





Identification and Development of BRD9 Chemical Probes

 ,
,  and
and 
Abstract
1. Introduction
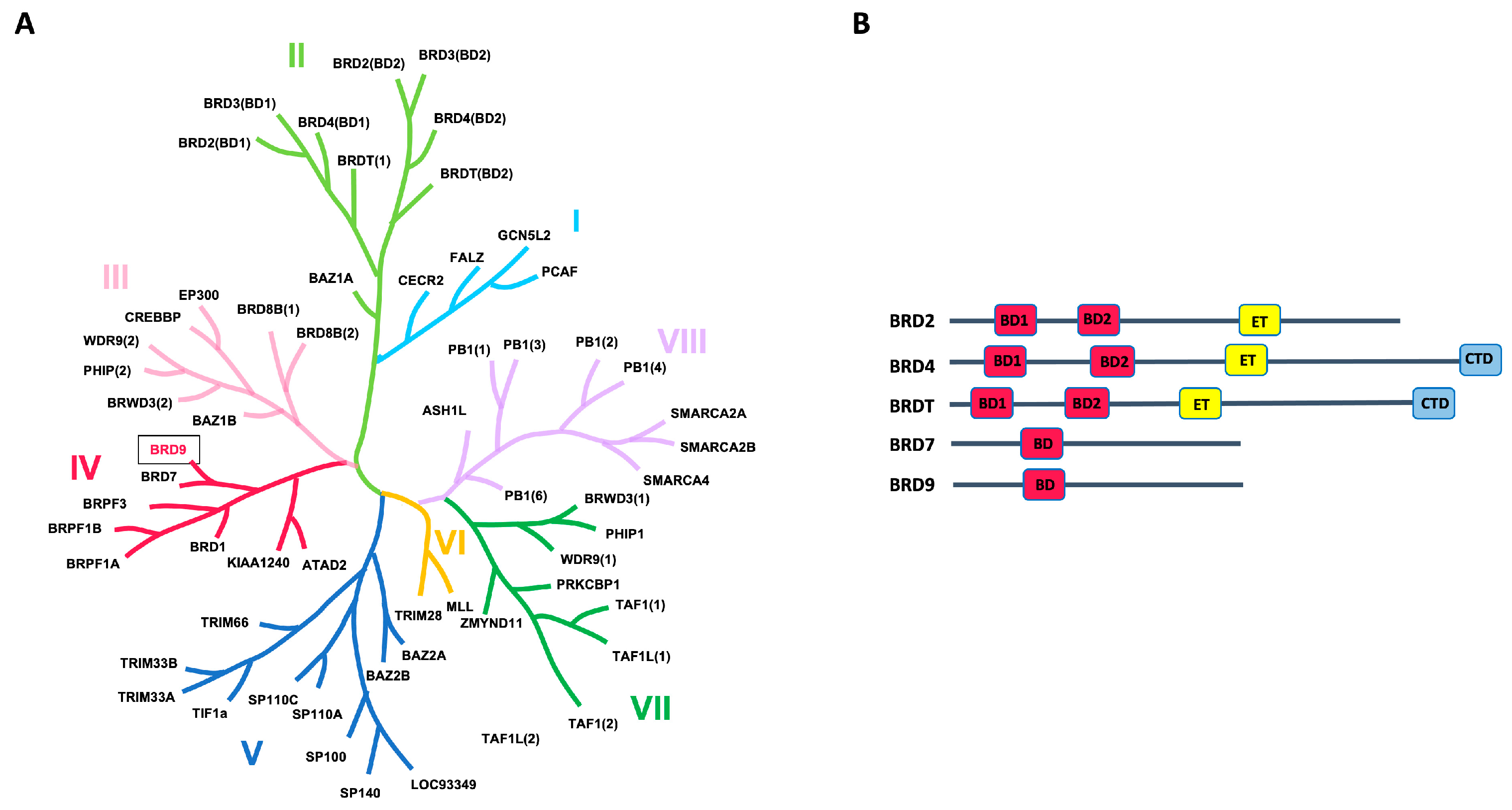
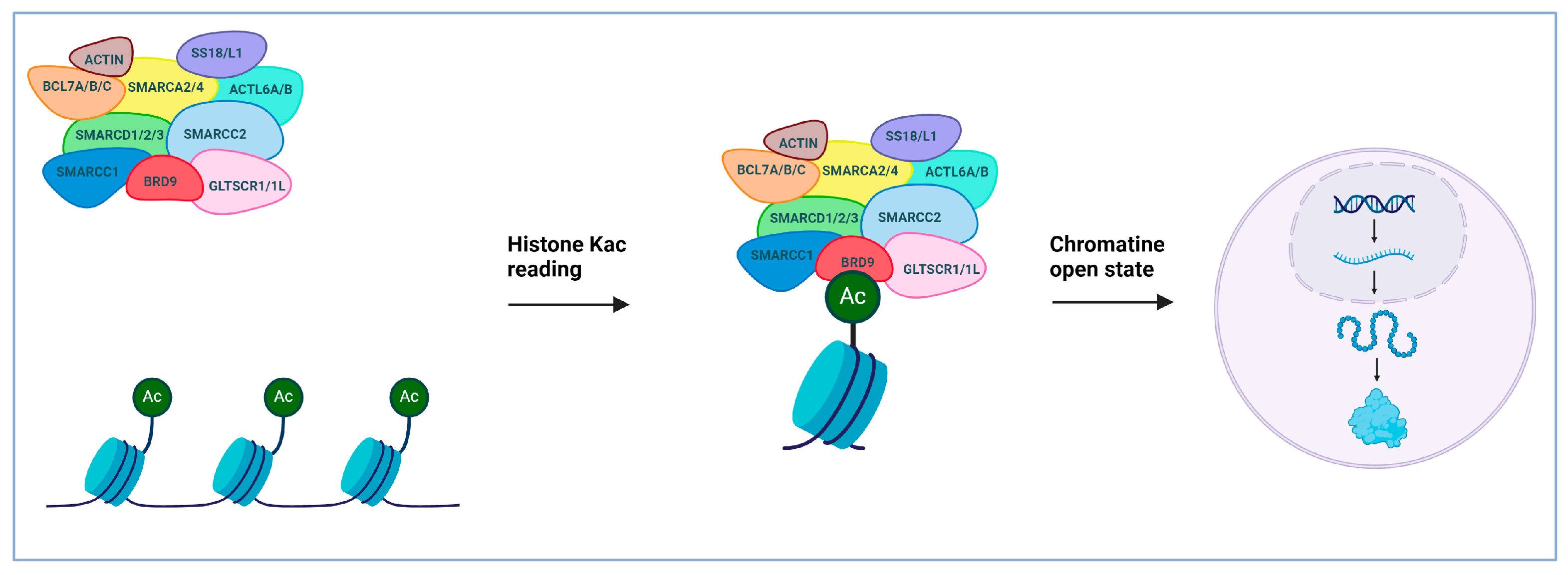
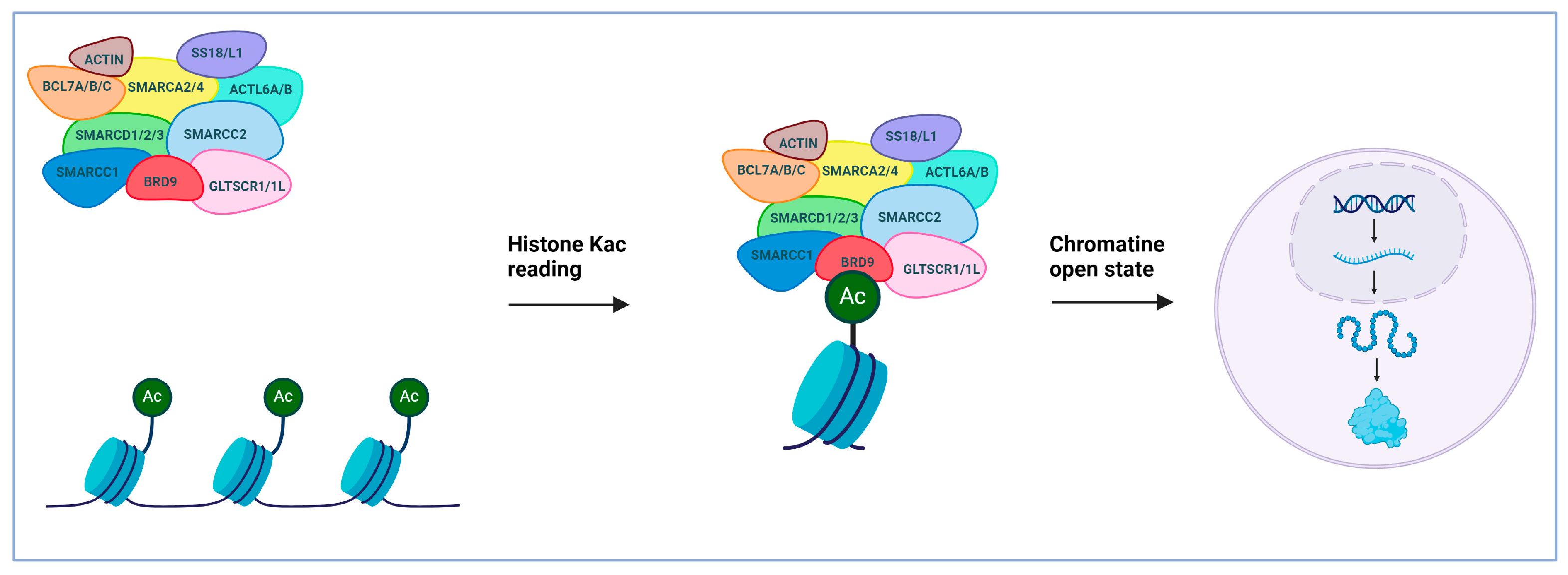
2. Bromodomain-Containing Protein 9 (BRD9): An Epigenetic Target Implicated in the Progression of Cancer
3. BRD9 Binders
3.1. Nonselective Binders
3.1.1. Quinolone Analogues
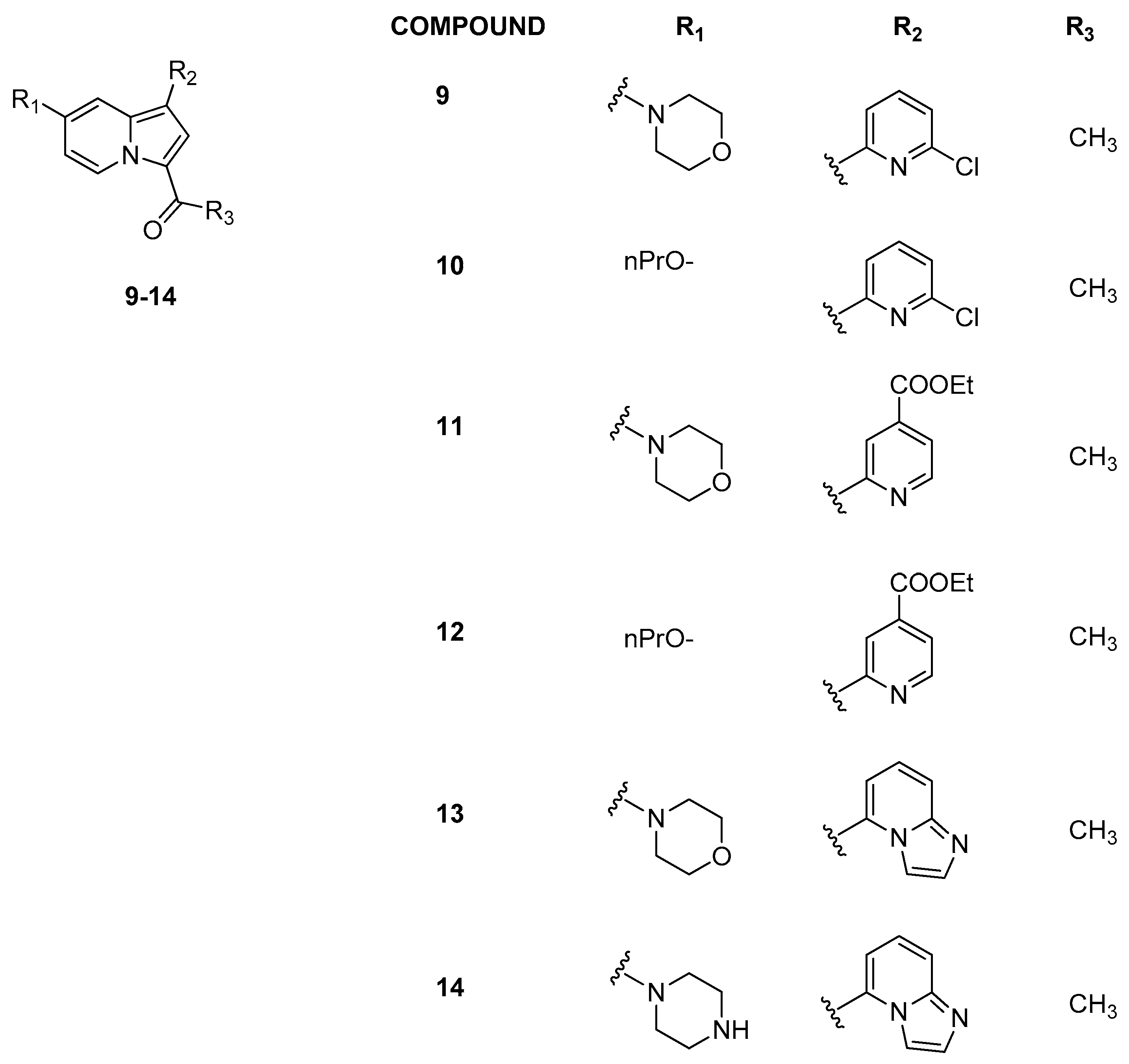
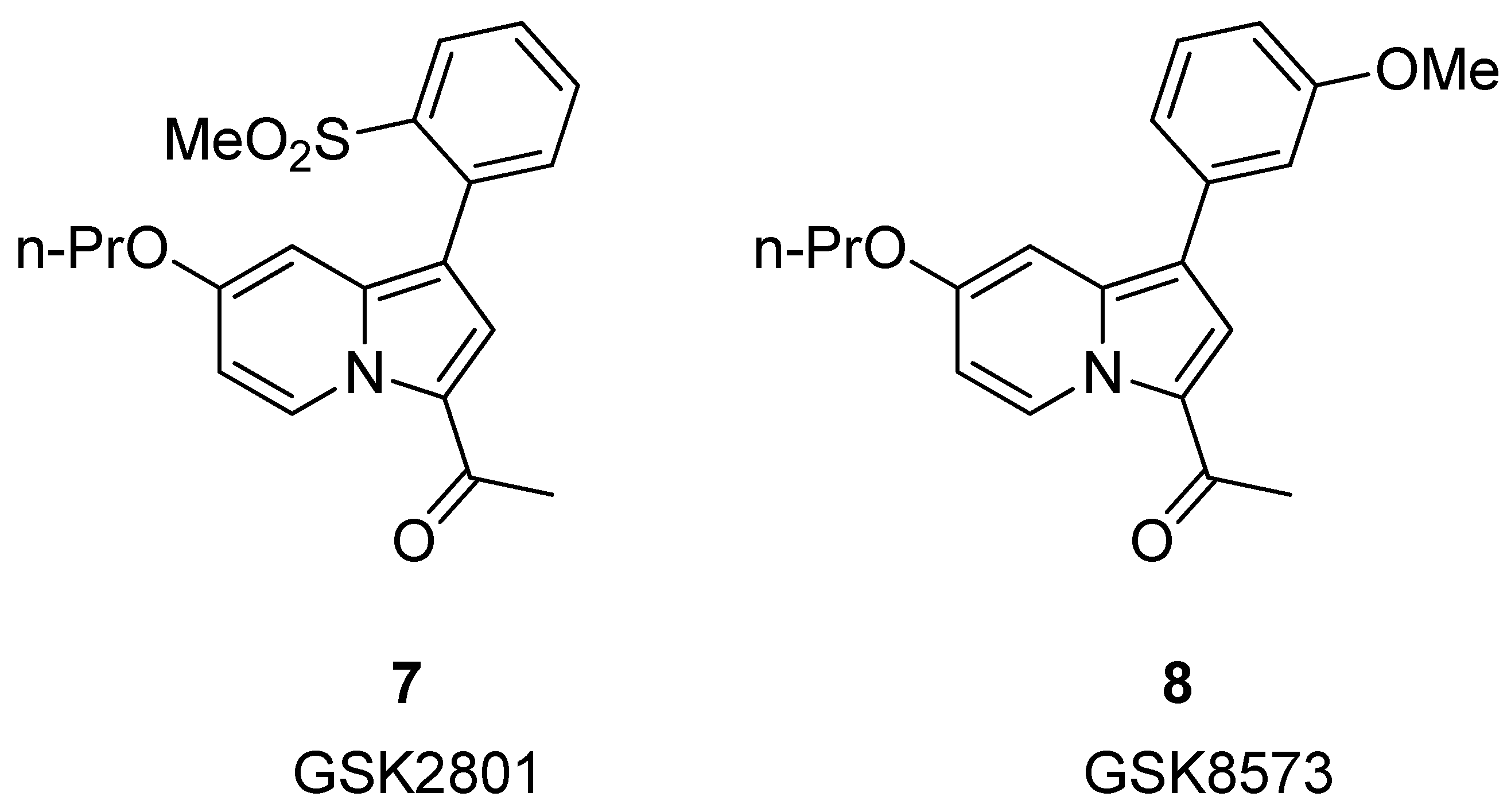
3.1.2. Indolizine Analogues
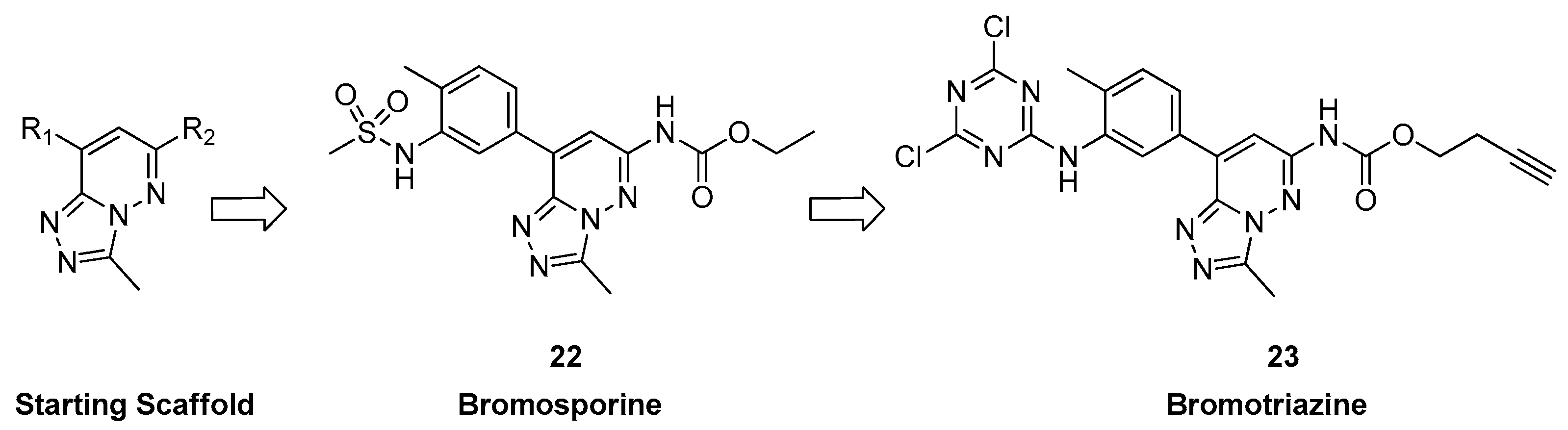
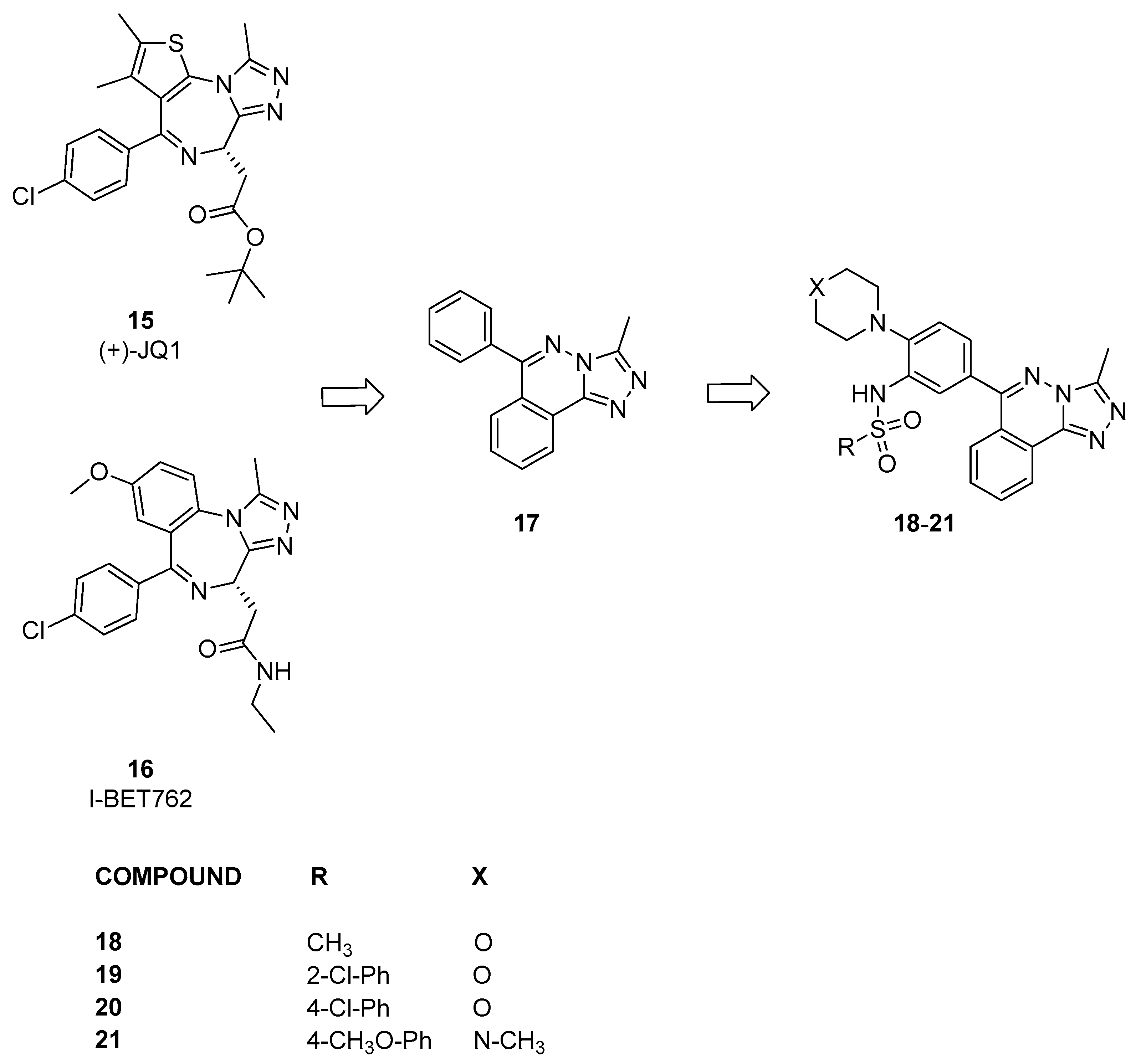
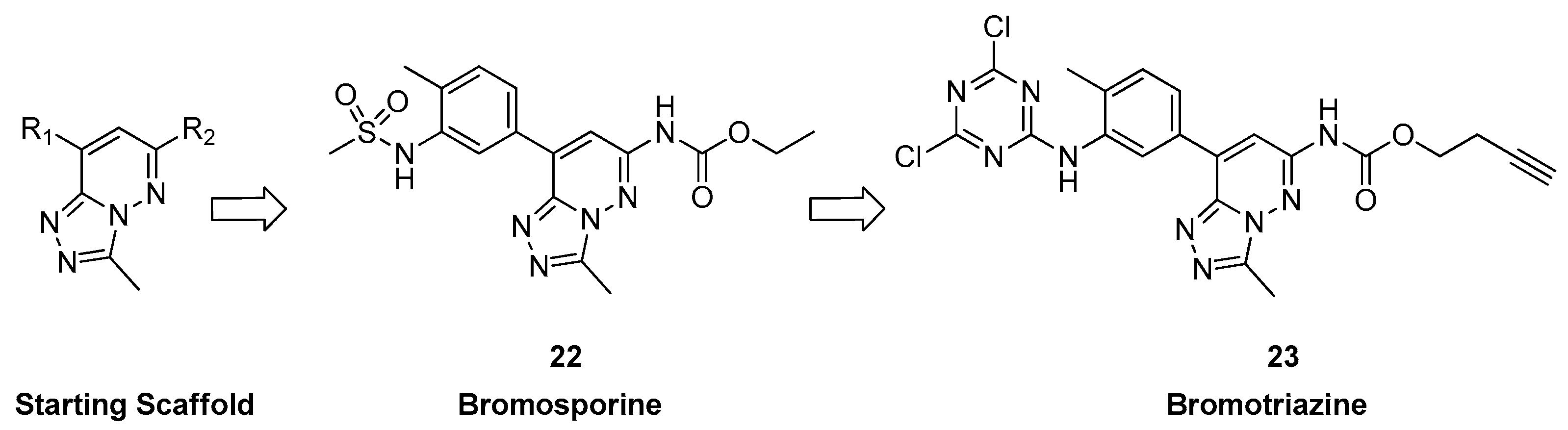
3.1.3. From [1,2,4]triazolo[4,3-a]phthalazine Analogues to Bromosporine and Bromotriazine
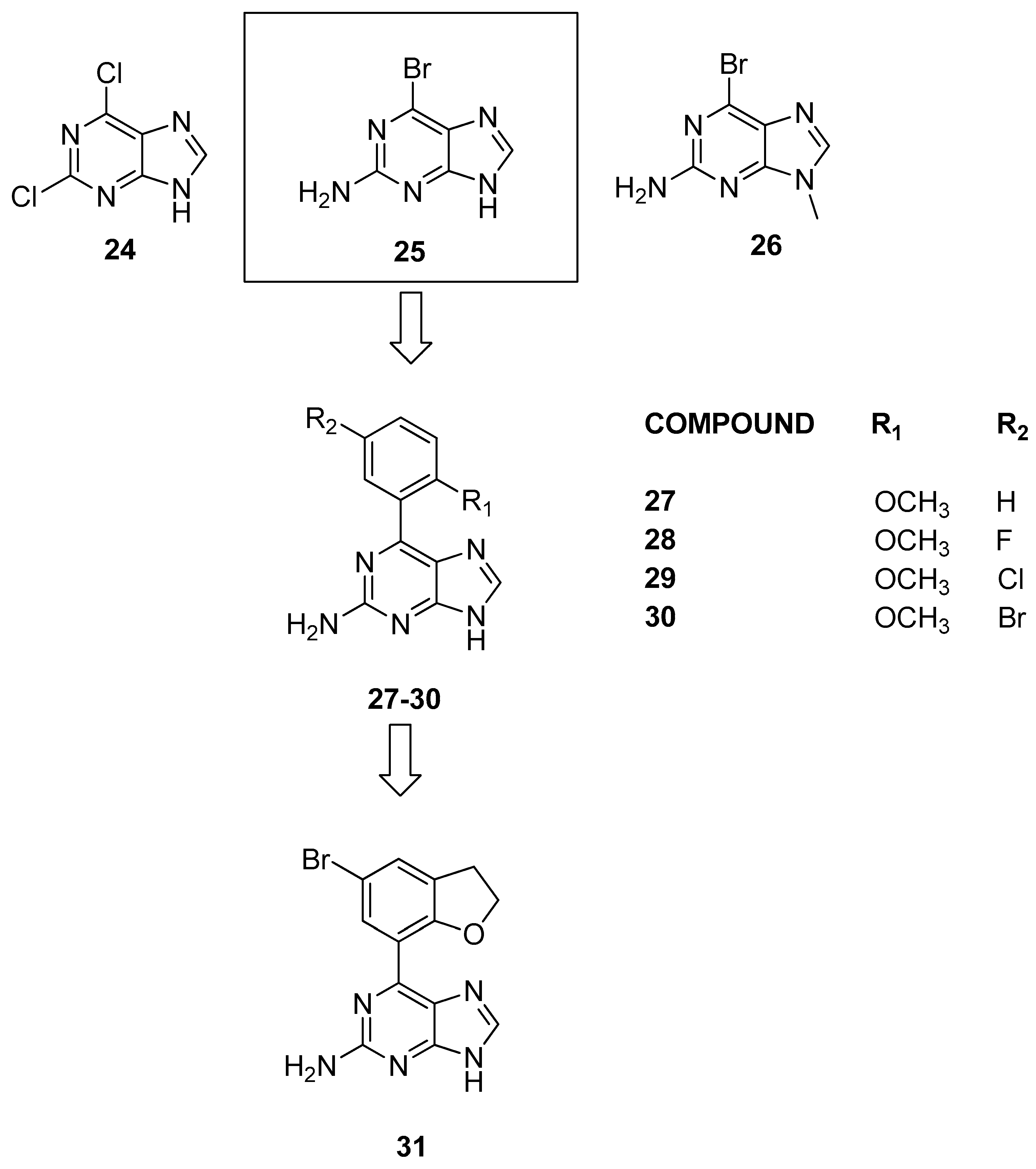
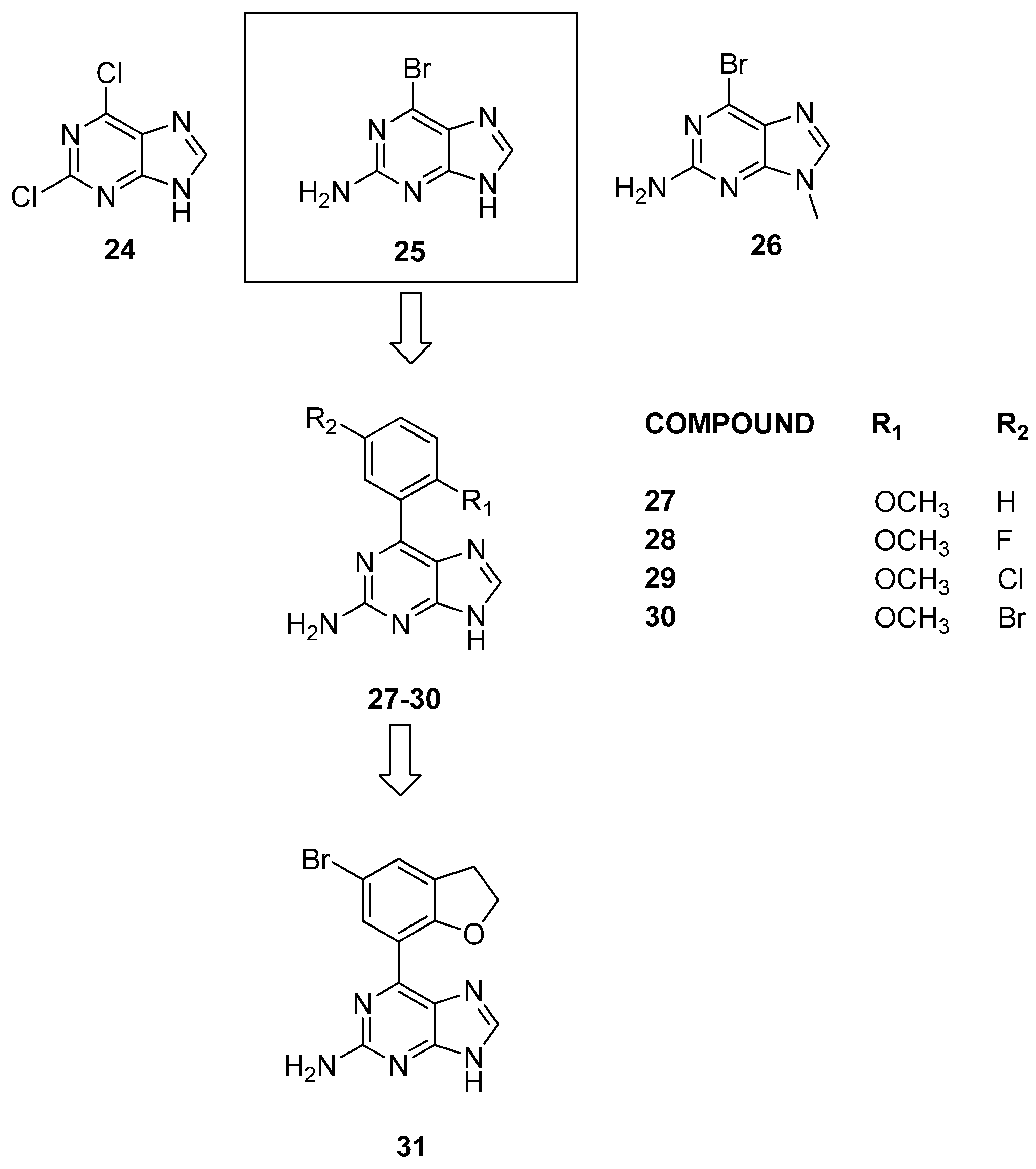
3.1.4. Purine Scaffolds
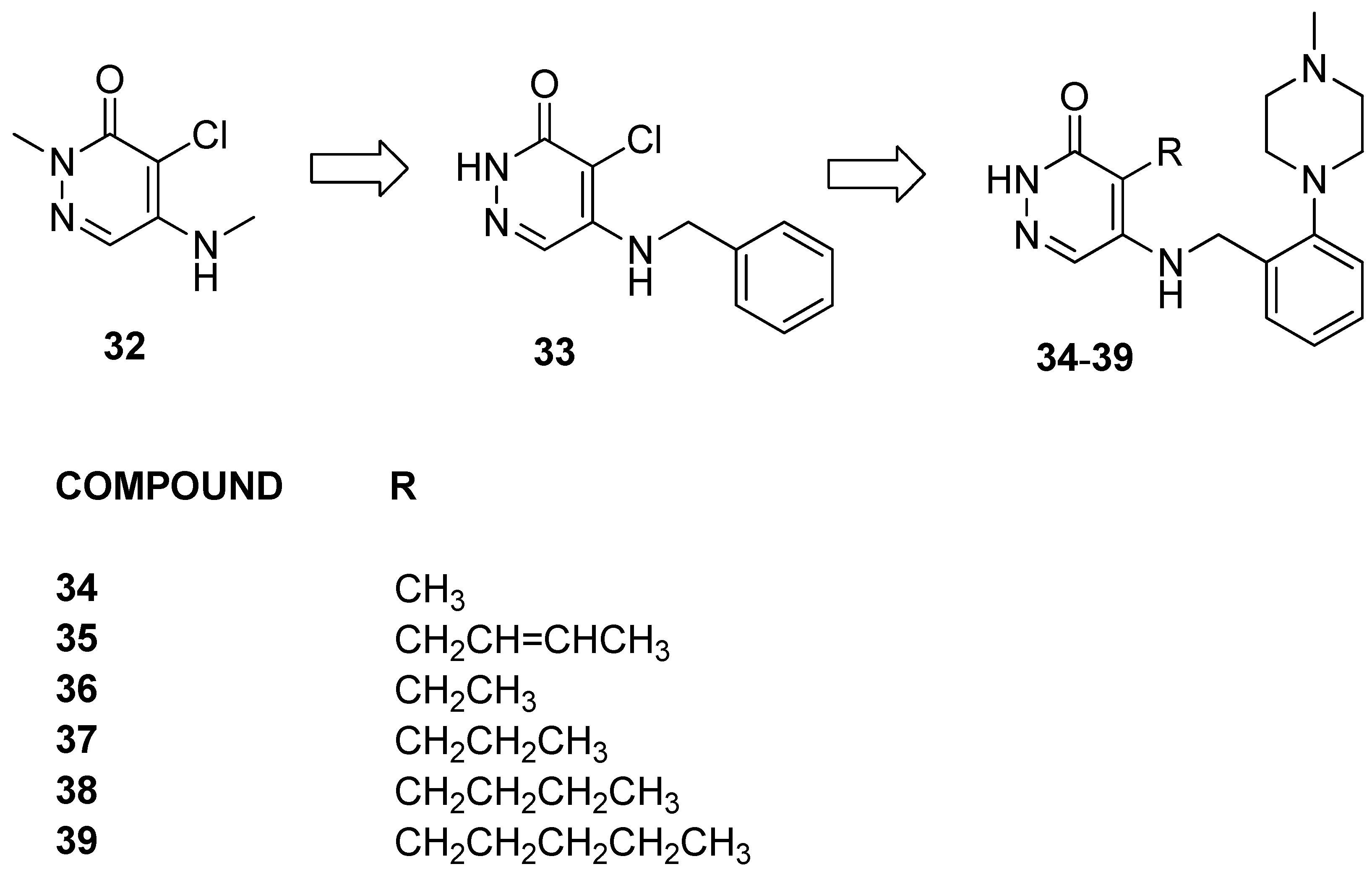
3.1.5. Alkyl-pyridazin-3(2H)-one Analogs
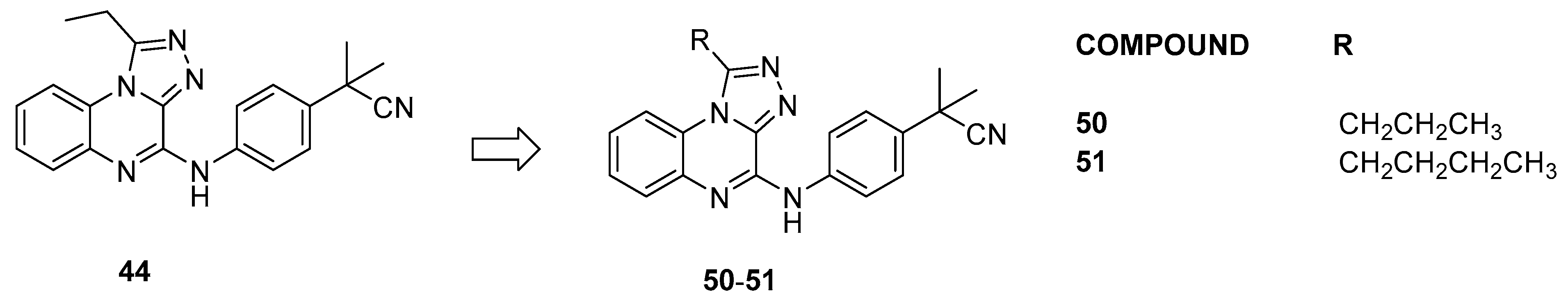
3.1.6. Triazoloquinoxaline Analogues
3.2. Selective Binders
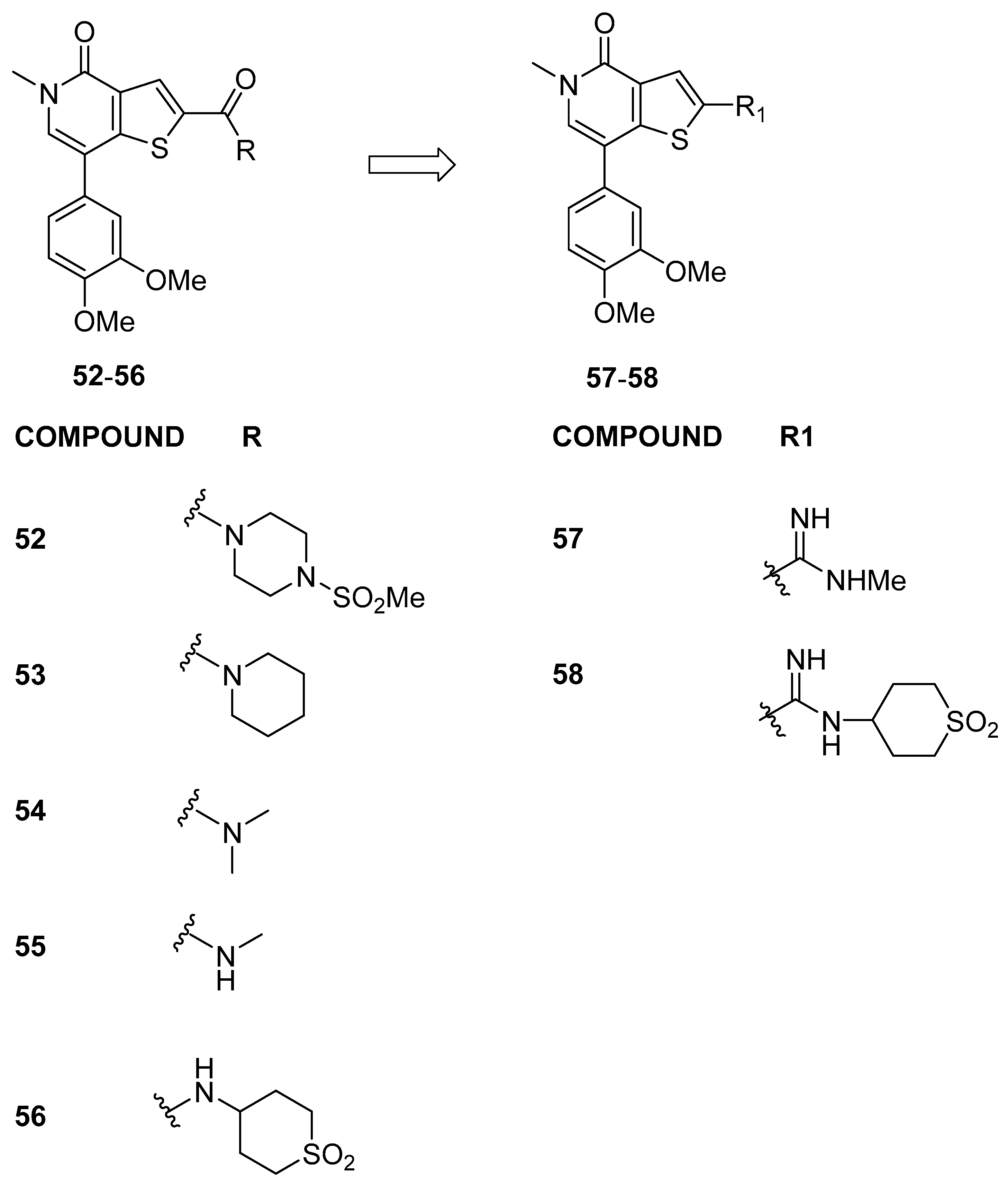
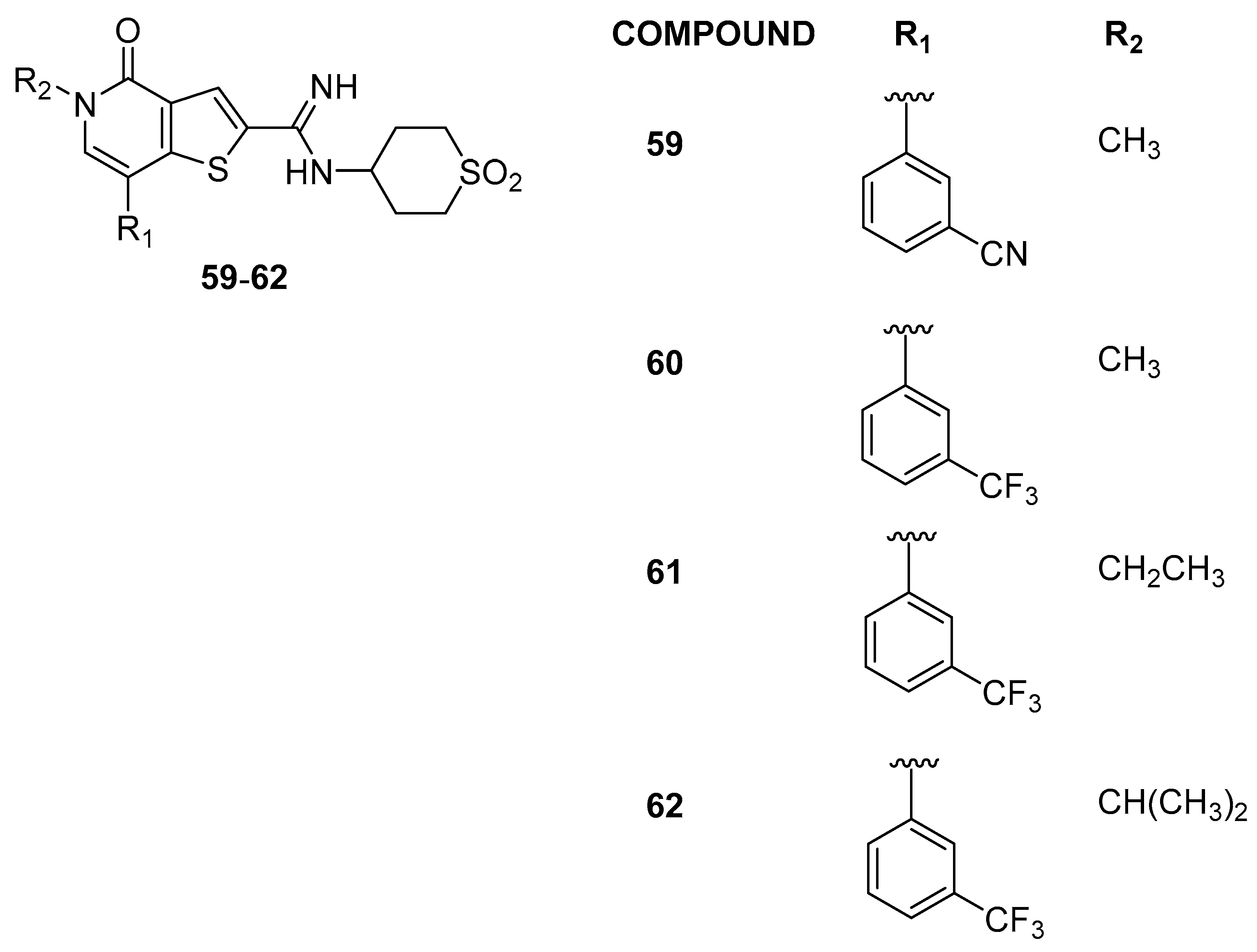
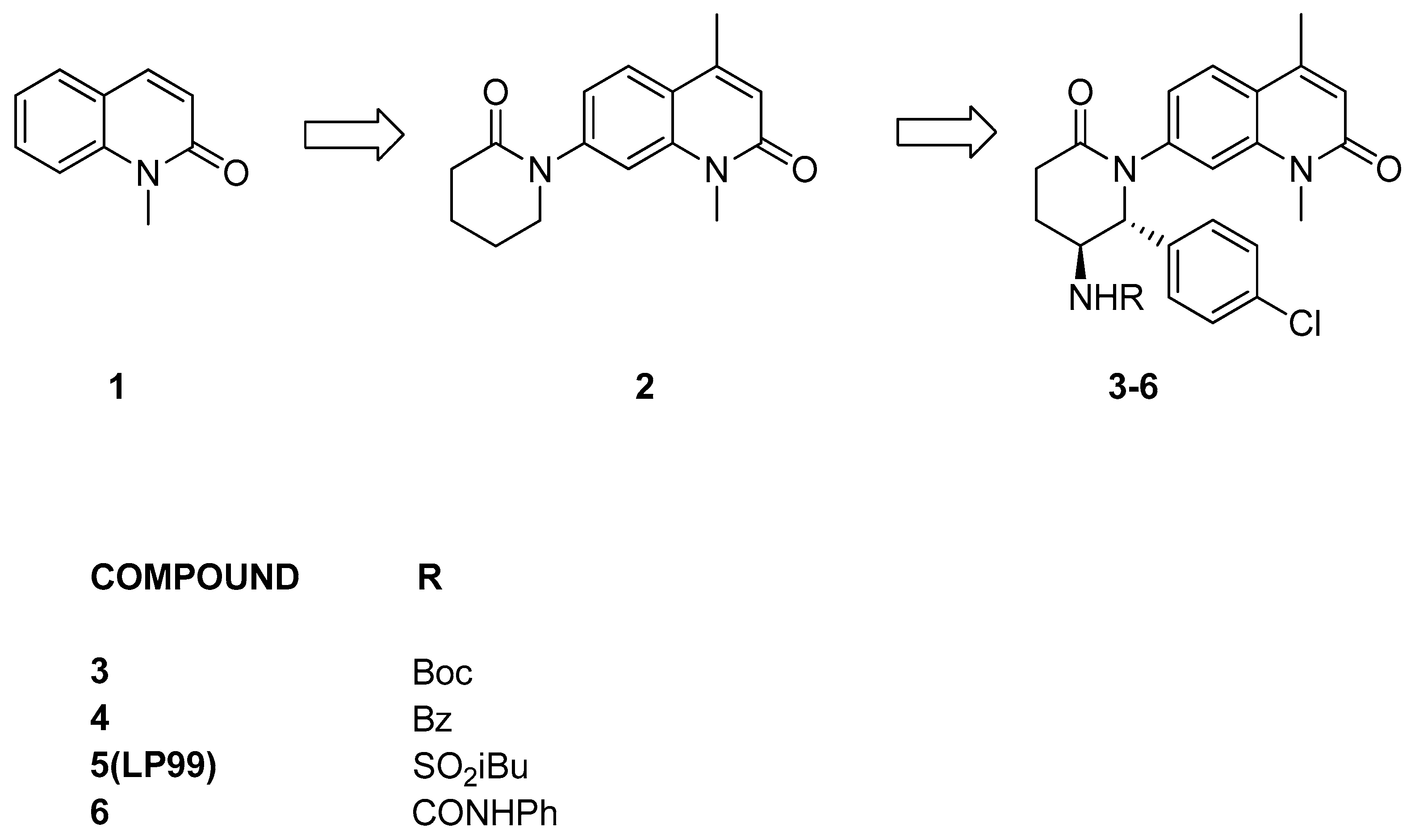
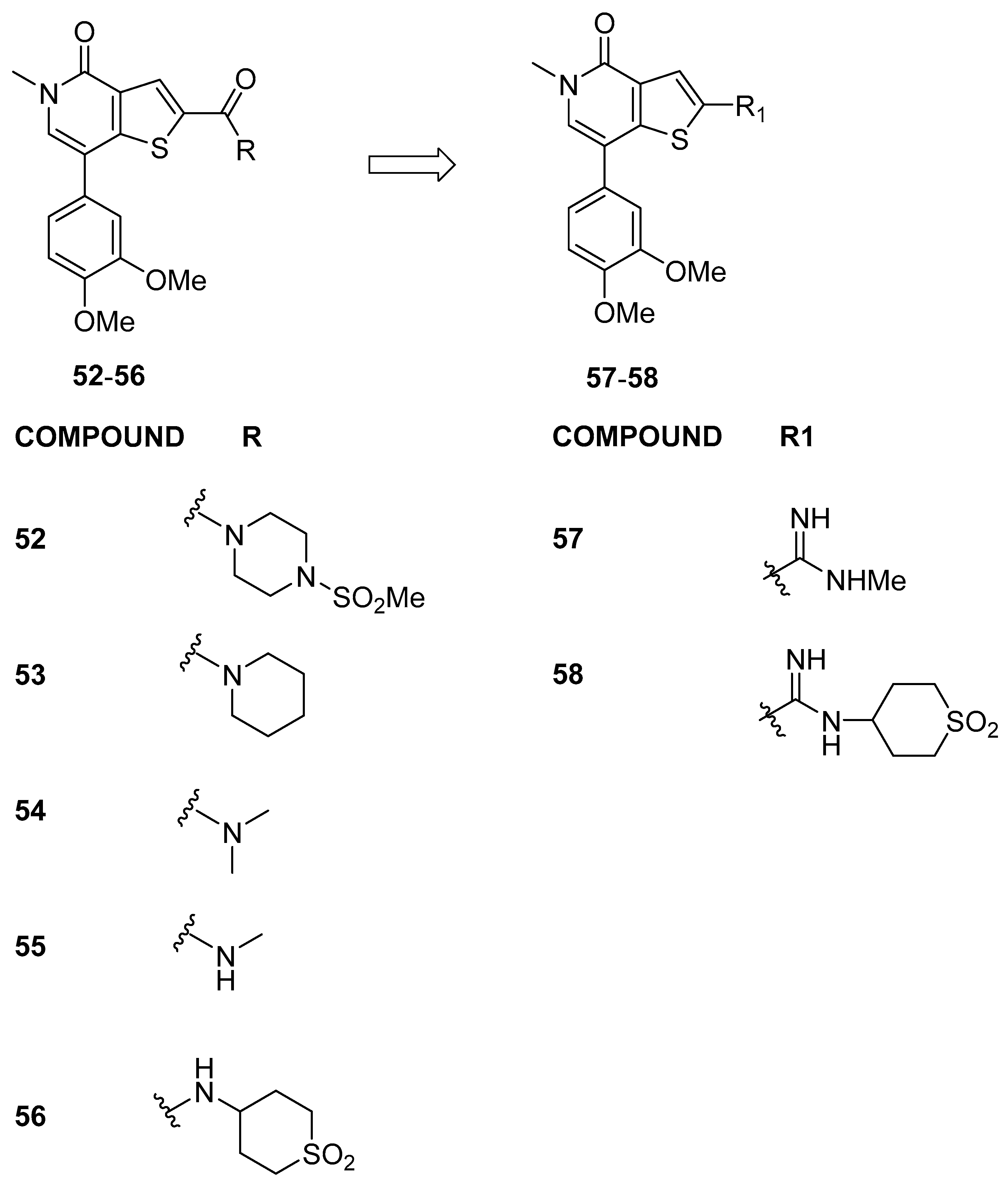
3.2.1. Thienopyridone Amidines and Their Amide Analogues
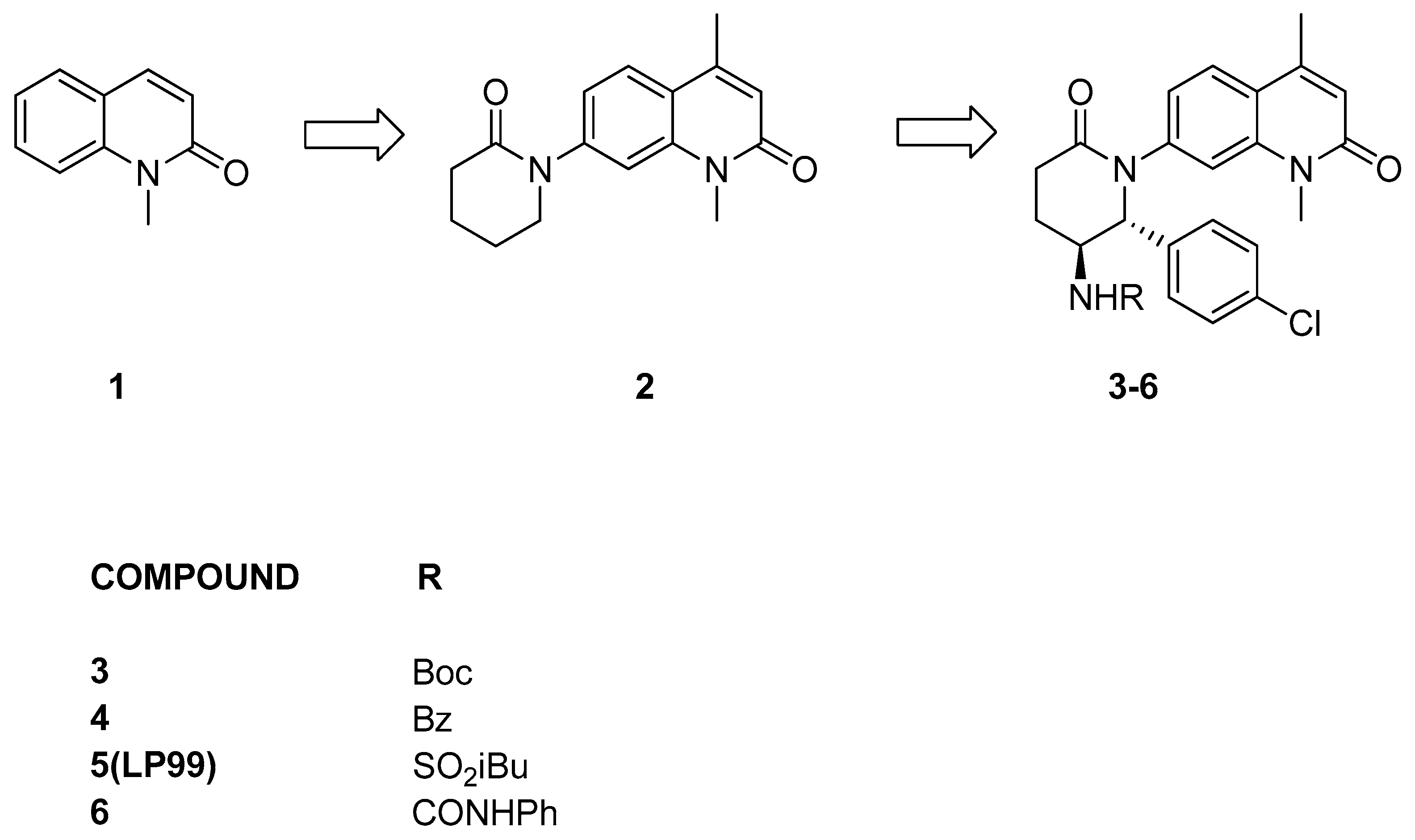
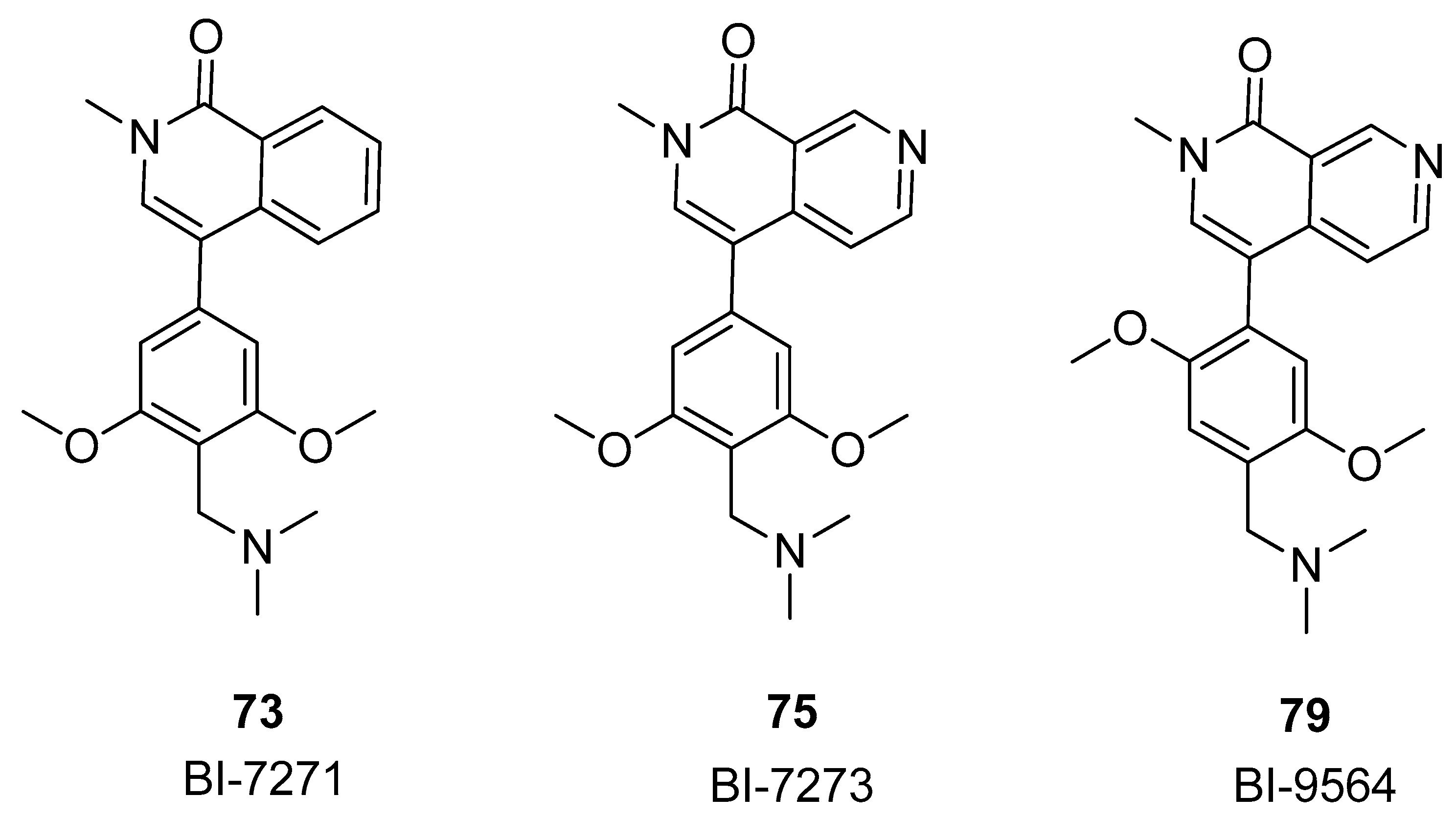
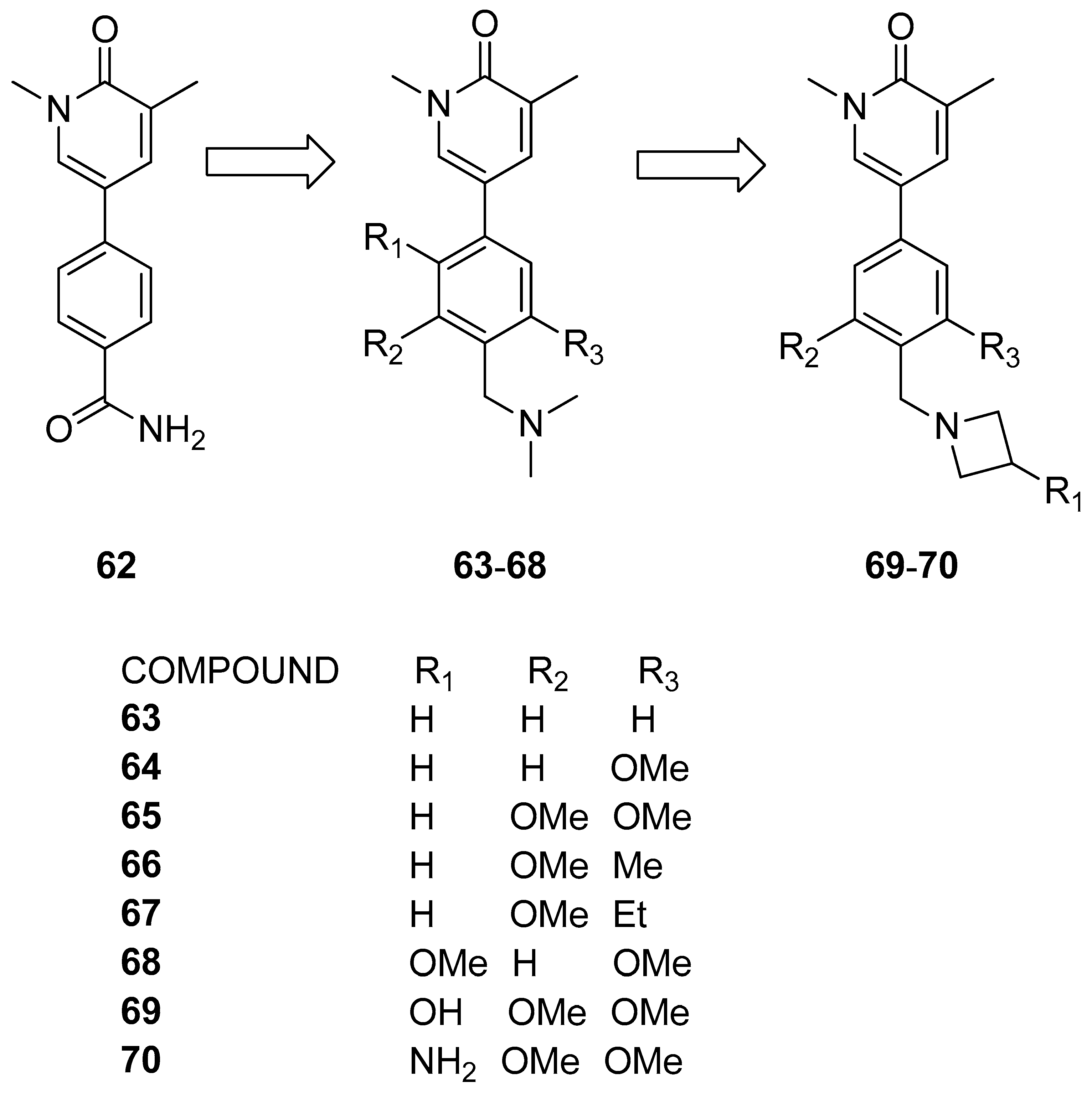
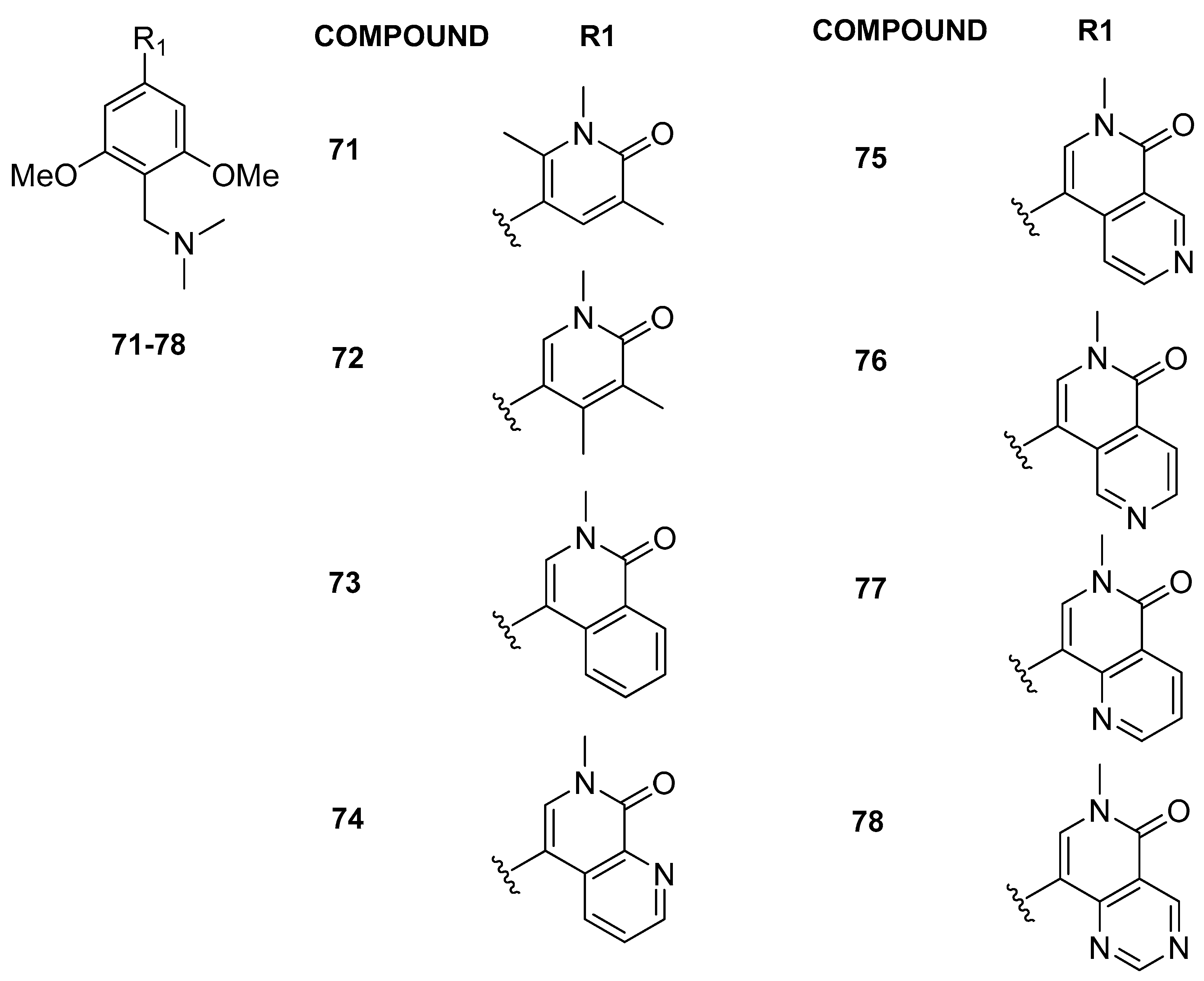
3.2.2. Isoquinolinone or Pyridinone Analogues
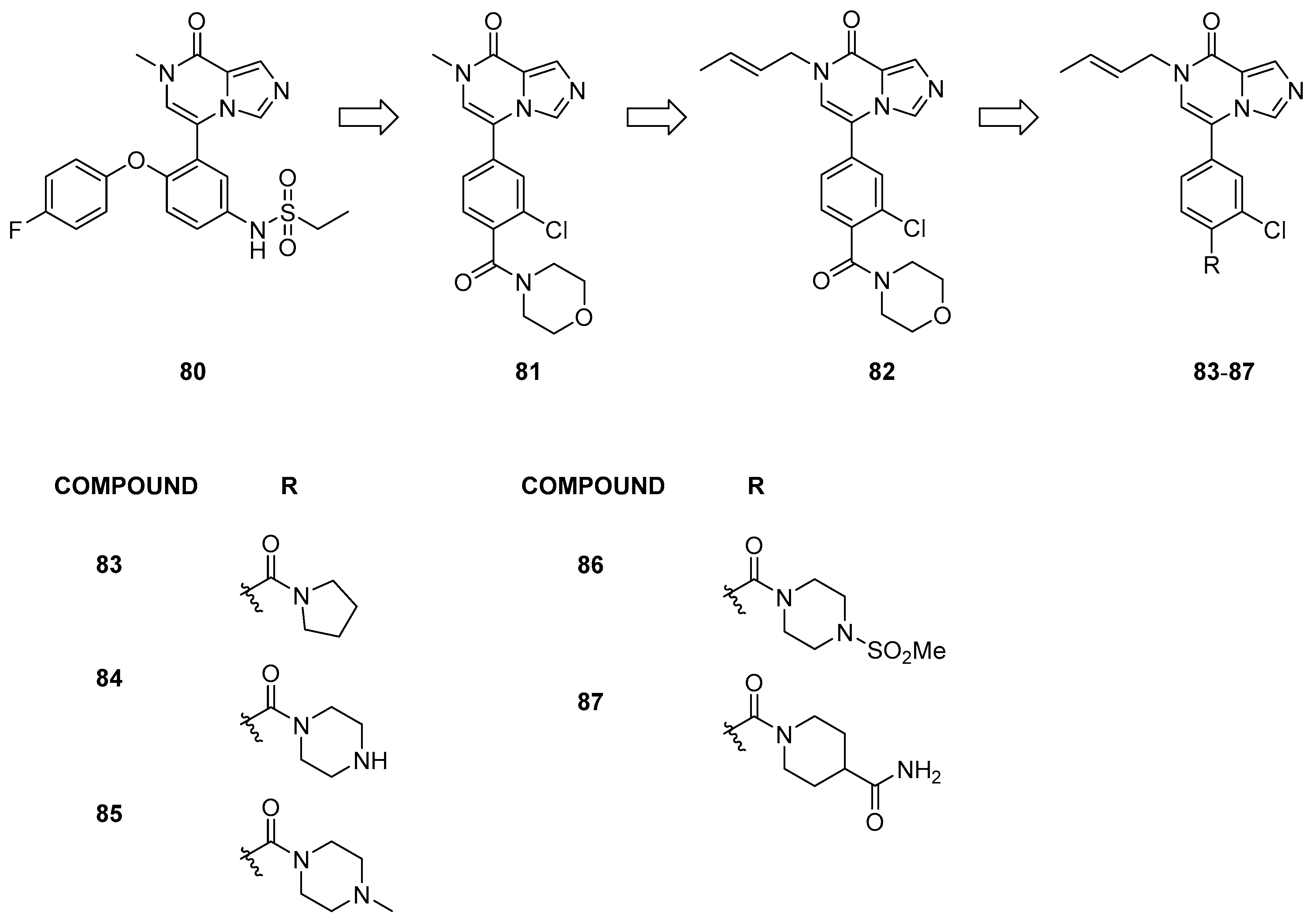
3.2.3. Imidazo[1,5-a]pyrazin-8(7H)-one Derivatives
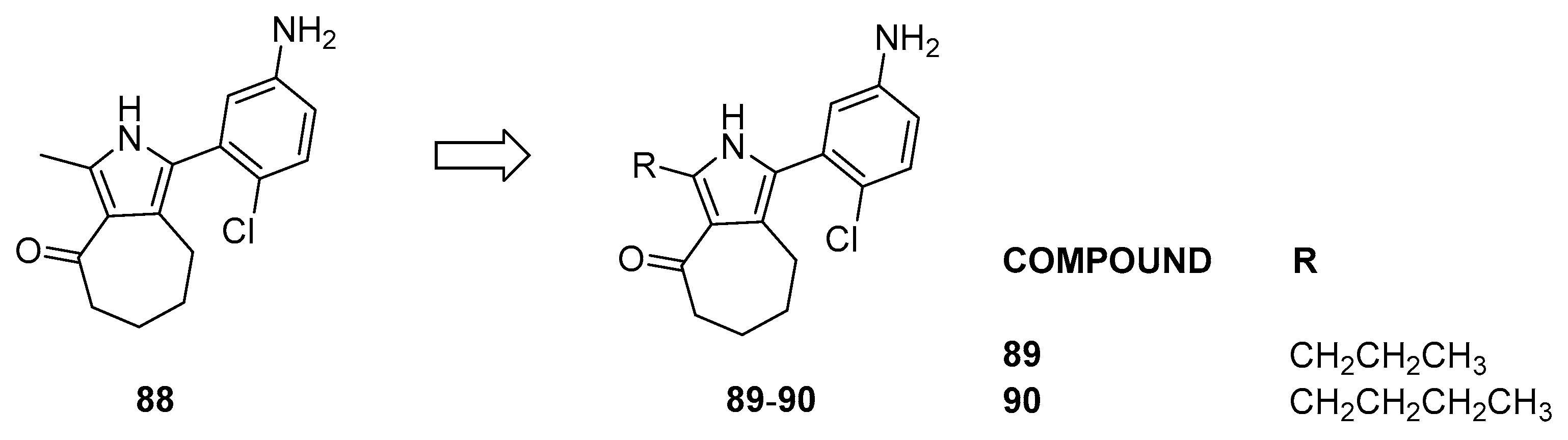
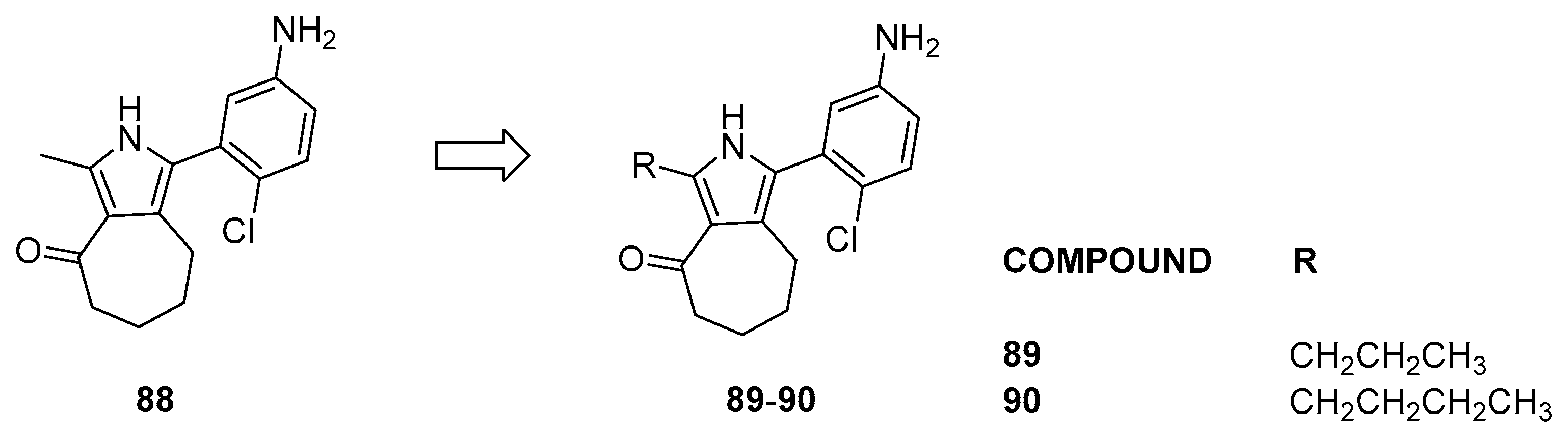
3.2.4. Pyrrole Analogs
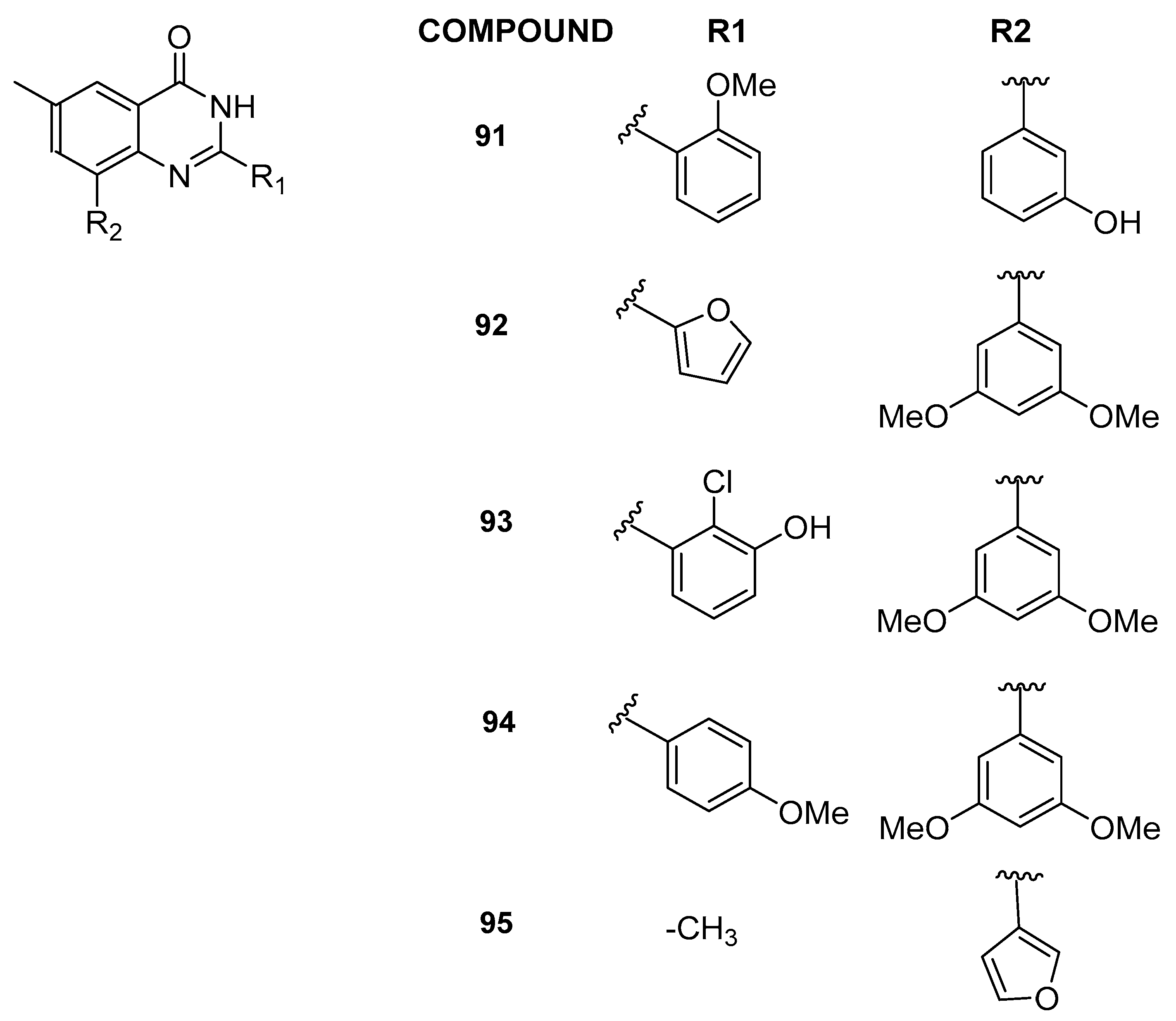
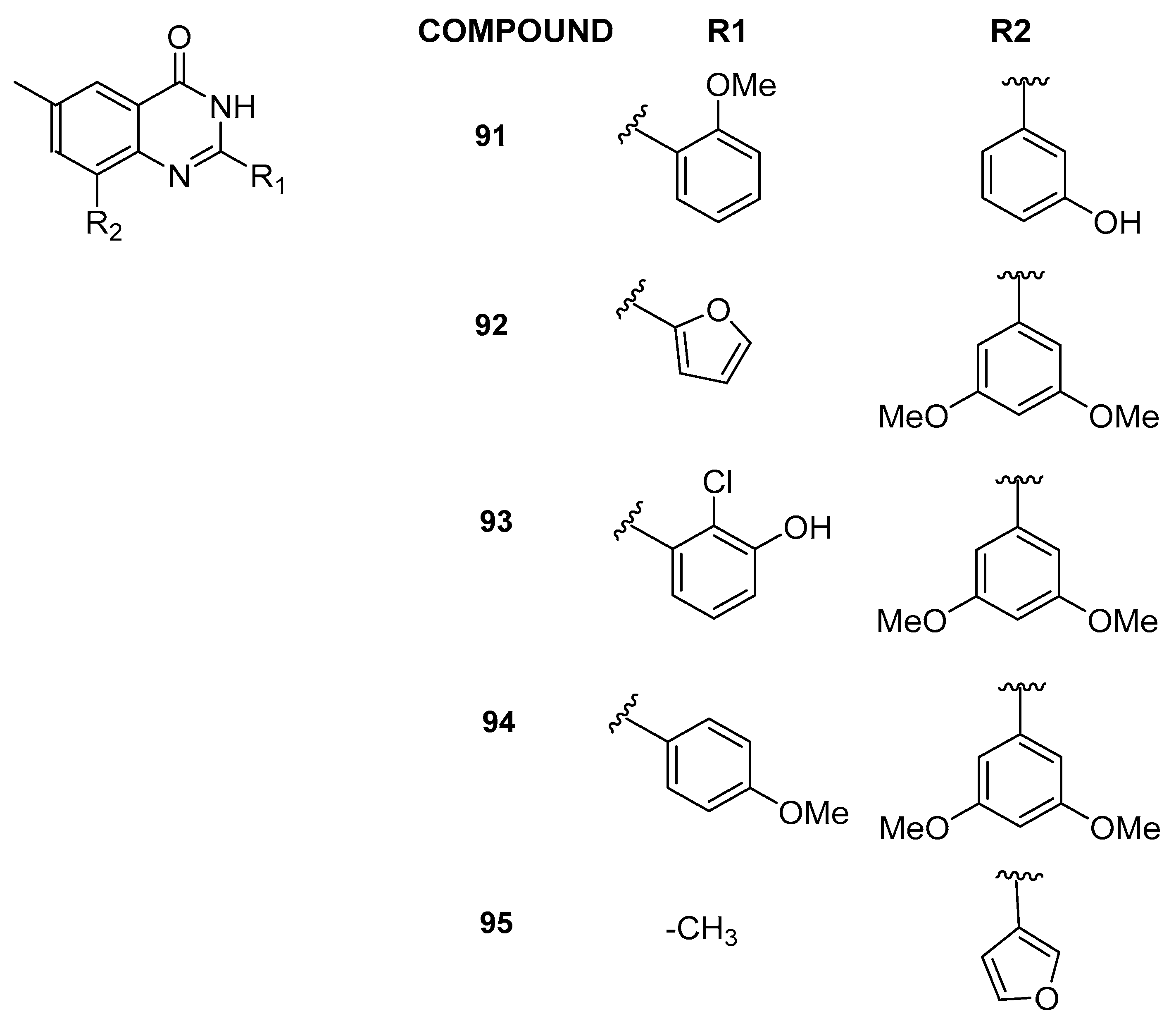
3.2.5. 6-Methylquinazolin-4(3H)-one Analogs
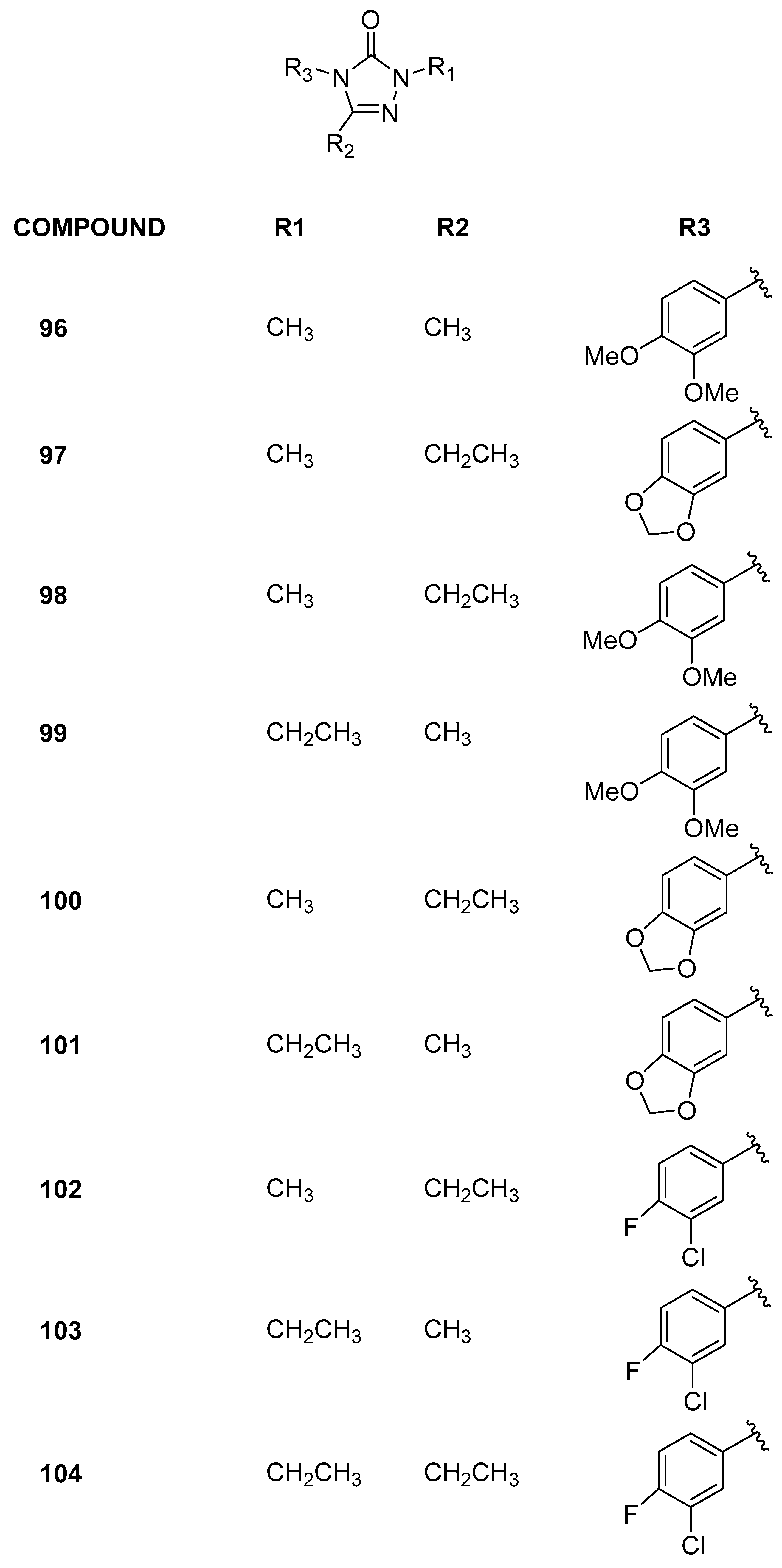
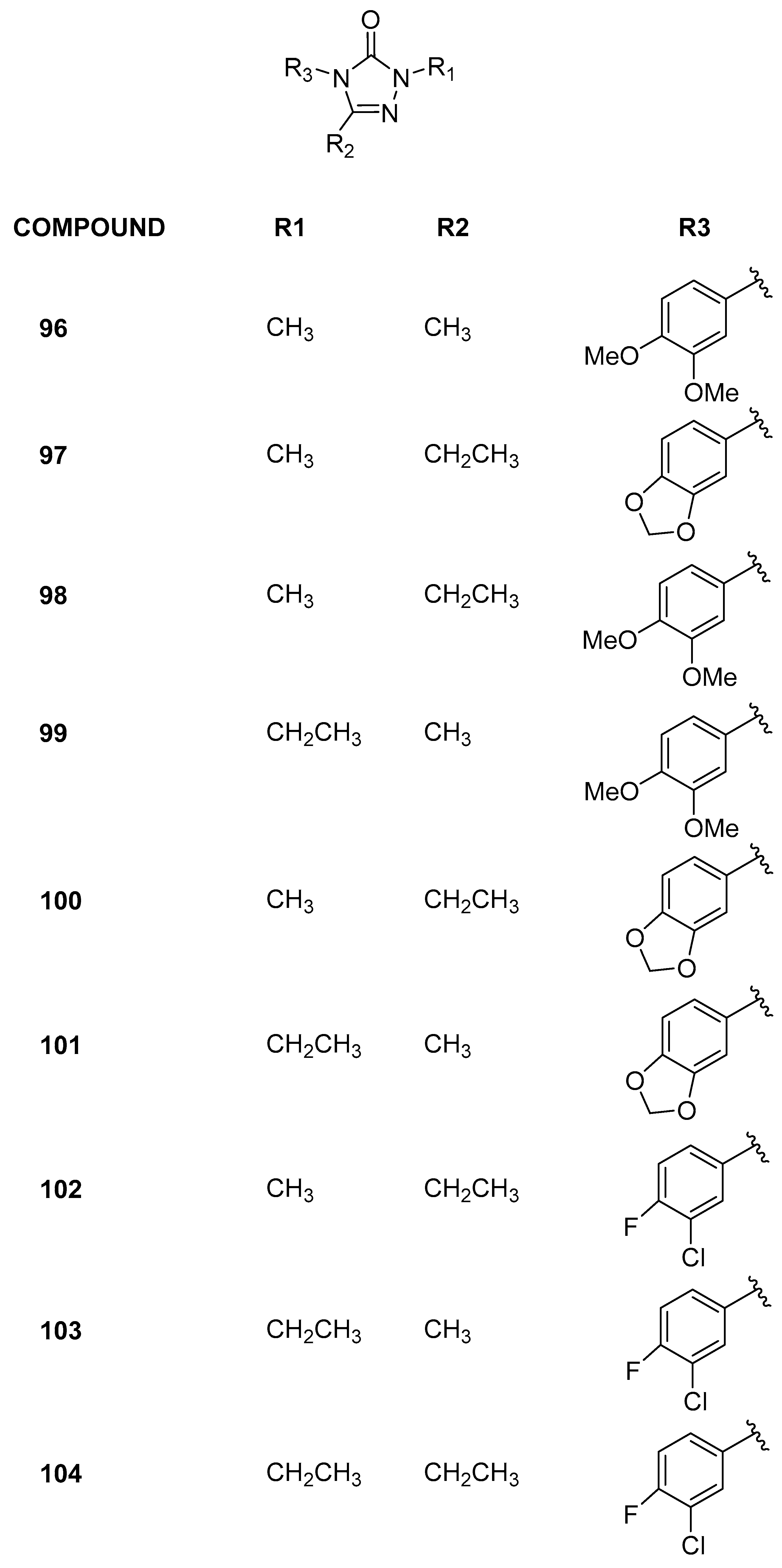
3.2.6. 2,4,5-Trisubstituted-2,4-dihydro-3H-1,2,4-triazol-3-one Analogs
3.3. Summary of Cellular Assays
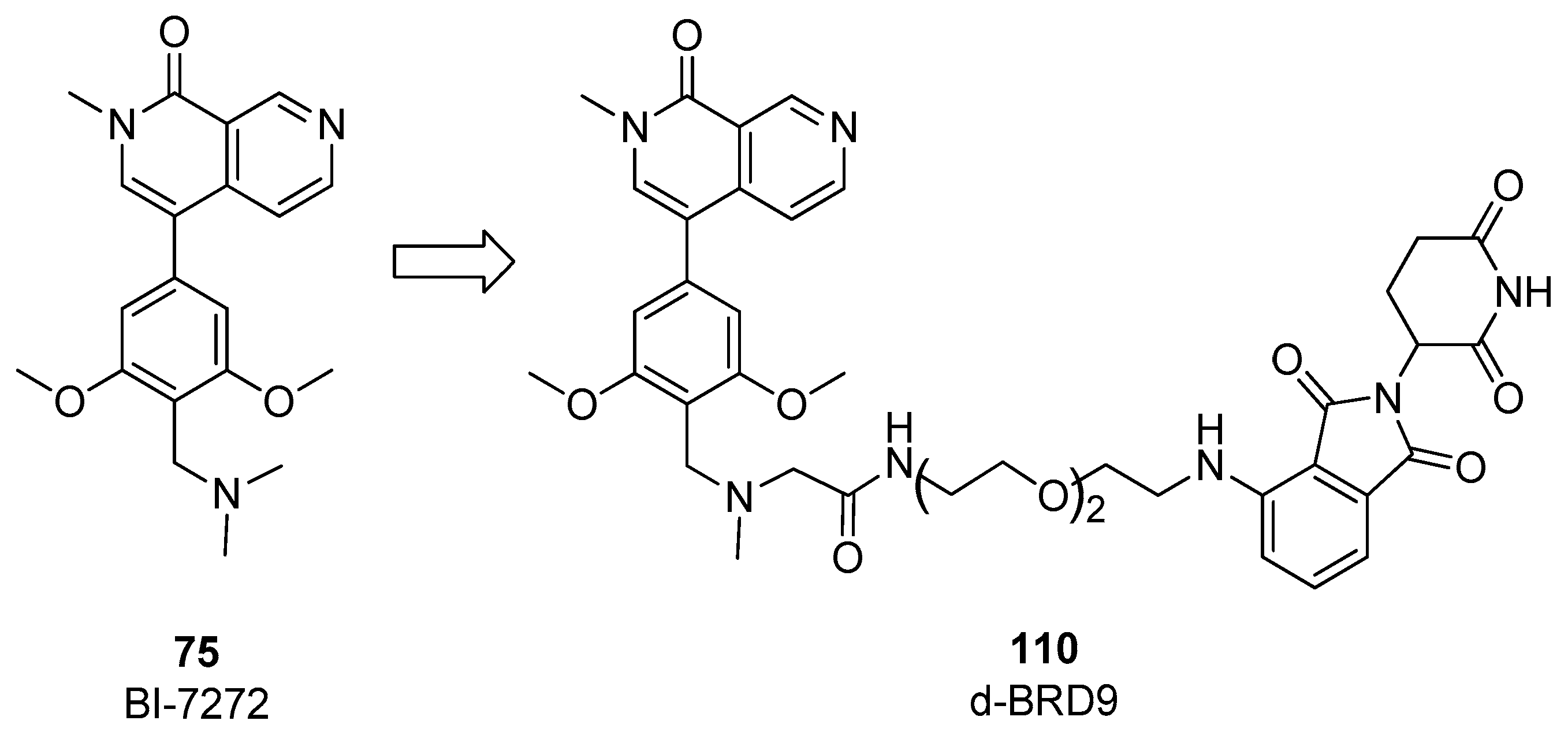
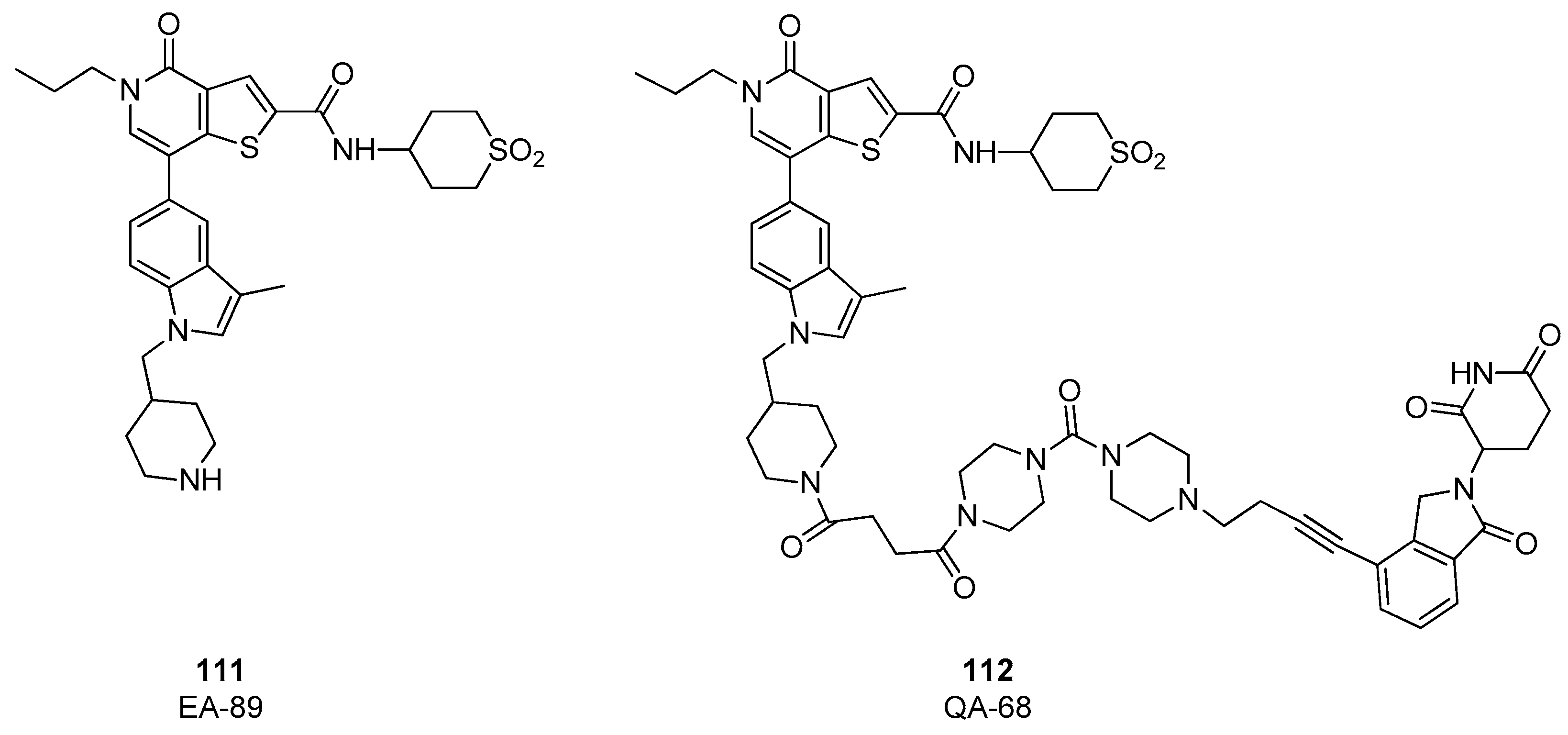
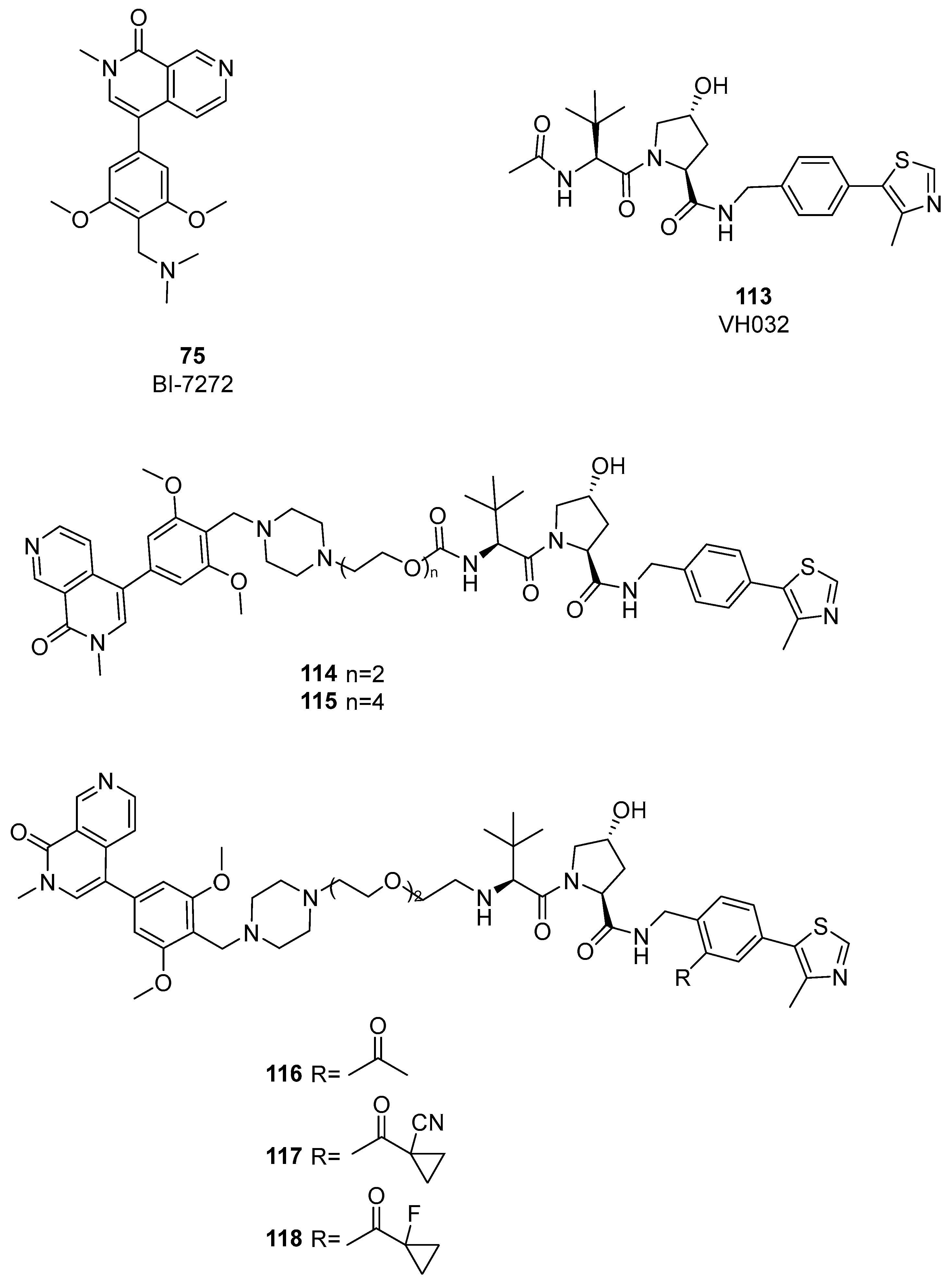
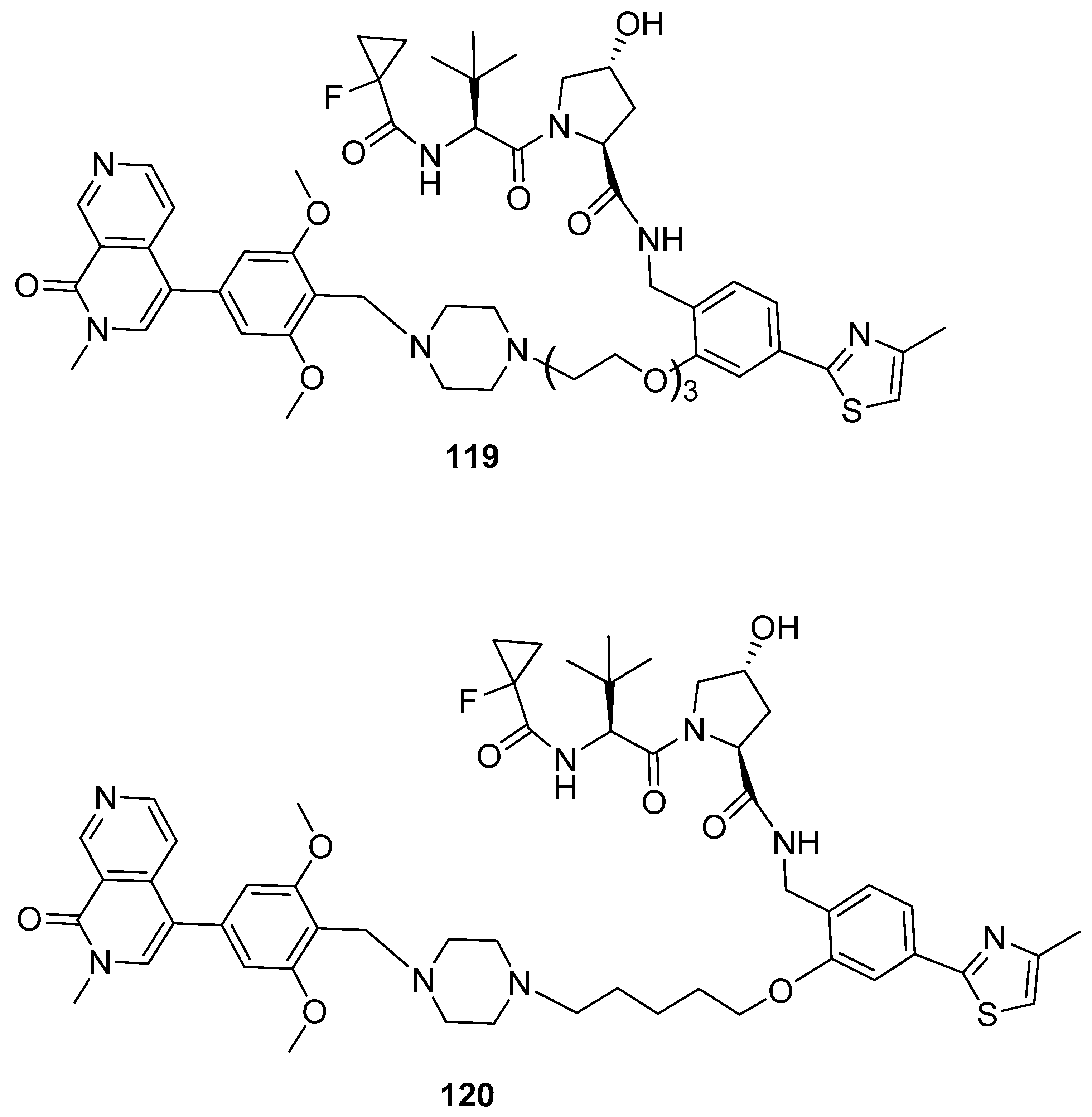
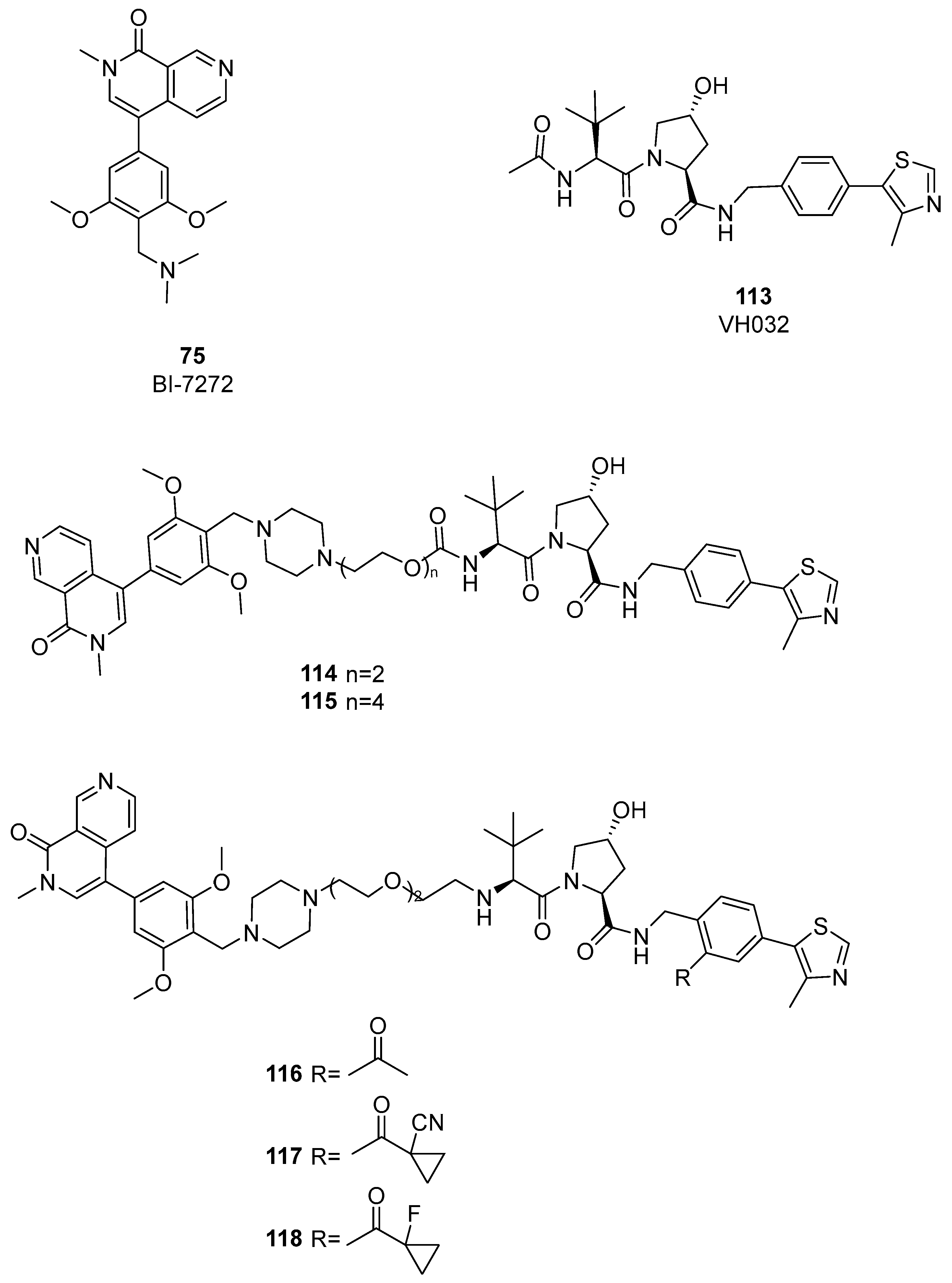
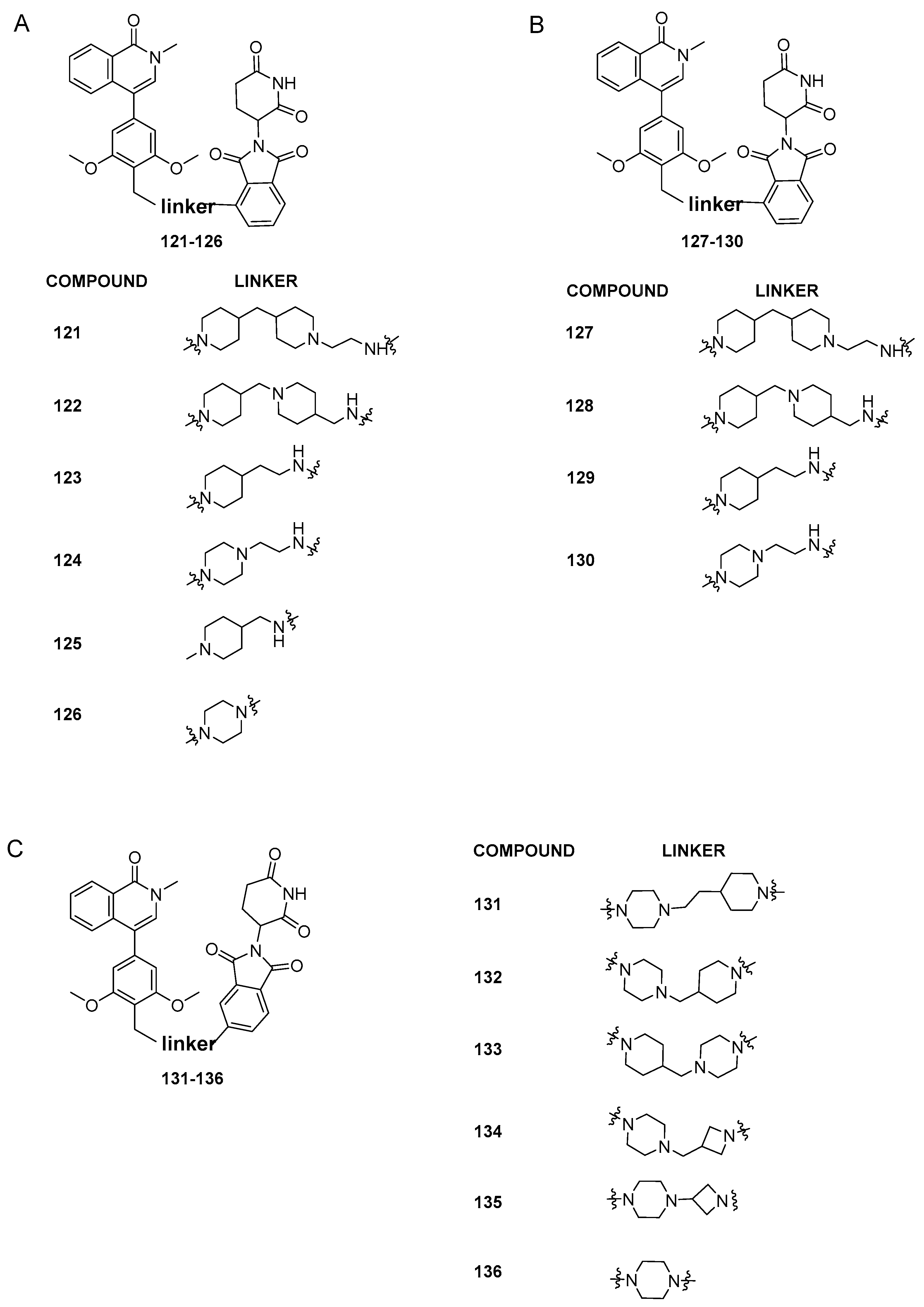
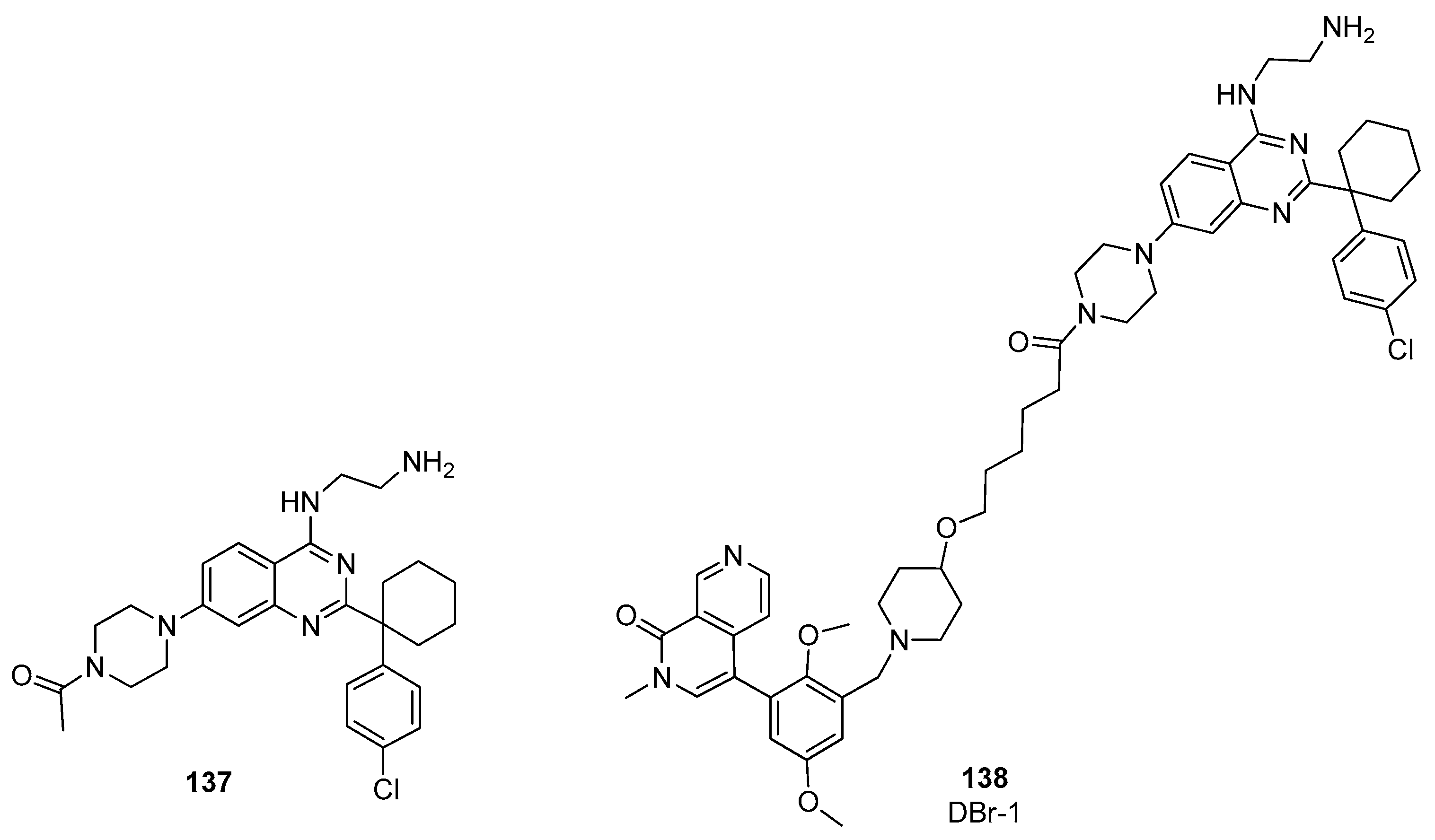
4. BRD9 PROteolysis TArgeting Chimeras (PROTACs)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. Brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849–1873. [Google Scholar] [CrossRef]
- Zhu, X.; Liao, Y.; Tang, L. Targeting BRD9 for Cancer Treatment: A New Strategy. Onco Targets Ther. 2020, 13, 13191–13200. [Google Scholar] [CrossRef] [PubMed]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Flynn, E.M.; Huang, O.W.; Poy, F.; Oppikofer, M.; Bellon, S.F.; Tang, Y.; Cochran, A.G. A subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure 2015, 23, 1801–1814. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Nayak, A.; Dutta, M.; Roychowdhury, A. Emerging oncogene ATAD2: Signaling cascades and therapeutic initiatives. Life Sci. 2021, 276, 119322. [Google Scholar] [CrossRef]
- Mishima, Y.; Miyagi, S.; Saraya, A.; Negishi, M.; Endoh, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Katsumoto, T.; Chiba, T.; et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 2011, 118, 2443–2453. [Google Scholar] [CrossRef]
- Sun, H.; Liu, J.; Zhang, J.; Shen, W.; Huang, H.; Xu, C.; Dai, H.; Wu, J.; Shi, Y. Solution structure of BRD7 bromodomain and its interaction with acetylated peptides from histone H3 and H4. Biochem. Biophys. Res. Commun. 2007, 358, 435–441. [Google Scholar] [CrossRef]
- Poplawski, A.; Hu, K.; Lee, W.; Natesan, S.; Peng, D.; Carlson, S.; Shi, X.; Balaz, S.; Markley, J.L.; Glass, K.C. Molecular insights into the recognition of N-terminal histone modifications by the BRPF1 bromodomain. J. Mol. Biol. 2014, 426, 1661–1676. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Vlassis, A.; Roques, C.; Lalonde, M.E.; González-Aguilera, C.; Lambert, J.P.; Lee, S.B.; Zhao, X.; Alabert, C.; Johansen, J.V.; et al. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016, 35, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Dreier, M.R.; De la Serna, I.L. SWI/SNF chromatin remodeling enzymes in melanoma. Epigenomes 2022, 6, 10–33. [Google Scholar] [CrossRef] [PubMed]
- Euskirchen, G.M.; Auerbach, R.K.; Davidov, E.; Gianoulis, T.A.; Zhong, G.; Rozowsky, J.; Bhardwaj, N.; Gerstein, M.B.; Snyder, M. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet. 2011, 7, e1002008. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, J.; Jiang, J.; Yuan, J.; Zhang, Y.; Wei, X.; Loo, N.; Wang, Y.; Pan, Y.; Zhang, T.; et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun. 2019, 10, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Subramaniyam, S.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; Schneider, A.; Arias-Pulido, H.; et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol. Cancer 2008, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.U.; Koo, S.H.; Kwon, K.C.; Park, J.W.; Kim, J.M. Gain at chromosomal region 5p15. 33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet. Cytogen. 2008, 182, 1–11. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. ELife 2018, 7, e41305. [Google Scholar] [CrossRef]
- Krämer, K.F.; Moreno, N.; Frühwald, M.C.; Kerl, K. BRD9 inhibition, alone or in combination with cytostatic compounds as a therapeutic approach in rhabdoid tumors. Int. J. Mol. Sci. 2017, 18, 1537–1549. [Google Scholar] [CrossRef]
- Clark, P.G.; Vieira, L.C.; Tallant, C.; Fedorov, O.; Singleton, D.C.; Rogers, C.M.; Monteiro, O.P.; Bennett, J.M.; Baronio, R.; Müller, S.; et al. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem. Int. Ed. 2015, 54, 6217–6221. [Google Scholar] [CrossRef]
- Hay, D.A.; Rogers, C.M.; Fedorov, O.; Tallant, C.; Martin, S.; Monteiro, O.P.; Müller, S.; Knapp, S.; Schofield, C.J.; Brennan, P.E. Design and synthesis of potent and selective inhibitors of BRD7 and BRD9 bromodomains. MedChemComm 2015, 6, 1381–1386. [Google Scholar] [CrossRef]
- Fedorov, O.; Lingard, H.; Wells, C.; Monteiro, O.P.; Picaud, S.; Keates, T.; Yapp, C.; Philpott, M.; Martin, S.J.; Felletar, I. [1, 2, 4] triazolo [4, 3-a] phthalazines: Inhibitors of diverse bromodomains. J. Med. Chem. 2014, 57, 462–476. [Google Scholar] [CrossRef]
- D’Ascenzio, M.; Pugh, K.M.; Konietzny, R.; Berridge, G.; Tallant, C.; Hashem, S.; Monteiro, O.; Thomas, J.R.; Schirle, M.; Knapp, S. An Activity-Based Probe Targeting Non-Catalytic, Highly Conserved Amino Acid Residues within Bromodomains. Angew. Chem. Int. Ed. 2019, 131, 1019–1024. [Google Scholar] [CrossRef]
- Picaud, S.; Strocchia, M.; Terracciano, S.; Lauro, G.; Mendez, J.; Daniels, D.L.; Riccio, R.; Bifulco, G.; Bruno, I.; Filippakopoulos, P. 9 H-purine scaffold reveals induced-fit pocket plasticity of the BRD9 bromodomain. J. Med. Chem. 2015, 58, 2718–2736. [Google Scholar] [CrossRef]
- Clegg, M.A.; Bamborough, P.; Chung, C.-W.; Craggs, P.D.; Gordon, L.; Grandi, P.; Leveridge, M.; Lindon, M.; Liwicki, G.M.; Michon, A.-M. Application of atypical acetyl-lysine methyl mimetics in the development of selective inhibitors of the bromodomain-containing protein 7 (BRD7)/bromodomain-containing protein 9 (BRD9) bromodomains. J. Med. Chem. 2020, 63, 5816–5840. [Google Scholar] [CrossRef]
- Pierri, M.; Gazzillo, E.; Chini, M.G.; Ferraro, M.G.; Piccolo, M.; Maione, F.; Irace, C.; Bifulco, G.; Bruno, I.; Terracciano, S.; et al. Introducing structure-based three-dimensional pharmacophore models for accelerating the discovery of selective BRD9 binders. Bioorg. Chem. 2022, 118, 105480. [Google Scholar] [CrossRef] [PubMed]
- Gazzillo, E.; Pierri, M.; Colarusso, E.; Chini, M.G.; Ferraro, M.G.; Piccolo, M.; Irace, C.; Bruno, I.; Bifulco, G.; Terracciano, S.; et al. Exploring the chemical space of functionalized [1, 2, 4] triazolo [4, 3-a] quinoxaline-based compounds targeting the bromodomain of BRD9. Bioorg. Chem. 2023, 139, 106677. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chaikuad, A.; Bamborough, P.; Bantscheff, M.; Bountra, C.; Chung, C.-W.; Fedorov, O.; Grandi, P.; Jung, D.; Lesniak, R.; et al. Discovery and characterization of GSK2801, a selective chemical probe for the bromodomains BAZ2A and BAZ2B. J. Med. Chem. 2016, 59, 1410–1424. [Google Scholar] [CrossRef]
- Picaud, S.; Leonards, K.; Lambert, J.P.; Dovey, O.; Wells, C.; Fedorov, O.; Monteiro, O.; Fujisawa, T.; Wang, C.Y.; Lingard, H.; et al. Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci. Adv. 2016, 2, e1600760. [Google Scholar] [CrossRef]
- Nakano, H.; Ōmura, S. Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J. Antibiot. 2009, 62, 17–26. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Shannon, D.A.; Banerjee, R.; Webster, E.R.; Bak, D.W.; Wang, C.; Weerapana, E. Investigating the proteome reactivity and selectivity of aryl halides. JACS 2014, 136, 3330–3333. [Google Scholar] [CrossRef]
- Humphreys, P.G.; Bamborough, P.; Chung, C.-W.; Craggs, P.D.; Gordon, L.; Grandi, P.; Hayhow, T.G.; Hussain, J.; Jones, K.L.; Lindon, M.; et al. Discovery of a potent, cell penetrant, and selective p300/CBP-associated factor (PCAF)/general control nonderepressible 5 (GCN5) bromodomain chemical probe. J. Med. Chem. 2017, 60, 695–709. [Google Scholar] [CrossRef]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.-L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-based design of an in vivo active selective BRD9 inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, Y.; Guo, Y.; Yang, L.; Zhang, J.; Zhou, J.; Zhang, H. Design, synthesis and biological evaluation of 7-methylimidazo [1, 5-a] pyrazin-8 (7H)-one derivatives as BRD4 inhibitors. Bioorg. Med. Chem. 2017, 25, 2482–2490. [Google Scholar] [CrossRef]
- Zheng, P.; Zhang, J.; Ma, H.; Yuan, X.; Chen, P.; Zhou, J.; Zhang, H. Design, synthesis and biological evaluation of imidazo [1, 5-a] pyrazin-8 (7H)-one derivatives as BRD9 inhibitors. Bioorg. Med. Chem. 2019, 27, 1391–1404. [Google Scholar] [CrossRef]
- Hügle, M.; Regenass, P.; Warstat, R.; Hau, M.; Schmidtkunz, K.; Lucas, X.; Wohlwend, D.; Einsle, O.; Jung, M.; Breit, B.; et al. 4-Acyl pyrroles as dual BET-BRD7/9 bromodomain inhibitors address BETi insensitive human cancer cell lines. J. Med. Chem. 2020, 63, 15603–15620. [Google Scholar] [CrossRef]
- Warstat, R.; Pervaiz, M.; Regenass, P.; Amann, M.; Schmidtkunz, K.; Einsle, O.; Jung, M.; Breit, B.; Hügle, M.; Günther, S. A novel pan-selective bromodomain inhibitor for epigenetic drug design. Eur. J. Med. Chem. 2023, 249, 115139. [Google Scholar] [CrossRef]
- Colarusso, E.; Gazzillo, E.; Boccia, E.; Giordano, A.; Chini, M.G.; Bifulco, G.; Lauro, G. 6-Methylquinazolin-4 (3H)-one Based Compounds as BRD9 Epigenetic Reader Binders: A Rational Combination of in silico Studies and Chemical Synthesis. EurJoc 2022, 2022, e202200868. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-1: Combiglide; Schrödinger LCC: New York, NY, USA, 2021.
- Bhat, M.; Belagali, S.; Mamatha, S.; Sagar, B.; Sekhar, E.V. Importance of quinazoline and quinazolinone derivatives in medicinal chemistry. Stud. Nat. Prod. Chem. 2021, 71, 185–219. [Google Scholar]
- Colarusso, E.; Ceccacci, S.; Monti, M.C.; Gazzillo, E.; Giordano, A.; Chini, M.G.; Ferraro, M.G.; Piccolo, M.; Ruggiero, D.; Irace, C.; et al. Identification of 2, 4, 5-trisubstituted-2, 4-dihydro-3H-1, 2, 4-triazol-3-one-based small molecules as selective BRD9 binders. Eur. J. Med. Chem. 2023, 247, 115018. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. Comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Ciulli, A.; Trainor, N. A beginner’s guide to PROTACs and targeted protein degradation. Biochemistry 2021, 43, 74–79. [Google Scholar] [CrossRef]
- Zhou, X.; Dong, R.; Zhang, J.-Y.; Zheng, X.; Sun, L.-P. PROTAC: A promising technology for cancer treatment. Eur. J. Med. Chem. 2020, 203, 112539. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Collins, I.; Wang, H.; Caldwell, J.J.; Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin–proteasome pathway. Biochem. J. 2017, 474, 1127–1147. [Google Scholar] [CrossRef]
- Salerno, A.; Seghetti, F.; Caciolla, J.; Uliassi, E.; Testi, E.; Guardigni, M.; Roberti, M.; Milelli, A.; Bolognesi, M.L. Enriching proteolysis targeting chimeras with a second modality: When two are better than one. J. Med. Chem. 2022, 65, 9507–9530. [Google Scholar] [CrossRef] [PubMed]
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.S.; Dastjerdi, S.; Wühr, M.; Dhe-Paganon, S.; Armstrong, S.A.; et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew. Chem. Int. Ed. 2017, 56, 5738–5743. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Chowdhury, B.; Meng, C.; Case, A.E.; Ni, W.; Garg, S.; Sattler, M.; Azab, A.K.; Sun, J.; Muz, B.; et al. BRD9 degraders as chemosensitizers in acute leukemia and multiple myeloma. Blood Cancer J. 2022, 12, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel–Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem. 2018, 62, 699–726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Duan, H.; Gui, R.; Wu, M.; Shen, L.; Jin, Y.; Pang, A.; Yu, X.; Zeng, S.; Zhang, B.; et al. Structure-based identification of new orally bioavailable BRD9-PROTACs for treating acute myelocytic leukemia. Eur. J. Med. Chem. 2023, 262, 115872. [Google Scholar] [CrossRef]
- Tao, Y.; Remillard, D.; Vinogradova, E.V.; Yokoyama, M.; Banchenko, S.; Schwefel, D.; Melillo, B.; Schreiber, S.L.; Zhang, X.; Cravatt, B.F. Targeted protein degradation by electrophilic PROTACs that stereoselectively and site-specifically engage DCAF1. J. Am. Chem. Soc. 2022, 144, 18688–18699. [Google Scholar] [CrossRef]
- Vulpetti, A.; Holzer, P.; Schmiedeberg, N.; Imbach-Weese, P.; Pissot-Soldermann, C.; Hollingworth, G.J.; Radimerski, T.; Thoma, C.R.; Stachyra, T.M.; Wojtynek, M.; et al. Discovery of New Binders for DCAF1, an Emerging Ligase Target in the Targeted Protein Degradation Field. ACS Med. Chem. Lett. 2023, 14, 949–954. [Google Scholar] [CrossRef]
- Schröder, M.; Renatus, M.; Liang, X.; Meili, F.; Zoller, T.; Ferrand, S.; Gauter, F.; Li, X.; Sigoillot, F.; Gleim, S.; et al. DCAF1-based PROTACs with activity against clinically validated targets overcoming intrinsic-and acquired-degrader resistance. Nat. Commun. 2024, 15, 275–294. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bromodomain | Substrate | Reference |
|---|---|---|
| ATAD2 | H3K9ac, H3K14ac, H4K5ac, H4K12ac | [8] |
| BRD1 | H3K14ac, H4K5acK8ac, H4K5prK8pr | [6,7,8] |
| BRD9 | H4K5acK8ac, H4K5prK8pr, H4K5buK8bu | [9] |
| BRD7 | H3K9ac H3K14ac H4K8ac H4K12ac H4K16ac | [10] |
| BRPF1 | H2AK5ac H3K14ac H4K8ac H4K5ac H4K12ac | [11] |
| BRPF3 | H3K14ac | [12] |
| KIAA1240 | Not determined |
| Compound | Cell Line | Conc (µM) | FRAP Recovery Time | Cytotoxicity |
|---|---|---|---|---|
| 5 (LP99) (a) | U2OS | 0.8 | 0.8 | - |
| 5 (LP99) (b) | U2OS | ≤33 | - | no |
| 13 (a) | U2OS | 1 | 0.8 | - |
| 19 (a) | U2OS | 1 | ~5 | - |
| 20 (a) | U2OS | 1 | ~5 | - |
| 21 (a) | U2OS | 1 | ~3 | - |
| 22 (Bromosporine) (a) | U2OS | 1 | 1 | - |
| 31 (b) | HEK293 | ≤33 | - | no |
| 75 (BI-7273) (a) | U2OS | 1 | ~0.70 | - |
| 79 (BI-9564) (a) | U2OS | 0.1 | ~0.80 | - |
| Compound | Cell Line | IC50 (µM) | EC50 (µM) | GI50 (µM) |
|---|---|---|---|---|
| 22 (Bromosporine) | MV4;11 | 0.5793 | n.d | n.d |
| KASUMI-1 | 0.2067 | n.d | n.d | |
| OCI-AML3 | 0.3990 | n.d | n.d | |
| 44 | CRF-CEM | 50 ± 5 | n.d | n.d |
| K-562 | 90 ± 5 | n.d | n.d | |
| HL-60 | >100 | n.d | n.d | |
| Kasumi1 | 97 ± 4 | n.d | n.d | |
| THP-1 | 95 ± 6 | n.d | n.d | |
| HaCaT | >100 | n.d | n.d | |
| 51 | CCRF-CEM | 35 ± 4 | n.d | n.d |
| K-562 | 65 ± 4 | n.d | n.d | |
| HL-60 | 81 ± 5.5 | n.d | n.d | |
| Kasumi1 | 72 ± 5 | n.d | n.d | |
| THP-1 | 60 ± 5 | n.d | n.d | |
| HaCaT | >100 | n.d | n.d | |
| 61 (I-BRD9) | NB4 | n.d | n.d | n.d |
| MV4-11 | n.d | n.d | n.d | |
| SU-DHL4 | n.d | n.d | n.d | |
| 75 (BI-7273) | EOL-1 | n.d | 1.4 | n.d |
| 79 (BI-9564) | EOL-1 | n.d | 0.8 | n.d |
| 84 | EOL-1 | 1.76 ± 0.05 | n.d | n.d |
| A549 | 6.12 ± 0.18 | n.d | n.d | |
| 88 | SNB-75 | n.d | n.d | 0.182 |
| UO-31 | n.d | n.d | 0.479 | |
| CAKI-1 | n.d | n.d | 0.708 | |
| DU-145 | n.d | n.d | 13.2 | |
| SW-620 | n.d | n.d | 12.0 | |
| NCI-H460 | n.d | n.d | 10.2 | |
| 96 | Jurkat | 110 ± 9 | n.d | n.d |
| MCF-7 | >500 | n.d | n.d | |
| A375 | 450 ± 16 | n.d | n.d | |
| Caco2 | 300 ± 14 | n.d | n.d | |
| HaCaT | >500 | n.d | n.d | |
| 97 | Jurkat | 145 ± 11 | n.d | n.d |
| MCF-7 | >500 | n.d | n.d | |
| A375 | 270 ± 18 | n.d | n.d | |
| Caco2 | 300 ± 12 | n.d | n.d | |
| HaCaT | >500 | n.d | n.d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colarusso, E.; Chini, M.G.; Bifulco, G.; Lauro, G.; Giordano, A. Identification and Development of BRD9 Chemical Probes. Pharmaceuticals 2024, 17, 392. https://doi.org/10.3390/ph17030392
Colarusso E, Chini MG, Bifulco G, Lauro G, Giordano A. Identification and Development of BRD9 Chemical Probes. Pharmaceuticals. 2024; 17(3):392. https://doi.org/10.3390/ph17030392
Chicago/Turabian StyleColarusso, Ester, Maria Giovanna Chini, Giuseppe Bifulco, Gianluigi Lauro, and Assunta Giordano. 2024. "Identification and Development of BRD9 Chemical Probes" Pharmaceuticals 17, no. 3: 392. https://doi.org/10.3390/ph17030392
APA StyleColarusso, E., Chini, M. G., Bifulco, G., Lauro, G., & Giordano, A. (2024). Identification and Development of BRD9 Chemical Probes. Pharmaceuticals, 17(3), 392. https://doi.org/10.3390/ph17030392