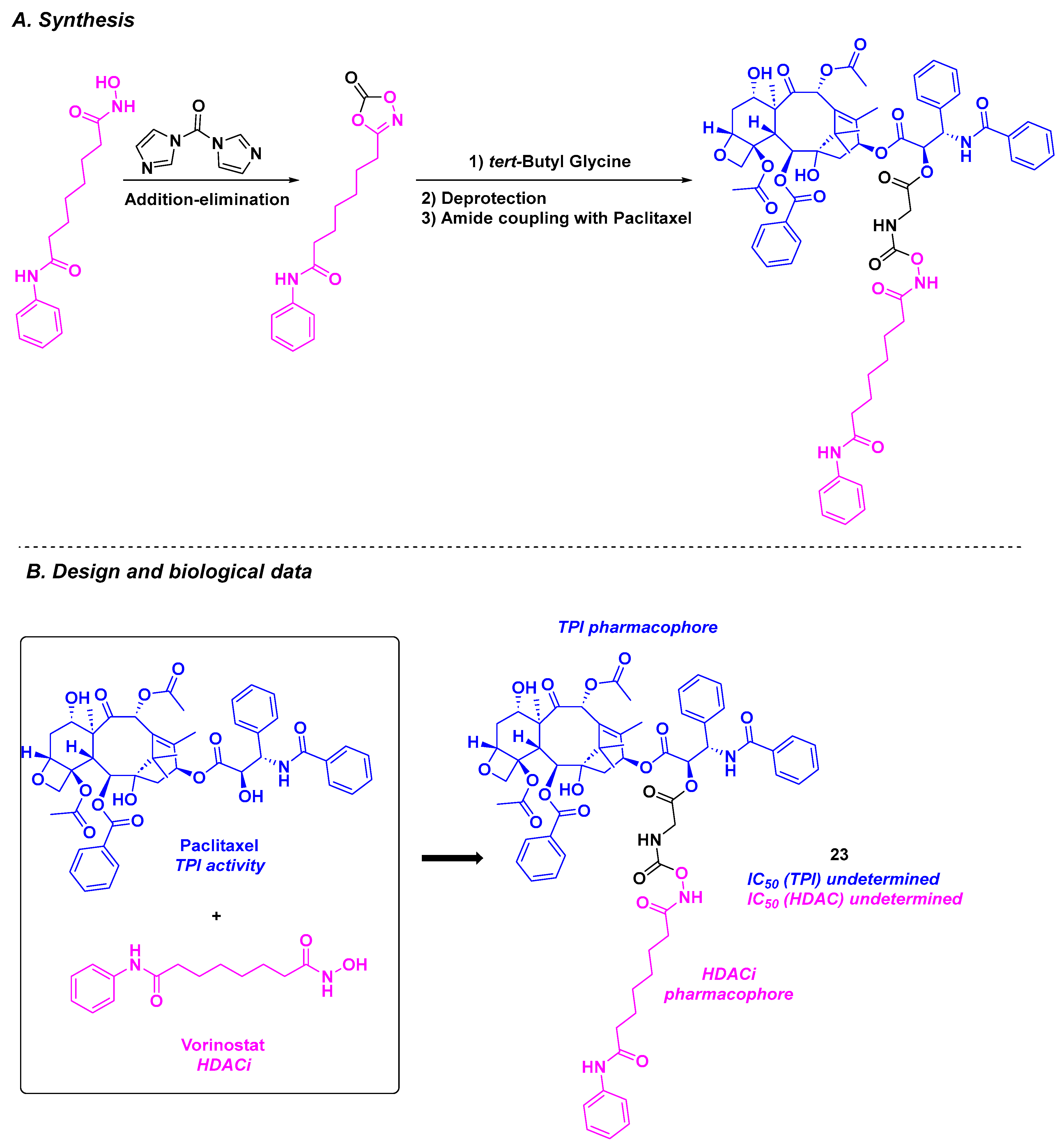
13. Paclitaxel Scaffold
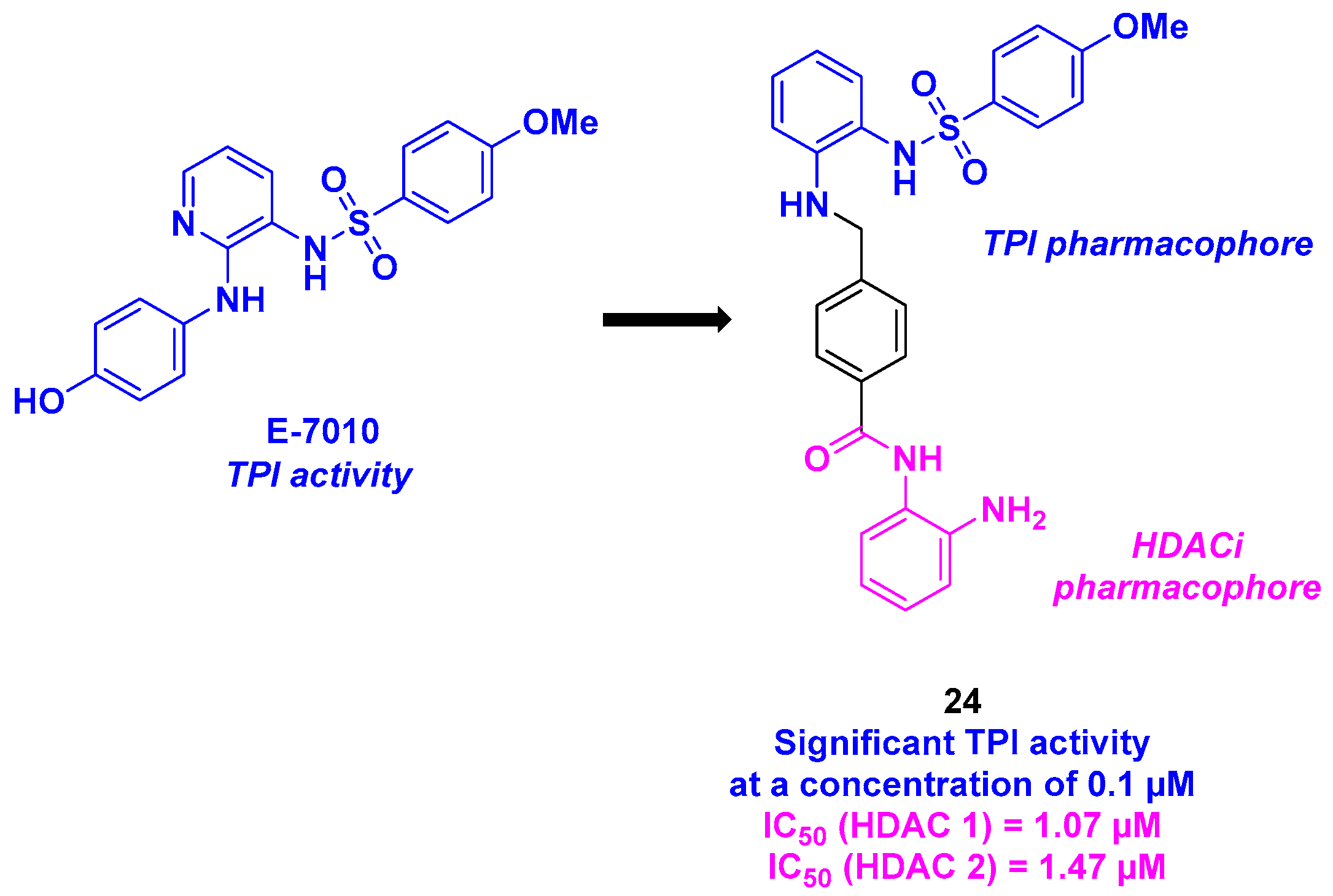
14. Sulfonamide Scaffold
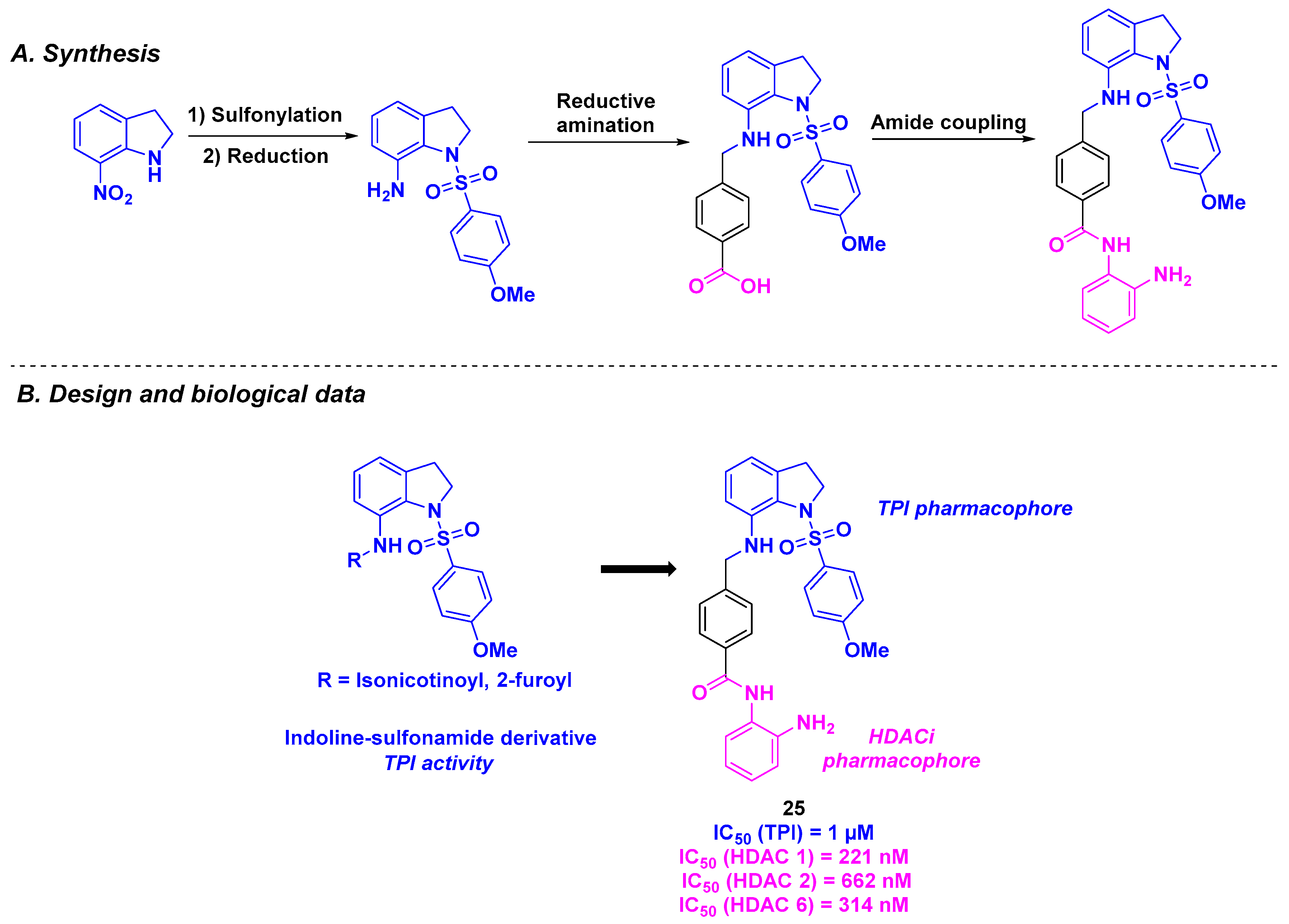
15. Indoline/Indole-Sulfonamide Scaffold
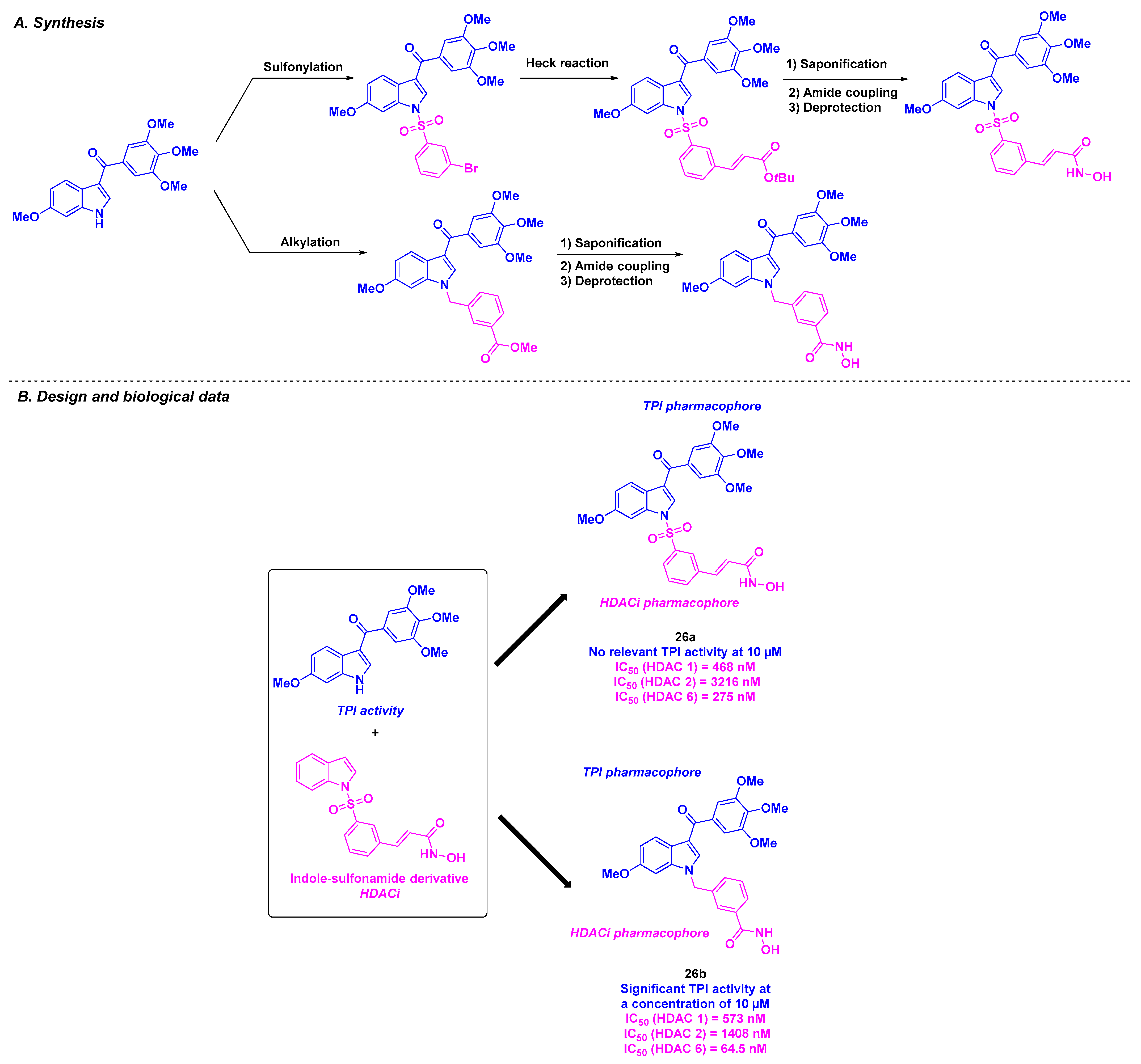
16. Dual HDAC/Tubulin Inhibitors Inspired by the HDACis
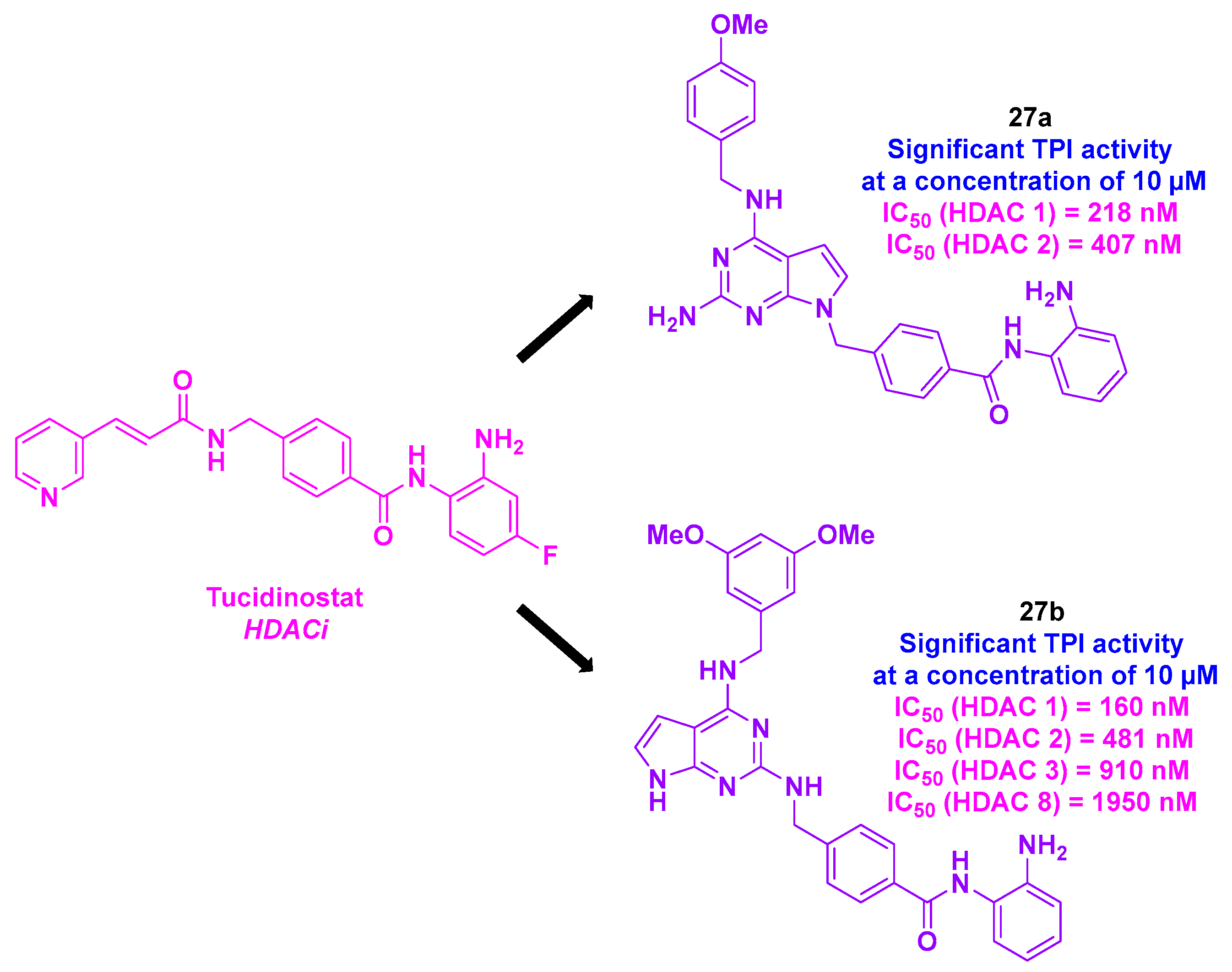
16.1. Pyrrolo [2,3-d]pyrimidine Skeleton
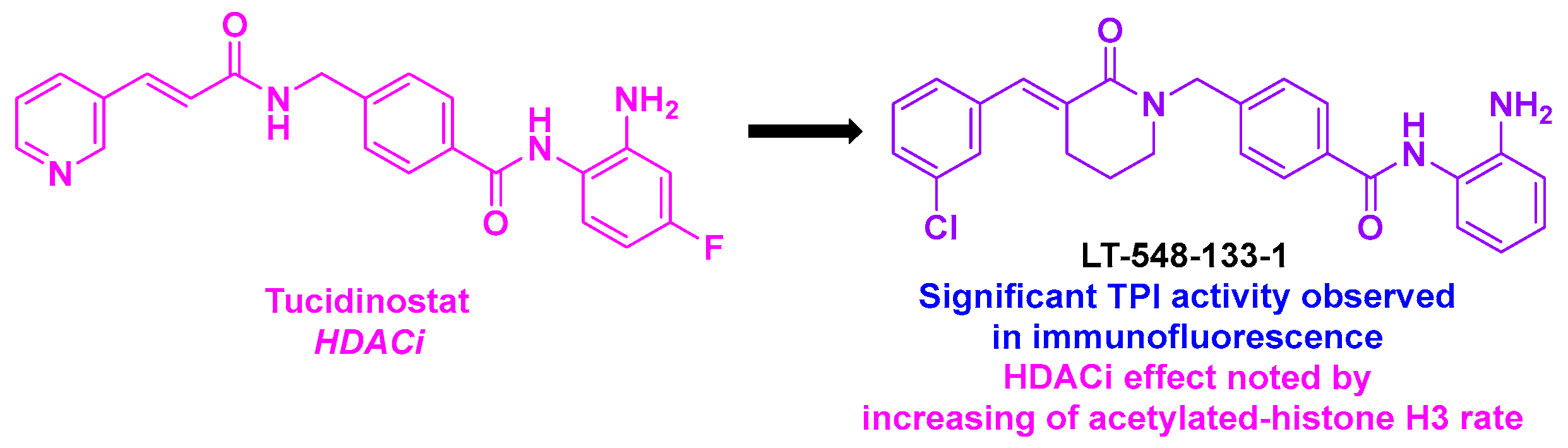
16.2. Benzamide Scaffold
16.3. Quinazoline Scaffold
16.4. Imidazolyl Motif
17. Conclusions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Huang, J.; Zheng, Y.; Sun, Q. Histone acetyltransferase inhibitors: An overview in synthesis, structure-activity relationship and molecular mechanism. Eur. J. Med. Chem. 2019, 178, 259–286. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- He, X.; Hui, Z.; Xu, L.; Bai, R.; Gao, Y.; Wang, Z.; Xie, T.; Ye, X.-Y. Medicinal chemistry updates of novel HDACs inhibitors (2020 to present). Eur. J. Med. Chem. 2022, 227, 113946. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal Structure of a Eukaryotic Zinc-Dependent Histone Deacetylase, Human HDAC8, Complexed with a Hydroxamic Acid Inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef]
- Squarzoni, A.; Scuteri, A.; Cavaletti, G. HDACi: The Columbus’ Egg in Improving Cancer Treatment and Reducing Neurotoxicity? Cancers 2022, 14, 5251. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Rai, S.; Kim, W.S.; Ando, K.; Choi, I.; Izutsu, K.; Tsukamoto, N.; Yokoyama, M.; Tsukasaki, K.; Kuroda, J.; Ando, J.; et al. Oral HDAC inhibitor tucidinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Phase IIb results. Haematologica 2023, 108, 811–821. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Qin, M.; Wu, H.-P.; Khamis, M.Y.; Li, Y.-H.; Ma, L.-Y.; Liu, H.-M. A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem. 2021, 64, 1362–1391. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur. J. Med. Chem. 2020, 208, 112831. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2008, 15, 91–99. [Google Scholar] [CrossRef]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Ecker, J.; Oehme, I.; Mazitschek, R.; Korshunov, A.; Kool, M.; Hielscher, T.; Kiss, J.; Selt, F.; Konrad, C.; Lodrini, M. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol. Commun. 2015, 3, 22. [Google Scholar] [CrossRef]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Xie, H.J.; Chang, Y.G.; Kim, M.G.; Park, H.; Lee, J.Y.; et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J. Cell. Biochem. 2012, 113, 2167–2177. [Google Scholar] [CrossRef]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Röcken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Mimori, K.; Nishida, N.; Kogo, R.; Iwaya, T.; Tanaka, F.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 2011, 26, 777–782. [Google Scholar] [PubMed]
- Jin, Z.; Jiang, W.; Jiao, F.; Guo, Z.; Hu, H.; Wang, L.; Wang, L. Decreased expression of histone deacetylase 10 predicts poor prognosis of gastric cancer patients. Int. J. Clin. Exp. Pathol. 2014, 7, 5872–5879. [Google Scholar]
- Xie, H.J.; Noh, J.H.; Kim, J.K.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Lee, J.Y.; Park, H.; et al. HDAC1 Inactivation Induces Mitotic Defect and Caspase-Independent Autophagic Cell Death in Liver Cancer. PLoS ONE 2012, 7, e34265. [Google Scholar] [CrossRef]
- Buurman, R.; Sandbothe, M.; Schlegelberger, B.; Skawran, B. HDAC inhibition activates the apoptosome via Apaf1 upregulation in hepatocellular carcinoma. Eur. J. Med. Res. 2016, 21, 26. [Google Scholar] [CrossRef]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef]
- Fan, J.; Lou, B.; Chen, W.; Zhang, J.; Lin, S.; Lv, F.F.; Chen, Y. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour Biol. 2014, 35, 11523–11532. [Google Scholar] [CrossRef]
- Feng, G.; Dong, L.; Shang, W.; Pang, X.; Li, J.; Liu, L.; Wang, Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 811–816. [Google Scholar]
- Lv, Z.; Weng, X.; Du, C.; Zhang, C.; Xiao, H.; Cai, X.; Ye, S.; Cheng, J.; Ding, C.; Xie, H.; et al. Downregulation of HDAC6 promotes angiogenesis in hepatocellular carcinoma cells and predicts poor prognosis in liver transplantation patients. Mol. Carcinog. 2016, 55, 1024–1033. [Google Scholar] [CrossRef]
- Fritsche, P.; Seidler, B.; Schüler, S.; Schnieke, A.; Göttlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef]
- Li, D.; Sun, X.; Zhang, L.; Yan, B.; Xie, S.; Liu, R.; Liu, M.; Zhou, J. Histone deacetylase 6 and cytoplasmic linker protein 170 function together to regulate the motility of pancreatic cancer cells. Protein Cell 2014, 5, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Ouaïssi, M.; Silvy, F.; Loncle, C.; Ferraz da Silva, D.; Martins Abreu, C.; Martinez, E.; Berthézene, P.; Cadra, S.; Le Treut, Y.P.; Hardwigsen, J.; et al. Further characterization of HDAC and SIRT gene expression patterns in pancreatic cancer and their relation to disease outcome. PLoS ONE 2014, 9, e108520. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.-C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I Histone Deacetylase Expression Has Independent Prognostic Impact in Human Colorectal Cancer: Specific Role of Class I Histone Deacetylases In vitro and In vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Stypula-Cyrus, Y.; Damania, D.; Kunte, D.P.; Cruz, M.D.; Subramanian, H.; Roy, H.K.; Backman, V. HDAC up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure. PLoS ONE 2013, 8, e64600. [Google Scholar] [CrossRef]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.-J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer-overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef]
- Hong, Y.R.; Song, B.J.; Jung, S.S.; Kang, B.J.; Kim, S.H.; Chae, B.J.; Seo, J.; Min, S.K.; Park, H.-R.; Kim, D.H. Expression of histone deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in invasive ductal carcinomas of the breast. J. Breast Cancer 2014, 19, 323–331. [Google Scholar]
- Song, C.; Zhu, S.; Wu, C.; Kang, J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J. Biol. Chem. 2013, 288, 28021–28033. [Google Scholar] [CrossRef]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer. 2010, 127, 1332–1346. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.; Niesporek, S.; Denkert, C.; Dietel, M. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.; de Paula Queiroz, R.; Valera, E.T.; da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kafeel, M.I.; Avezbakiyev, B.; Chen, C.; Sun, Y.; Rathnasabapathy, C.; Kalavar, M.; He, Z.; Burton, J.; Lichter, S. Histone Deacetylase in Chronic Lymphocytic Leukemia. Oncology 2012, 81, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Min, S.K.; Koh, Y.H.; Park, Y.; Kim, H.J.; Seo, J.; Park, H.-R.; Cho, S.J.; Kim, I.S. Expression of HAT1 and HDAC1, 2, 3 in diffuse large B-cell lymphomas, peripheral T-cell lymphomas, and NK/T-cell lymphomas. Korean J. Pathol. 2012, 46, 142. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Yoo, C.; Im, S.; Jung, J.H.; Choi, H.J.; Yoo, J. Expression of histone deacetylases in diffuse large B-cell lymphoma and its clinical significance. Int. J. Med. Sci. 2014, 11, 994–1000. [Google Scholar] [CrossRef]
- Marquard, L.; Poulsen, C.B.; Gjerdrum, L.M.; de Nully Brown, P.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Johansen, P.; Ralfkiaer, E. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 2009, 54, 688–698. [Google Scholar] [CrossRef]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef]
- Marquard, L.; Gjerdrum, L.; Christensen, I.J.; Jensen, P.; Sehested, M.; Ralfkiaer, E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 2008, 53, 267–277. [Google Scholar] [CrossRef]
- Adams, H.; Fritzsche, F.R.; Dirnhofer, S.; Kristiansen, G.; Tzankov, A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 2010, 14, 577–584. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef]
- de Lera, A.R.; Ganesan, A. Epigenetic polypharmacology: From combination therapy to multitargeted drugs. Clin. Epigenet. 2016, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Soltan, O.M.; Shoman, M.E.; Abdel-Aziz, S.A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of Chimeric Histone Deacetylase- and Tyrosine Kinase-Inhibitors: A Series of Imatinib Hybrides as Potent Inhibitors of Wild-Type and Mutant BCR-ABL, PDGF-Rβ, and Histone Deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Mustafa, N.; Tan, E.C.; Poulsen, A.; Singh, P.; Duong-Thi, M.-D.; Lee, J.X.T.; Ramanujulu, P.M.; Chng, W.J.; Yen, J.J.Y.; et al. Design and Synthesis of Ligand Efficient Dual Inhibitors of Janus Kinase (JAK) and Histone Deacetylase (HDAC) Based on Ruxolitinib and Vorinostat. J. Med. Chem. 2017, 60, 8336–8357. [Google Scholar] [CrossRef]
- Yu, Y.; Ran, D.; Jiang, J.; Pan, T.; Dan, Y.; Tang, Q.; Li, W.; Zhang, L.; Gan, L.; Gan, Z. Discovery of novel 9H-purin derivatives as dual inhibitors of HDAC1 and CDK2. Bioorg. Med. Chem. Lett. 2019, 29, 2136–2140. [Google Scholar] [CrossRef]
- Millis, S.Z.; Ikeda, S.; Reddy, S.; Gatalica, Z.; Kurzrock, R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncol. 2016, 2, 1565–1573. [Google Scholar] [CrossRef]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-one-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Attina, G.; Triarico, S.; Romano, A.; Maurizi, P.; Ruggiero, A. The DNA-topoisomerase inhibitors in cancer therapy. Biomed. Pharmacol. J. 2022, 15, 553–562. [Google Scholar] [CrossRef]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Yao, L.-P.; Hood, R.; Oyelere, A.K. Dual-acting histone deacetylase-topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3283–3287. [Google Scholar] [CrossRef]
- Chen, W.; Dong, G.; Wu, Y.; Zhang, W.; Miao, C.; Sheng, C. Dual NAMPT/HDAC Inhibitors as a New Strategy for Multitargeting Antitumor Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; He, L.; Zheng, L.; Huang, L.; Zhou, Y.; Wang, T.; Chen, Y.; Shen, M.; Wang, F.; Yang, Z.; et al. Structure-based design, synthesis and in vitro antiproliferative effects studies of novel dual BRD4/HDAC inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hu, W.; Wang, Z.; Gou, S. Platinum(IV) prodrugs multiply targeting genomic DNA, histone deacetylases and PARP-1. Eur. J. Med. Chem. 2017, 141, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of Histone Deacetylase 6 Acetylates and Disrupts the Chaperone Function of Heat Shock Protein 90: A Novel Basis for Antileukemia Activity of Histone Deacetylase Inhibitors*. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- Ojha, R.; Huang, H.-L.; HuangFu, W.-C.; Wu, Y.-W.; Nepali, K.; Lai, M.-J.; Su, C.-J.; Sung, T.-Y.; Chen, Y.-L.; Pan, S.-L.; et al. 1-Aroylindoline-hydroxamic acids as anticancer agents, inhibitors of HSP90 and HDAC. Eur. J. Med. Chem. 2018, 150, 667–677. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef]
- Li, N.; Guan, Q.; Hong, Y.; Zhang, B.; Li, M.; Li, X.; Li, B.; Wu, L.; Zhang, W. Discovery of 6-aryl-2-(3,4,5-trimethoxyphenyl)thiazole [3,2-b][1,2,4]triazoles as potent tubulin polymerization inhibitors. Eur. J. Med. Chem. 2023, 256, 115402. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The Novel Microtubule-Destabilizing Drug BAL27862 Binds to the Colchicine Site of Tubulin with Distinct Effects on Microtubule Organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Gao, M.; Zhang, H.; Guan, Q.; Xu, J.; Li, Y.; Qi, H.; Li, Z.; Zuo, D.; Zhang, W.; et al. BZML, a novel colchicine binding site inhibitor, overcomes multidrug resistance in A549/Taxol cells by inhibiting P-gp function and inducing mitotic catastrophe. Cancer Lett. 2017, 402, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More Than Just Vascular Targeting Agents? J. Pharmacol. Exp. Therapeut. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Arnst, K.E.; Wang, Y.; Lei, Z.-N.; Hwang, D.-J.; Kumar, G.; Ma, D.; Parke, D.N.; Chen, Q.; Yang, J.; White, S.W.; et al. Colchicine Binding Site Agent DJ95 Overcomes Drug Resistance and Exhibits Antitumor Efficacy. Mol. Pharmacol. 2019, 96, 73–89. [Google Scholar] [CrossRef]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic Antitumor Effects of Novel HDAC Inhibitors and Paclitaxel In Vitro and In Vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef]
- Chao, M.-W.; Lai, M.-J.; Liou, J.-P.; Chang, Y.-L.; Wang, J.-C.; Pan, S.-L.; Teng, C.-M. The synergic effect of vincristine and vorinostat in leukemia in vitro and in vivo. J. Hematol. Oncol. 2015, 8, 82. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendal, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef]
- Lin, C.M.; Ho, H.H.; Pettit, G.R.; Hamel, E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: Studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry 1989, 28, 6984–6991. [Google Scholar] [CrossRef]
- McGown, A.T.; Fox, B.W. Differential cytotoxicity of combretastatins A1 and A4 in two daunorubicin-resistant P388 cell lines. Cancer Chemother. Pharmacol. 1990, 26, 79–81. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Li, C.-Y.; Chiu, C.-C.; Hu, H.-T.; Han, C.-H.; Chen, Y.-L.; Tzeng, C.-C. Combretastatin A-4 derivatives: Synthesis and evaluation of 2,4,5-triaryl-1H-imidazoles as potential agents against H1299 (non-small cell lung cancer cell). Mol. Divers. 2012, 16, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, X.; Gao, J.; Su, L.; Zhang, L.; Xu, H.; Luan, Y. Anti-tumor activity evaluation of novel tubulin and HDAC dual-targeting inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 2638–2645. [Google Scholar] [CrossRef] [PubMed]
- Mourad, A.A.E.; Mourad, M.A.E.; Jones, P.G. Novel HDAC/Tubulin Dual Inhibitor: Design, Synthesis and Docking Studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity. Drug Des. Devel. Ther. 2020, 14, 3111–3130. [Google Scholar] [CrossRef]
- Sylvie, D. Antimitotic Chalcones and Related Compounds as Inhibitors of Tubulin Assembly. Anticancer Agents Med. Chem. 2009, 9, 336–347. [Google Scholar]
- Belluti, S.; Orteca, G.; Semeghini, V.; Rigillo, G.; Parenti, F.; Ferrari, E.; Imbriano, C. Potent Anti-Cancer Properties of Phthalimide-Based Curcumin Derivatives on Prostate Tumor Cells. Int. J. Mol. Sci. 2019, 20, 28. [Google Scholar] [CrossRef]
- Khelifi, I.; Naret, T.; Renko, D.; Hamze, A.; Bernadat, G.; Bignon, J.; Lenoir, C.; Dubois, J.; Brion, J.-D.; Provot, O.; et al. Design, synthesis and anticancer properties of IsoCombretaQuinolines as potent tubulin assembly inhibitors. Eur. J. Med. Chem. 2017, 127, 1025–1034. [Google Scholar] [CrossRef]
- Lamaa, D.; Lin, H.-P.; Zig, L.; Bauvais, C.; Bollot, G.; Bignon, J.; Levaique, H.; Pamlard, O.; Dubois, J.; Ouaissi, M.; et al. Design and Synthesis of Tubulin and Histone Deacetylase Inhibitor Based on iso-Combretastatin A-4. J. Med. Chem. 2018, 61, 6574–6591. [Google Scholar] [CrossRef]
- Hauguel, C.; Ducellier, S.; Provot, O.; Ibrahim, N.; Lamaa, D.; Balcerowiak, C.; Letribot, B.; Nascimento, M.; Blanchard, V.; Askenatzis, L.; et al. Design, synthesis and biological evaluation of quinoline-2-carbonitrile-based hydroxamic acids as dual tubulin polymerization and histone deacetylases inhibitors. Eur. J. Med. Chem. 2022, 240, 114573. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, M.; Wang, Y.; Qin, J.; Zhang, Y.; Pang, Y.; Yao, Y.; Yang, H.; Duan, Y. Discovery of novel tubulin/HDAC dual-targeting inhibitors with strong antitumor and antiangiogenic potency. Eur. J. Med. Chem. 2021, 225, 113790. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, W.; Liu, Y.; Gao, T.; Zhu, J.; Tan, Y.; Hu, H.; Liang, W.; Zhao, L.; Chen, J.; et al. Synthesis and bioevaluation of novel stilbene-based derivatives as tubulin/HDAC dual-target inhibitors with potent antitumor activities in vitro and in vivo. Eur. J. Med. Chem. 2023, 257, 115529. [Google Scholar] [CrossRef]
- Li, Y.-R.; Liu, F.-F.; Liu, W.-B.; Zhang, Y.-F.; Tian, X.-Y.; Fu, X.-J.; Xu, Y.; Song, J.; Zhang, S.-Y. A novel aromatic amide derivative SY-65 co-targeted tubulin and histone deacetylase 1 with potent anticancer activity in vitro and in vivo. Biochem. Pharmacol. 2022, 201, 115070. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.; Gosch, L.C.; Dittmer, A.; Rothemund, M.; Mueller, T.; Schobert, R.; Biersack, B.; Volkamer, A.; Höpfner, M. Oxazole-Bridged Combretastatin A-4 Derivatives with Tethered Hydroxamic Acids: Structure–Activity Relations of New Inhibitors of HDAC and/or Tubulin Function. Int. J. Mol. Sci. 2019, 20, 383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Liang, Y.; Zhang, H.; Jiang, H.; Feng, K.; Xu, P.; Wang, J.; Wang, X.; Ding, K.; Luo, C.; et al. Design, synthesis, biological evaluation and cocrystal structures with tubulin of chiral β-lactam bridged combretastatin A-4 analogues as potent antitumor agents. Eur. J. Med. Chem. 2018, 144, 817–842. [Google Scholar] [CrossRef]
- Tang, H.; Liang, Y.; Yu, M.; Cai, S.; Ding, K.; Wang, Y. Discovery of chiral 1,4-diarylazetidin-2-one-based hydroxamic acid derivatives as novel tubulin polymerization inhibitors with histone deacetylase inhibitory activity. Bioorg. Med. Chem. 2023, 92, 117437. [Google Scholar] [CrossRef]
- Tang, H.; Liang, Y.; Shen, H.; Cai, S.; Yu, M.; Fan, H.; Ding, K.; Wang, Y. Discovery of a 2,6-diarylpyridine-based hydroxamic acid derivative as novel histone deacetylase 8 and tubulin dual inhibitor for the treatment of neuroblastoma. Bioorg. Chem. 2022, 128, 106112. [Google Scholar] [CrossRef]
- Zheng, S.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.; LeMelle, E.; McFerrin, H.; Wang, G. Design, Synthesis, and Biological Evaluation of Novel Pyridine-Bridged Analogues of Combretastatin-A4 as Anticancer Agents. J. Med. Chem. 2014, 57, 3369–3381. [Google Scholar] [CrossRef]
- El-Zoghbi, M.S.; Bass, A.K.A.; A Abuo-Rahma, G.E.-D.; Mohamed, M.F.A.; Badr, M.; Al-Ghulikah, H.A.; Abdelhafez, E.-S.M.N. Design, Synthesis and Mechanistic Study of New Dual Targeting HDAC/Tubulin Inhibitors. Future Med. Chem. 2024, 16, 601–622. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Tong, L.; Luo, Y.; Su, M.; Zang, Y.; Li, J.; Lu, W.; Chen, Y. The discovery of colchicine-SAHA hybrids as a new class of antitumor agents. Bioorg. Med. Chem. 2013, 21, 3240–3244. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, Y.; Zhang, J.; Su, M.; Zhou, Y.; Zang, Y.; Li, J.; Chen, Y.; Fang, Y.; Zhang, X.; et al. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur. J. Med. Chem. 2015, 95, 127–135. [Google Scholar] [CrossRef]
- Zhu, H.; Li, W.; Shuai, W.; Liu, Y.; Yang, L.; Tan, Y.; Zheng, T.; Yao, H.; Xu, J.; Zhu, Z.; et al. Discovery of novel N-benzylbenzamide derivatives as tubulin polymerization inhibitors with potent antitumor activities. Eur. J. Med. Chem. 2021, 216, 113316. [Google Scholar] [CrossRef]
- Zhu, H.; Tan, Y.; He, C.; Liu, Y.; Duan, Y.; Zhu, W.; Zheng, T.; Li, D.; Xu, J.; Yang, D.-H.; et al. Discovery of a Novel Vascular Disrupting Agent Inhibiting Tubulin Polymerization and HDACs with Potent Antitumor Effects. J. Med. Chem. 2022, 65, 11187–11213. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; Aldhahrani, A.; Althobaiti, F.; Fayad, E.; Abu Ali, O.A.; Albogami, S.; Abu Almaaty, A.H.; Khedr, A.I.M.; Bukhari, S.N.A.; Zaki, I. Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells. Molecules 2022, 27, 3960. [Google Scholar] [CrossRef] [PubMed]
- Korolyov, A.; Dorbes, S.; Azéma, J.; Guidetti, B.; Danel, M.; Lamoral-Theys, D.; Gras, T.; Dubois, J.; Kiss, R.; Martino, R.; et al. Novel lipophilic 7H-pyrido [1,2,3-de]-1,4-benzoxazine-6-carboxylic acid derivatives as potential antitumor agents: Improved synthesis and in vitro evaluation. Bioorg. Med. Chem. 2010, 18, 8537–8548. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, X.; Sun, S.; Liu, Y. Synthesis and biological evaluation of novel quinolone derivatives dual targeting histone deacetylase and tubulin polymerization as antiproliferative agents. RSC Adv. 2018, 8, 16494–16502. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Tolomeo, M.; Grimaudo, S.; Cristina, A.D.; Pipitone, M.R.; Balzarini, J.; et al. Design, synthesis and structure–activity relationship of 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan derivatives as a novel class of inhibitors of tubulin polymerization. Bioorg. Med. Chem. 2009, 17, 6862–6871. [Google Scholar] [CrossRef]
- Mariotto, E.; Canton, M.; Marchioro, C.; Brancale, A.; Hamel, E.; Varani, K.; Vincenzi, F.; De Ventura, T.; Padroni, C.; Viola, G.; et al. Synthesis and Biological Evaluation of Novel 2-Aroyl Benzofuran-Based Hydroxamic Acids as Antimicrotubule Agents. Int. J. Mol. Sci. 2024, 25, 7519. [Google Scholar] [CrossRef]
- Singh, A.; Fatima, K.; Singh, A.; Behl, A.; Mintoo, M.J.; Hasanain, M.; Ashraf, R.; Luqman, S.; Shanker, K.; Mondhe, D.M.; et al. Anticancer activity and toxicity profiles of 2-benzylidene indanone lead molecule. Eur. J. Pharm. Sci. 2015, 76, 57–67. [Google Scholar] [CrossRef]
- Saxena, H.O.; Faridi, U.; Srivastava, S.; Kumar, J.K.; Darokar, M.P.; Luqman, S.; Chanotiya, C.S.; Krishna, V.; Negi, A.S.; Khanuja, S.P.S. Gallic acid-based indanone derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2008, 18, 3914–3918. [Google Scholar] [CrossRef]
- Negi, A.S.; Prakasham, A.P.; Saxena, A.K.; Luqman, S.; Chanda, D.; Kaur, T.; Gupta, A. Anticancer and Tubulin Polymerisation Activity of Benzylidene Indanones and the Process of Preparing the Same. US Patent US8633242 B2, 21 January 2014. [Google Scholar]
- Kumar, K.; Das, R.; Thapa, B.; Rakhecha, B.; Srivastava, S.; Savita, K.; Israr, M.; Chanda, D.; Banerjee, D.; Shanker, K.; et al. Dual targeted 2-Benzylideneindanone pendant hydroxamic acid group exhibits selective HDAC6 inhibition along with tubulin stabilization effect. Bioorg. Med. Chem. 2023, 86, 117300. [Google Scholar] [CrossRef]
- Li, L.; Quan, D.; Chen, J.; Ding, J.; Zhao, J.; Lv, L.; Chen, J. Design, synthesis, and biological evaluation of 1-substituted -2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. [Google Scholar] [CrossRef]
- Peng, X.; Chen, J.; Li, L.; Sun, Z.; Liu, J.; Ren, Y.; Huang, J.; Chen, J. Efficient Synthesis and Bioevaluation of Novel Dual Tubulin/Histone Deacetylase 3 Inhibitors as Potential Anticancer Agents. J. Med. Chem. 2021, 64, 8447–8473. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure–Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Sinatra, L.; Vogelmann, A.; Friedrich, F.; Tararina, M.A.; Neuwirt, E.; Colcerasa, A.; König, P.; Toy, L.; Yesiloglu, T.Z.; Hilscher, S.; et al. Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation. J. Med. Chem. 2023, 66, 14787–14814. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, Y.; Qin, J.; Ba, M.; Yao, Y.; Duan, Y.; Liu, H.; Yu, D. Synthesis and biological evaluation of new 2-methoxyestradiol derivatives: Potent inhibitors of angiogenesis and tubulin polymerization. Bioorg. Chem. 2021, 113, 104988. [Google Scholar] [CrossRef]
- Sun, M.; Qin, J.; Kang, Y.; Zhang, Y.; Ba, M.; Yang, H.; Duan, Y.; Yao, Y. 2-Methoxydiol derivatives as new tubulin and HDAC dual-targeting inhibitors, displaying antitumor and antiangiogenic response. Bioorg. Chem. 2022, 120, 105625. [Google Scholar] [CrossRef]
- Xie, S.; Leng, J.; Zhao, S.; Zhu, L.; Zhang, M.; Ning, M.; Zhao, B.; Kong, L.; Yin, Y. Design and biological evaluation of dual tubulin/HDAC inhibitors based on millepachine for treatment of prostate cancer. Eur. J. Med. Chem. 2024, 268, 116301. [Google Scholar] [CrossRef]
- Zhang, X.; Bao, B.; Yu, X.; Tong, L.; Luo, Y.; Huang, Q.; Su, M.; Sheng, L.; Li, J.; Zhu, H.; et al. The discovery and optimization of novel dual inhibitors of topoisomerase II and histone deacetylase. Bioorg. Med. Chem. 2013, 21, 6981–6995. [Google Scholar] [CrossRef]
- Yong, Y.; Shin, S.Y.; Lee, Y.H.; Lim, Y. Antitumor activity of deoxypodophyllotoxin isolated from Anthriscus sylvestris: Induction of G2/M cell cycle arrest and caspase-dependent apoptosis. Bioorg. Med. Chem. Lett. 2009, 19, 4367–4371. [Google Scholar] [CrossRef]
- Shin, S.Y.; Yong, Y.; Kim, C.G.; Lee, Y.H.; Lim, Y. Deoxypodophyllotoxin induces G2/M cell cycle arrest and apoptosis in HeLa cells. Cancer Lett. 2010, 287, 231–239. [Google Scholar] [CrossRef]
- Chen, S.-W.; Gao, Y.-Y.; Zhou, N.-N.; Liu, J.; Huang, W.-T.; Hui, L.; Jin, Y.; Jin, Y.-X. Carbamates of 4′-demethyl-4-deoxypodophyllotoxin: Synthesis, cytotoxicity and cell cycle effects. Bioorg. Med. Chem. Lett. 2011, 21, 7355–7358. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Su, M.; Zhou, Y.; Chen, Y.; Li, J.; Lu, W. Design, synthesis and biological evaluation of 4′-demethyl-4-deoxypodophyllotoxin derivatives as novel tubulin and histone deacetylase dual inhibitors. RSC Adv. 2014, 4, 40444–40448. [Google Scholar] [CrossRef]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, K.; Zhu, Q.; Shen, Q.; Zhang, Q.; Yu, J.; Chen, Y.; Lu, W. Synthesis and biological evaluation of paclitaxel and vorinostat co-prodrugs for overcoming drug resistance in cancer therapy in vitro. Bioorg. Med. Chem. 2019, 27, 1405–1413. [Google Scholar] [CrossRef]
- Yoshimatsu, K.; Yamaguchi, A.; Yoshino, H.; Koyanagi, N.; Kitoh, K. Mechanism of Action of E7010, an Orally Active Sulfonamide Antitumor Agent: Inhibition of Mitosis by Binding to the Colchicine Site of Tubulin. Cancer Res. 1997, 57, 3208–3213. [Google Scholar]
- Wu, W.-C.; Liu, Y.-M.; Lin, M.-H.; Liao, Y.-H.; Lai, M.-J.; Chuang, H.-Y.; Hung, T.-Y.; Chen, C.-H.; Liou, J.-P. Design, synthesis, and evaluation of N-phenyl-4-(2-phenylsulfonamido)-benzamides as microtubule-targeting agents in drug-resistant cancer cells, displaying HDAC inhibitory response. Eur. J. Med. Chem. 2020, 192, 112158. [Google Scholar] [CrossRef]
- Chang, J.-Y.; Hsieh, H.-P.; Chang, C.-Y.; Hsu, K.-S.; Chiang, Y.-F.; Chen, C.-M.; Kuo, C.-C.; Liou, J.-P. 7-Aroyl-aminoindoline-1-sulfonamides as a Novel Class of Potent Antitubulin Agents. J. Med. Chem. 2006, 49, 6656–6659. [Google Scholar] [CrossRef]
- Lai, M.-J.; Ojha, R.; Lin, M.-H.; Liu, Y.-M.; Lee, H.-Y.; Lin, T.E.; Hsu, K.-C.; Chang, C.-Y.; Chen, M.-C.; Nepali, K.; et al. 1-Arylsulfonyl indoline-benzamides as a new antitubulin agents, with inhibition of histone deacetylase. Eur. J. Med. Chem. 2019, 162, 612–630. [Google Scholar] [CrossRef]
- Kuo, C.-C.; Hsieh, H.-P.; Pan, W.-Y.; Chen, C.-P.; Liou, J.-P.; Lee, S.-J.; Chang, Y.-L.; Chen, L.-T.; Chen, C.-T.; Chang, J.-Y. BPR0L075, a Novel Synthetic Indole Compound with Antimitotic Activity in Human Cancer Cells, Exerts Effective Antitumoral Activity in Vivo. Cancer Res. 2004, 64, 4621–4628. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Tsai, A.-C.; Chen, M.-C.; Shen, P.-J.; Cheng, Y.-C.; Kuo, C.-C.; Pan, S.-L.; Liu, Y.-M.; Liu, J.-F.; Yeh, T.-K.; et al. Azaindolylsulfonamides, with a More Selective Inhibitory Effect on Histone Deacetylase 6 Activity, Exhibit Antitumor Activity in Colorectal Cancer HCT116 Cells. J. Med. Chem. 2014, 57, 4009–4022. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Lee, J.-F.; Kumar, S.; Wu, Y.-W.; HuangFu, W.-C.; Lai, M.-J.; Li, Y.-H.; Huang, H.-L.; Kuo, F.-C.; Hsiao, C.-J.; et al. 3-Aroylindoles display antitumor activity in vitro and in vivo: Effects of N1-substituents on biological activity. Eur. J. Med. Chem. 2017, 125, 1268–1278. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Hsu, K.-C.; Lee, H.-Y.; Huang, T.-C.; Lin, T.E.; Chen, Y.-L.; Sung, T.-Y.; Liou, J.-P.; Hwang-Verslues, W.W.; Pan, S.-L.; et al. A Novel Dual HDAC6 and Tubulin Inhibitor, MPT0B451, Displays Anti-tumor Ability in Human Cancer Cells in Vitro and in Vivo. Front. Pharmacol. 2018, 9, 205. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivodestabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef]
- Nepali, K.; Chang, T.-Y.; Lai, M.-J.; Hsu, K.-C.; Yen, Y.; Lin, T.E.; Lee, S.-B.; Liou, J.-P. Purine/purine isoster based scaffolds as new derivatives of benzamide class of HDAC inhibitors. Eur. J. Med. Chem. 2020, 196, 112291. [Google Scholar] [CrossRef]
- Schemies, J.; Sippl, W.; Jung, M. Histone deacetylase inhibitors that target tubulin. Cancer Lett. 2009, 280, 222–232. [Google Scholar] [CrossRef]
- Singh, A.; Chang, T.-Y.; Kaur, N.; Hsu, K.-C.; Yen, Y.; Lin, T.E.; Lai, M.-J.; Lee, S.-B.; Liou, J.-P. CAP rigidification of MS-275 and chidamide leads to enhanced antiproliferative effects mediated through HDAC1, 2 and tubulin polymerization inhibition. Eur. J. Med. Chem. 2021, 215, 113169. [Google Scholar] [CrossRef]
- Xue, J.; Wu, G.; Ejaz, U.; Akhtar, F.; Wan, X.; Zhu, Y.; Geng, A.; Chen, Y.; He, S. A novel histone deacetylase inhibitor LT-548-133-1 induces apoptosis by inhibiting HDAC and interfering with microtubule assembly in MCF-7 cells. Investig. New Drugs 2021, 39, 1222–1231. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, T.; Wang, F.; Niu, T.; Liu, Z.; Chen, X.; Long, C.; Tang, M.; Cao, D.; Wang, X.; et al. Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem. 2016, 59, 1455–1470. [Google Scholar] [CrossRef]
- Wang, F.; Zheng, L.; Yi, Y.; Yang, Z.; Qiu, Q.; Wang, X.; Yan, W.; Bai, P.; Yang, J.; Li, D.; et al. SKLB-23bb, A HDAC6-Selective Inhibitor, Exhibits Superior and Broad-Spectrum Antitumor Activity via Additionally Targeting Microtubules. Mol. Cancer Ther. 2018, 17, 763–775. [Google Scholar] [CrossRef]
- Wang, X.-F.; Guan, F.; Ohkoshi, E.; Guo, W.; Wang, L.; Zhu, D.-Q.; Wang, S.-B.; Wang, L.-T.; Hamel, E.; Yang, D.; et al. Optimization of 4-(N-Cycloamino)phenylquinazolines as a Novel Class of Tubulin-Polymerization Inhibitors Targeting the Colchicine Site. J. Med. Chem. 2014, 57, 1390–1402. [Google Scholar] [CrossRef]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted Cancer Therapy: Giving Histone Deacetylase Inhibitors All They Need To Succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef]
- Ning, Z.-Q.; Li, Z.-B.; Newman, M.J.; Shan, S.; Wang, X.-H.; Pan, D.-S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.-P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef]
- Vasudevan, A.; Ji, Z.; Frey, R.R.; Wada, C.K.; Steinman, D.; Heyman, H.R.; Guo, Y.; Curtin, M.L.; Guo, J.; Li, J.; et al. Heterocyclic ketones as inhibitors of histone deacetylase. Bioorg. Med. Chem. Lett. 2003, 13, 3909–3913. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Al-Hamashi, A.A.; Koranne, R.; Dlamini, S.; Alqahtani, A.; Karaj, E.; Rashid, M.S.; Knoff, J.R.; Dunworth, M.; Pflum, M.K.H.; Casero, R.A.; et al. A new class of cytotoxic agents targets tubulin and disrupts microtubule dynamics. Bioorg. Chem. 2021, 116, 105297. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 for Cancer Cells | IC50 for Normal Cells | IC50 (TPI) | IC50 (HDAC) | In Vivo Results | Water Solubility | References | |
|---|---|---|---|---|---|---|---|---|
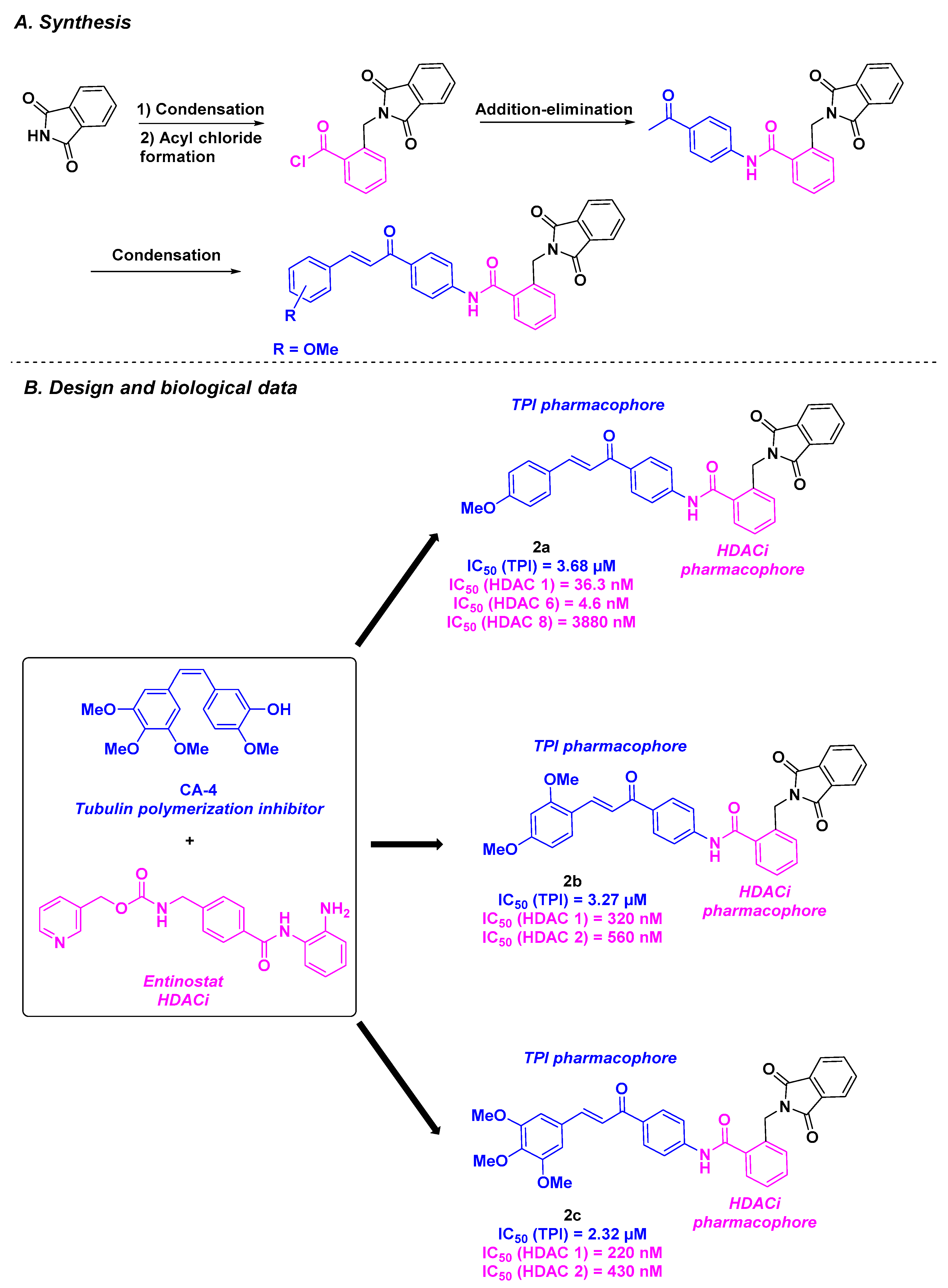
| CA-4 derivatives | Chalcone derivative 1a–c | Up to 550 nM for A549 cells | n. d. | Up to 5.0 µM | 4.6–4910 nM (HDACs 1, 6 and 8) | n. d. | n. d. | [82] |
| Chalcone derivative 2a–c | From 1.62 to 2.21 µM against MCF-7 and Hep G2 cells | n. d. | Up to 2.32 µM | 4.6–3880 nM (HDACs 1, 6 and 8) | n. d. | n. d. | [83] | |
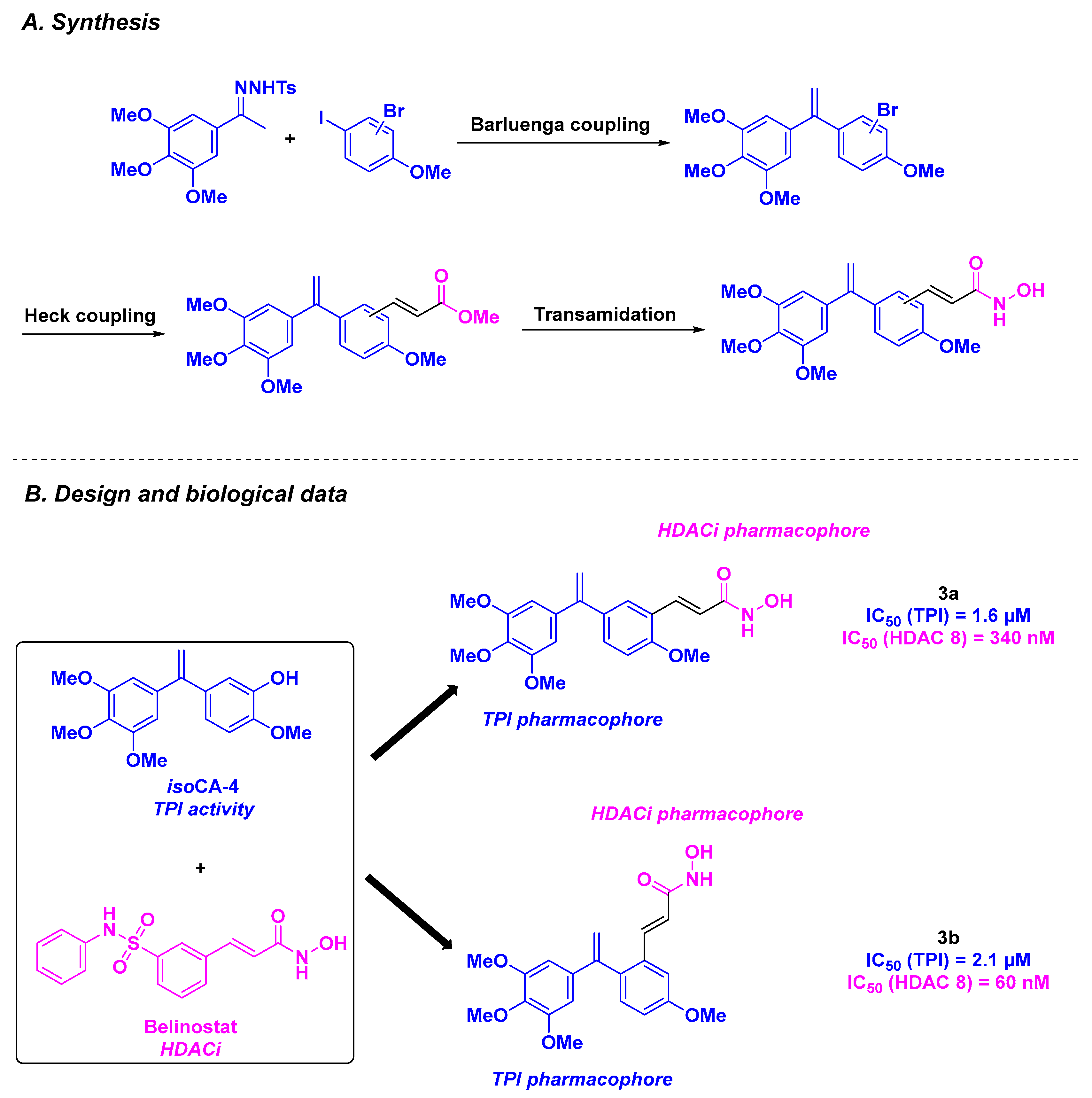
| Stilbene derivative 3a–b | From 0.4 to 5.1 nM for A549, K562, K562R, PC3, U87-MG, MCF7, BXPC3, MiaPaca2, and HT29 cells | 7 µM | Up to 1.6 µM | 30–340 nM (HDAC 8) | n. d. | n. d. | [87] | |
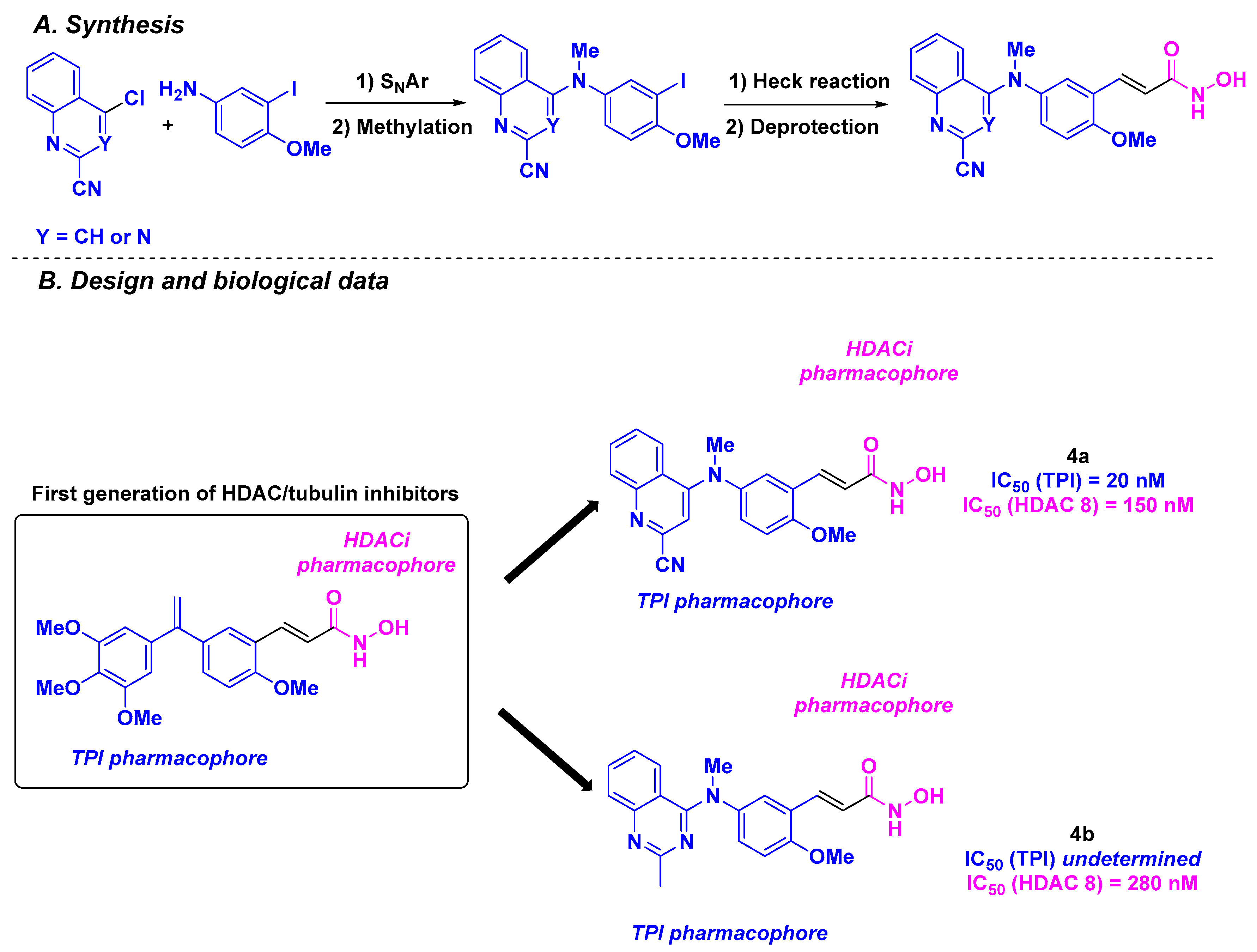
| Stilbene derivative 4a–b | ca. 0.65 nM for NCI-N87, K562, K562R, MiaPaca2, SKOV3, A549, MCF7, MDA MB231 and HT-29 cells | n. d. | Up to 20 nM | 150–280 nM (HDAC 8) | Significant tumor decrease for 0.25–0.50 mg/kg doses of 4a | From 68.7 to 65 µg/mL | [88] | |
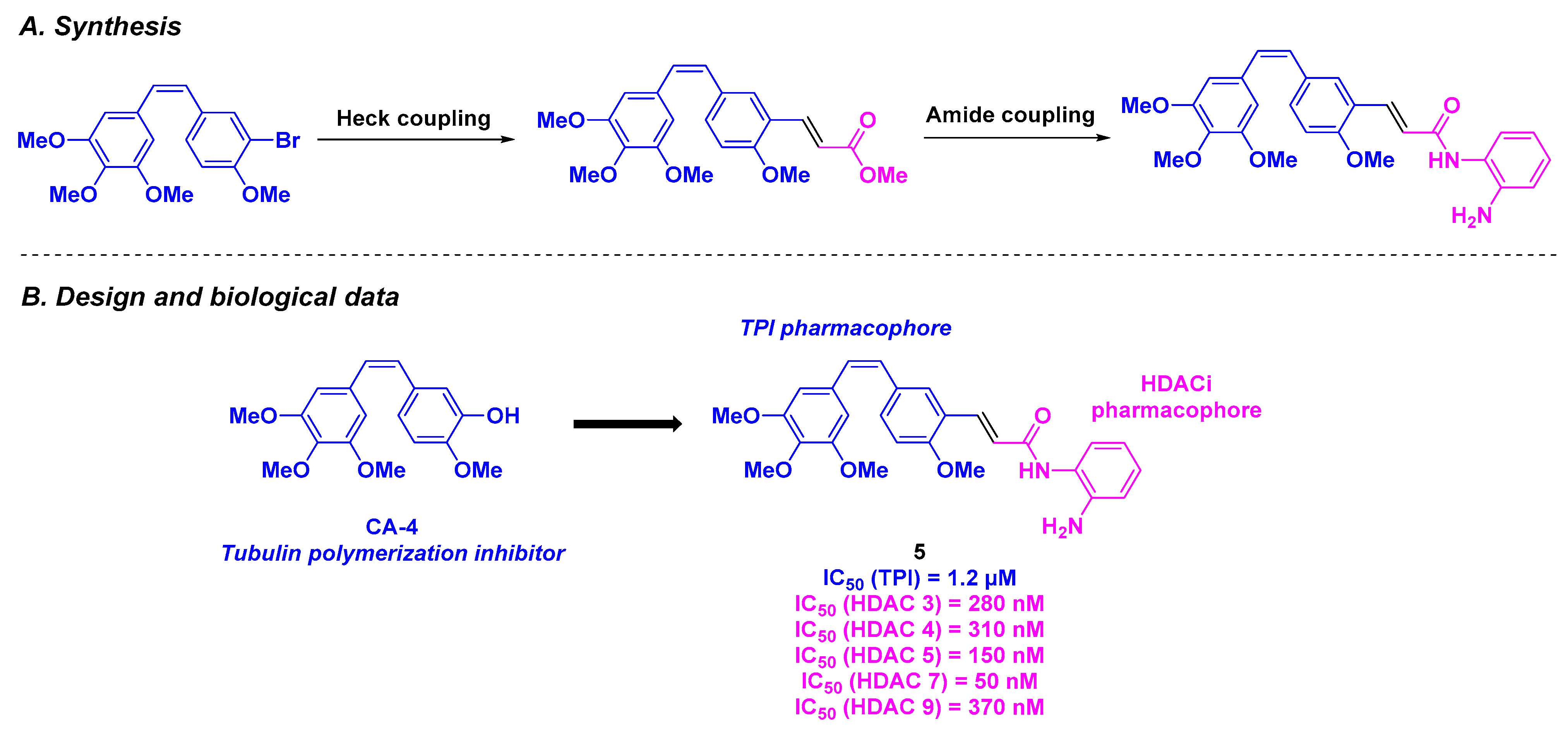
| Stilbene derivative 5 | From 16 to 305 nM for MGC-803, MCF-7, U937, A549, HepG2 and HeLa | 195–556 nM | 1.2 µM | 50–370 nM (HDACs 3–9) | Significant tumor decrease at a concentration of 100 nM | n. d. | [89] | |
| Stilbene derivative 6 | From 3 to 36 nM for K562, MCF-7, MDA-MB-231, A549, B16F10 and A2780 cells | 230 nM | 3.84 µM | 403–591 nM (HDACs 1, 2 and 6) | TGI = 73.12% for daily 20 mg/kg dose | n. d. | [90] | |
| Amine derivative SY-65 | From 39 to 254 nM for MGC-803, HGC-27, SGC-7901, HCT-116 and KYSE450 cells | n. d. | 3.64 µM | 525–5270 nM (HDACs 1–3) | Inhibition of the tumor growth at a 30 mg/kg dose | n. d. | [91] | |
| Oxazole derivatives 7a–c | From 0.11 to 14.2 µM for 518A2, 518A2, HT-29, DLD-1, HCT-116, KB-V1Vbl and MCF-7Topo cells | n. d. | TPI activity at 10 µM of 7a | Down to 320–13,800 nM (HDAC 1 and 6) | Low toxicity at up to 200 mg/kg of 7a in nude mice model | n. d. | [92] | |
| 1,4-Diarylazetidin-2-one derivatives 8a–b | From 16 to 56 nM for BE-(2)-C, A549, U87MG and HCT-116 cells | n. d. | 5.4 µM for 8b | Down to 155–2405 nM (HDACs 1, 6 and 8) | TGI = 67% for 25 mg/kg dose of 8b every 3 days | 102.2 µg/mL | [94] | |
| Arylpyridine derivative 9 | From 17 to 90 nM for BE-(2)–C, A549, U87MG and HCT-116 cells | 170 nM | 2.4 µM | 117–5839 nM (HDACs 1, 6 and 8) | TGI = 61% for 50 mg/kg dose every 3 days | 138 µg/mL | [95] | |
| Arylpyridine derivative 10a–b | For 10a: 3.52 µM for UO-31 cells and 5.50 µM for T47D cells | n. d. | Up to 3.73 µM | 57–84 nM (HDACs 1 and 6) | n. d. | n. d. | [97] | |
| Colchicine derivatives | 11a–b | For 11b: from 0.242 to 4.672 µM for A431, A549, HCT-116, MCF-7 and PC-3 cells | n. d. | n. d. | 440–3430 nM (HDACs 1, 3 and 6) | n. d. | n. d. | [98] |
| 12a–b | From 2 to 106 nM for A549, HCT-116, SW620, Hep3B, HeoG2, MHCC97H, SNU-5, SNU-16, MKN-45, PANC-1 and SJSA-1 cells | n. d. | TPI activity at 10 µM | 0.19–12.5 µM (HDACs 1–3) | n. d. | n. d. | [99] | |
| Aminobenzamide derivative | 13 | From 26 to 30 nM for HepG2, HCT-116, MDA-MB-231, H22 and MCF-7 cells | 134 nM | 4.06 µM | 115–242 nM (HDACs 1–6) | Reduction of the tumor weights by 82% for daily 20 mg/kg dose | n. d. | [101] |
| Amide derivatives | 14a–b | From 0.65 to 0.92 µM for HepG2 | From 9.62 and 11.09 µM for normal liver HL-7022 cells | 270 nM for 14a | For 14a: 47 nM (HDAC 1) and 86 nM (HDAC 2) | n. d. | n. d. | [102] |
| Quinolone derivative | 15 | From 0.3 to 4.9 µM for A549, HepG2, MCF-7, PC-3 and HeLa cells | >100 µM | 1.79 µM | 21–41 nM (HDACs 1, 2 and 6) | n. d. | n. d. | [104] |
| Benzofuran derivatives | 16a–d | From 0.4 to 23.5 nM for HeLa, MDA-MB-231, A549, HT-29, and MCF-7 cells | n. d. | From 420 to 580 nM | From 6 to >30 µM (HDACs 1, 6 and 10) | n. d. | n. d. | [106] |
| 2-Benzylideneindanone derivatives | 17a–b | From 0.36 to 49.67 µM for MCF-7, MD-AMB-231 and K562 cells | From 47.23 to 100.32 µM for Vero cells | TPI activity at 5 µM | Residual activity for HDAC 6 from 0.03 to 0.09 | Low toxicity at up to 1000 mg/kg of 17a | 1.76 µg/mL for 17b | [110] |
| Aminothiazole derivatives | 18 | From 30 to 140 nM for HCT-116, B16-F10, Jurkat and A549 cells | n. d. | 12.2 µM | 30 nM (HDAC 3) | TGI = 70% for daily 10 mg/kg dose | n. d. | [112] |
| 19 | EC50 from 12.9 to 30.1 µM for HGC27, W1, MCF-7, and PC-3M-luc cells | n. d. | 320 nM (for Sirt2) | 43 nM (HDAC 6) | n. d. | n. d. | [115] | |
| 2-Methoxyestradiol derivative | 20 | From 0.371 to 4.840 µM for MCF-7, MGC-803, HeLa, A549, HepG2 and U937 cells | 8.14–10.89 µM | TPI activity at 4 µM | 60 nM (HDAC 2) and 12 nM (HDAC 6) | Antitumor activity 1 to 4 µM 3-day incubation | n. d. | [117] |
| Millepachine derivative | 21 | From 13 nM to 340 nM for MDA-MB-231, A549, PC-3, U251 and MCF-7 cells | n. d. | 4.82 µM | 430–730 nM (HDACs 1, 2 and 6) | TGI = 90% for 20 mg/kg dose every two days | n. d. | [118] |
| Deoxypodophyllotoxin derivatives | 22a–b | From 36 to 40 nM for A549 and HCT-116 cells | n. d. | n. d. | 0.75–11.09 µM (HDACs 1–3) | n. d. | n. d. | [123] |
| Paclitaxel derivative | 23 | 1384 nM for MCF-7/ADR cells | n. d. | n. d. | n. d. | n. d. | n. d. | [125] |
| Sulfonamide derivative | 24 | GI50 from 12 to 22 nM for KB and drug-resistant KB cells | n. d. | TPI activity at 0.1 µM | 1.07 µM (HDAC 1) and 1.47 µM (HDAC 2) | n. d. | n. d. | [127] |
| Indoline/indole-sulfonamide derivative | 25 | From 44 to 79 nM for KB, A549, MKN45, KBVIN10, KB-S15 and KB-7D cells | n. d. | 1 µM | 221–662 nM (HDACs 1, 2 and 6) | TGI = 62.9% for daily 25 mg/kg dose | n. d. | [129] |
| 26a–b | From 179.26 to 6684.85 nM for A549, HCT-116 and PC-3 cells | n. d. | TPI activity at 10 µM for 26b | 64.5–3216 nM (HDACs 1, 2 and 6) | TGI = 68.5% for daily 200 mg/kg dose of 26b | n. d. | [132] | |
| Pyrrolo [2,3-d]pyrimidine derivative | 27a–b | From 50 to 1150 nM for MDA-MB-231, MDA-MB-468, HeLa, DLD-1, HCT-116, H661, H1299 and A549 cells | n. d. | TPI activity at 10 µM | 160–1950 nM (HDACs 1, 2, 3 and 8) | n. d. | n. d. | [139] |
| Benzamide derivative | LT-548-133-1 | 2.1 mM for MCF-7 cells | n. d. | TPI activity in immunofluorescence | HDACi effect with increase of acetylated-histone H3 rate | n. d. | n. d. | [140] |
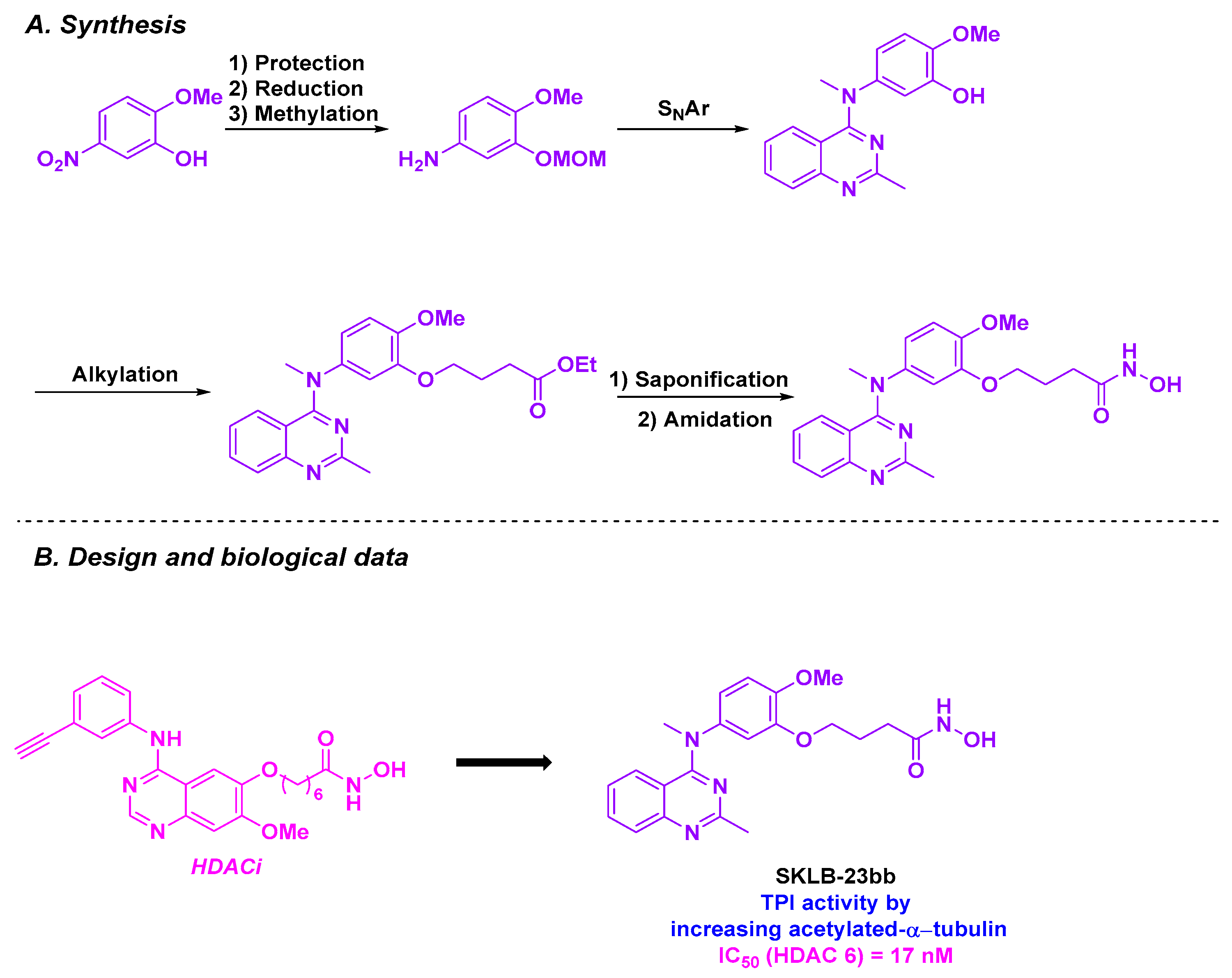
| Quinazoline derivative | SKLB-23bb | From 36.68 to 116.56 nM for 23 cancer cell lines | n. d. | TPI activity with acetylated α-tubulin | 17 nM (HDAC 6) | TGI = 58.22% for a 40 mg/kg dose three times a week | n. d. | [142] |
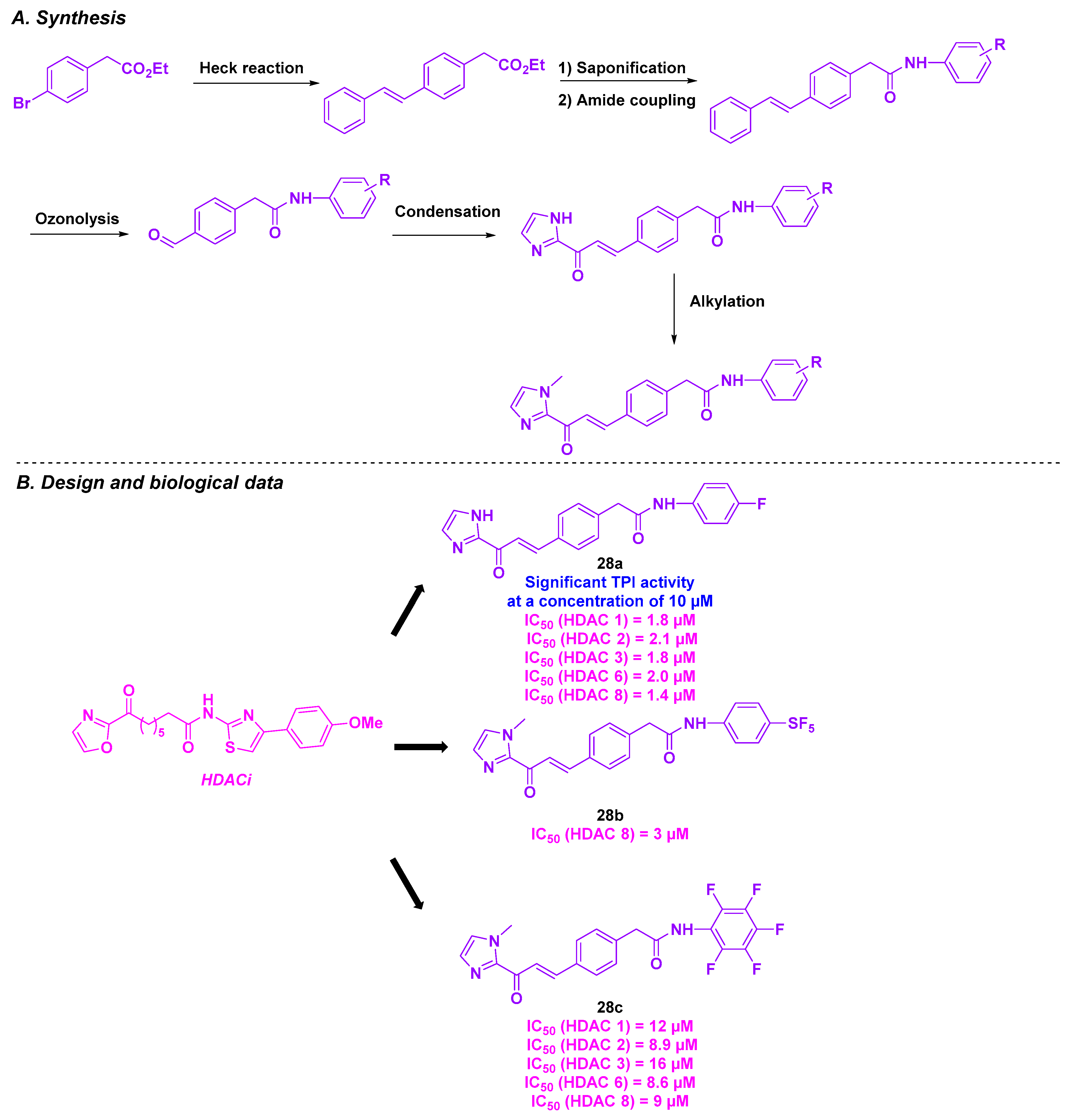
| Imidazolyl derivative | 28a–c | 5.14–6.95 µM for HCT 116 cells, GI50 with micromolar values for a wide panel of cancer cells | n. d. | TPI activity at 10 µM for 28a | 1.4–12 µM (HDACs 1, 2, 3, 6 and 8) | n. d. | n. d. | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, C.; Hamze, A. Recent Advancements in the Development of HDAC/Tubulin Dual-Targeting Inhibitors. Pharmaceuticals 2025, 18, 341. https://doi.org/10.3390/ph18030341
Tran C, Hamze A. Recent Advancements in the Development of HDAC/Tubulin Dual-Targeting Inhibitors. Pharmaceuticals. 2025; 18(3):341. https://doi.org/10.3390/ph18030341
Chicago/Turabian StyleTran, Christine, and Abdallah Hamze. 2025. "Recent Advancements in the Development of HDAC/Tubulin Dual-Targeting Inhibitors" Pharmaceuticals 18, no. 3: 341. https://doi.org/10.3390/ph18030341
APA StyleTran, C., & Hamze, A. (2025). Recent Advancements in the Development of HDAC/Tubulin Dual-Targeting Inhibitors. Pharmaceuticals, 18(3), 341. https://doi.org/10.3390/ph18030341