
A Monocarbonyl Curcuminoid Derivative Inhibits the Activity of Human Glutathione Transferase A4-4 and Chemosensitizes Glioblastoma Cells to Temozolomide

 , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
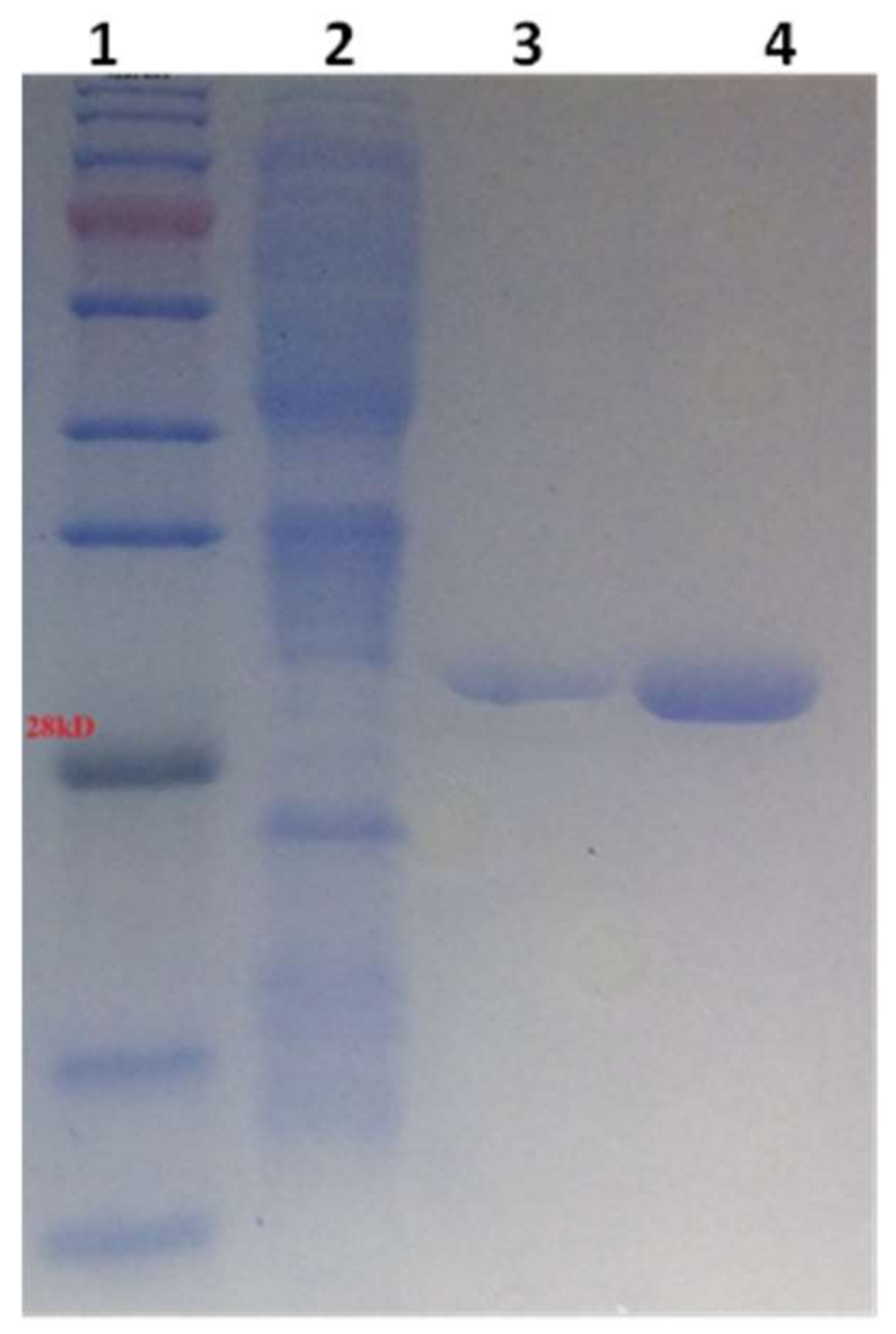
2.1. Cloning, Expression, and Purification of hGSTA4-4 from Recombinant E. coli Cells
2.2. Screening of Natural Polyphenols as hGSTA4-4 Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Products | Enzyme Inhibition (%) | Natural Products | Enzyme Inhibition (%) |
|---|---|---|---|
Polydatin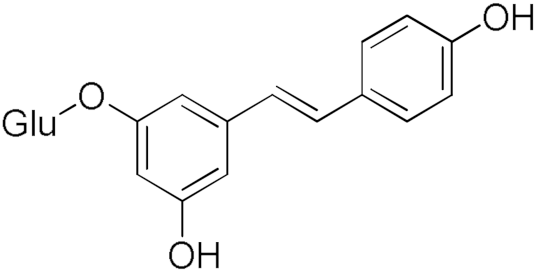 | 32.30 | Resveratrol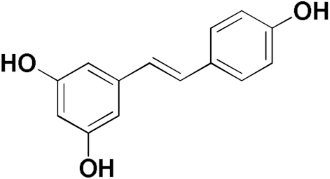 | 8.87 |
Taxifolin hydrate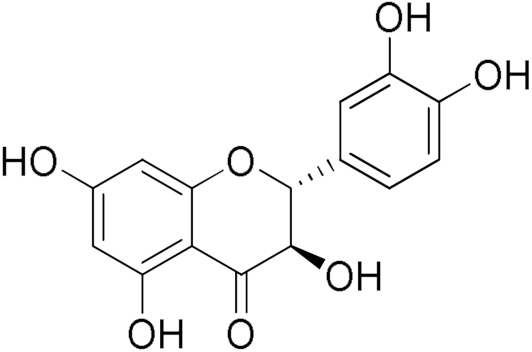 | 40.37 | Curcumin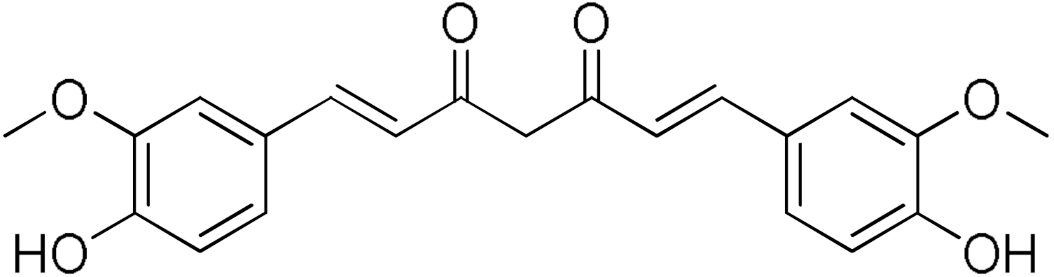 | 80.92 |
Naringenin | 28.49 | Safranal | 21.84 |
| Epigallocatechin gallate 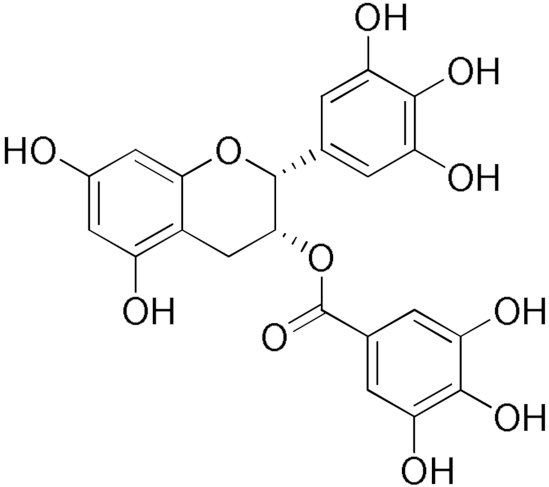 | 86.39 | Gallic acid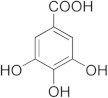 | 20.81 |
Quercetin | 84.65 | Coumaric Acid | 27.3 |
Colchicine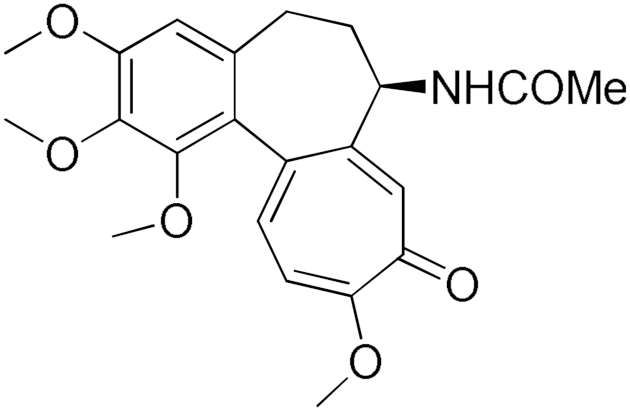 | 17.1 | Ellagic Acid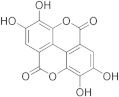 | 95.1 |
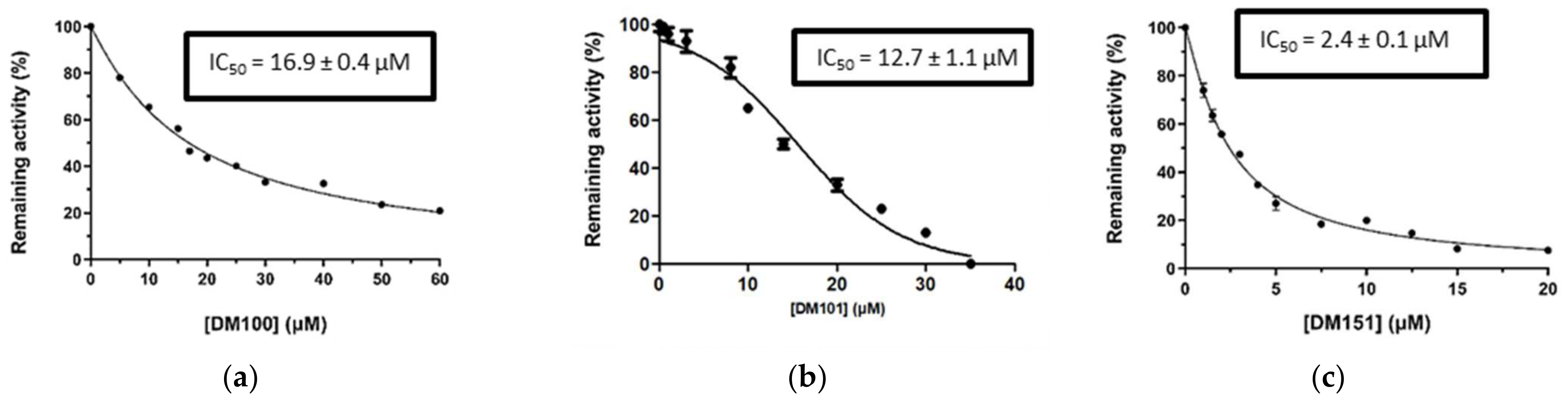
2.3. Screening of Curcumin Derivatives as hGSTA4-4 Inhibitors
| Compound | Enzyme Inhibition (%) | Compound | Enzyme Inhibition (%) |
|---|---|---|---|
DM148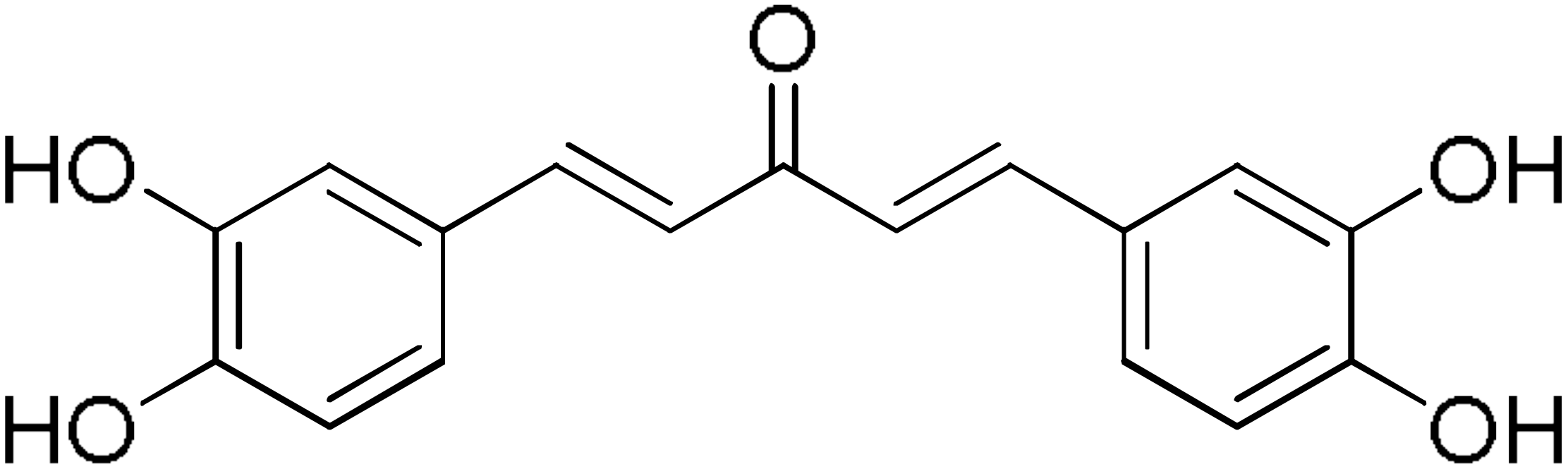 | 4.6 | DM62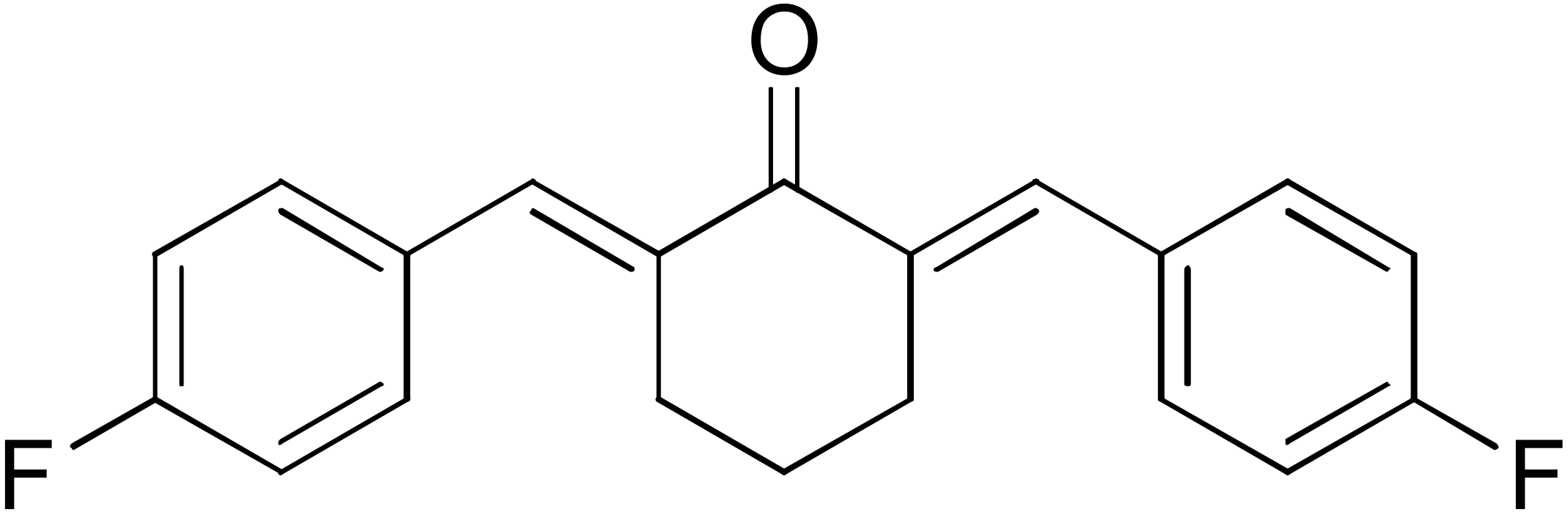 | 74.9 |
DM95 | 54.8 | DM96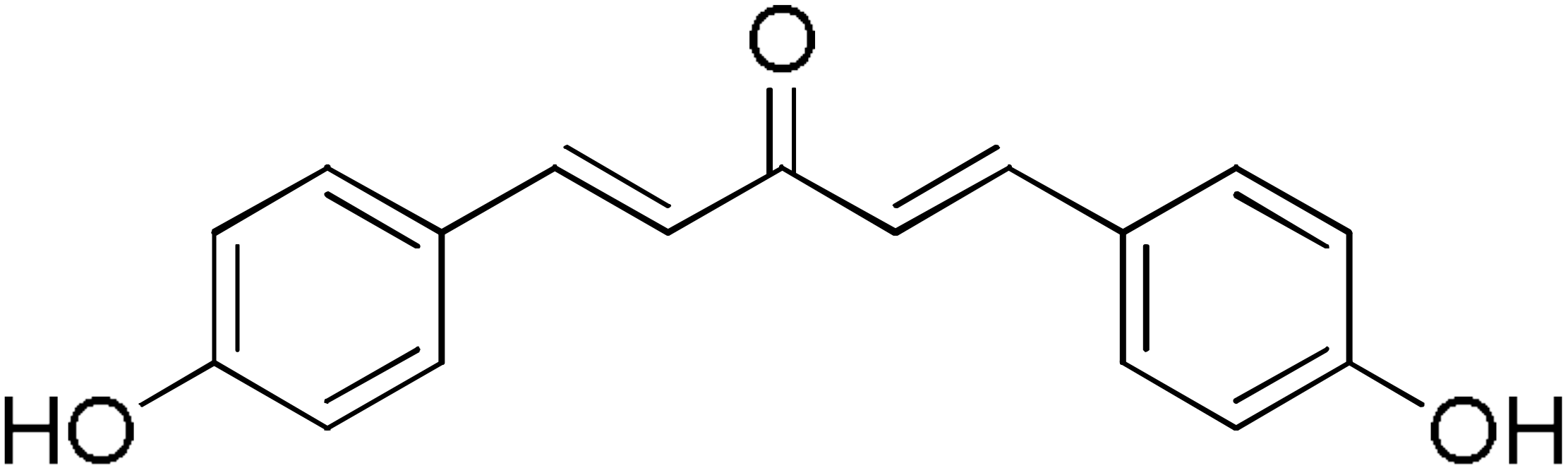 | 81.8 |
DM57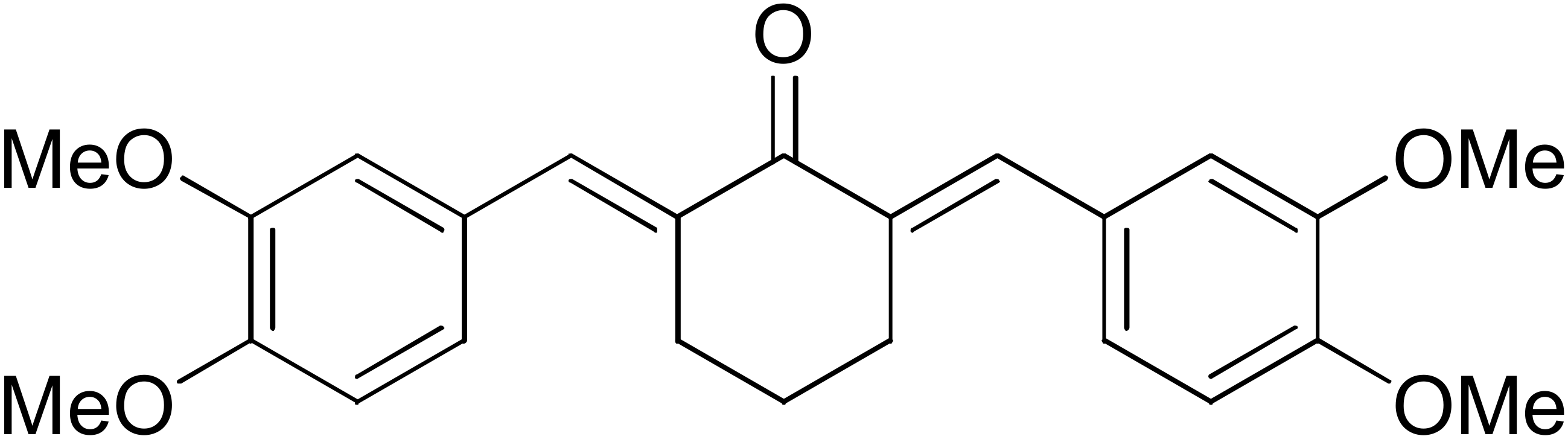 | 55.7 | DM151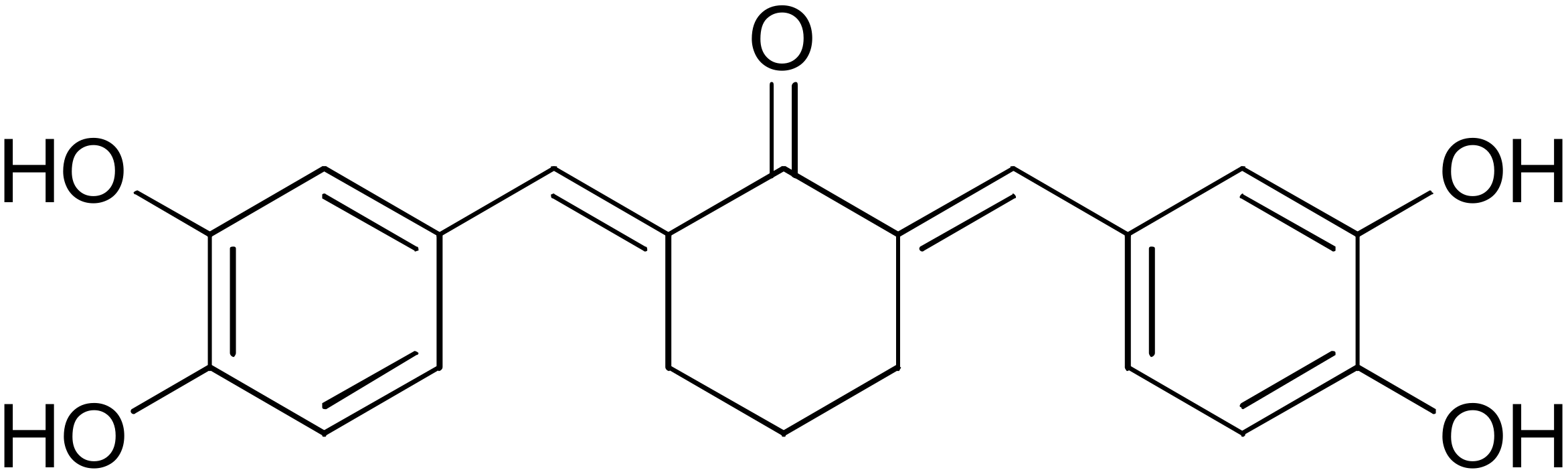 | 91.6 |
DM46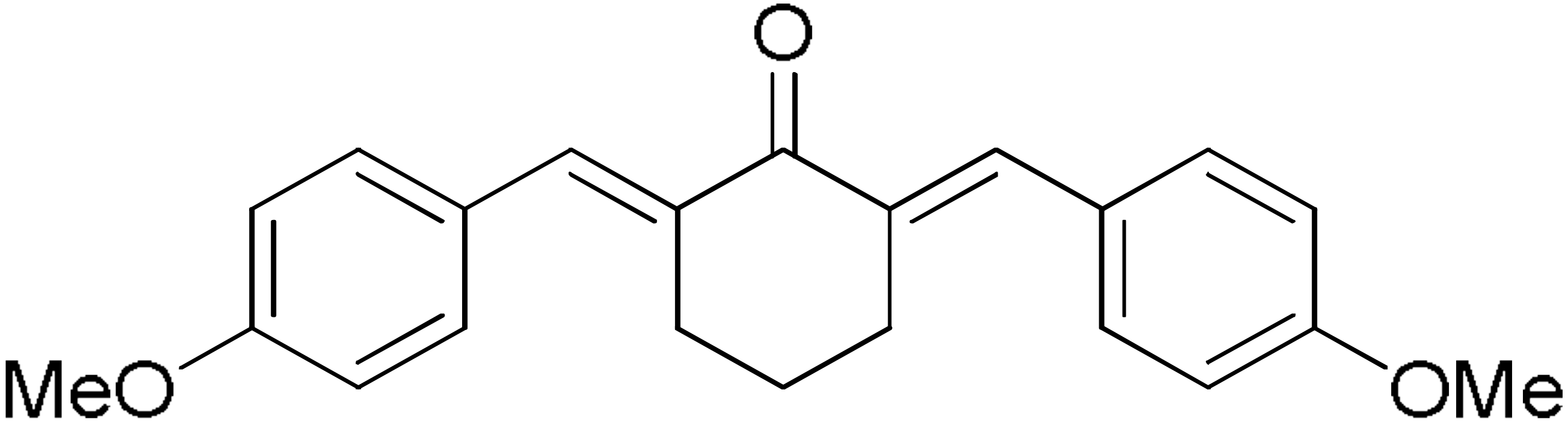 | 64.9 | DM101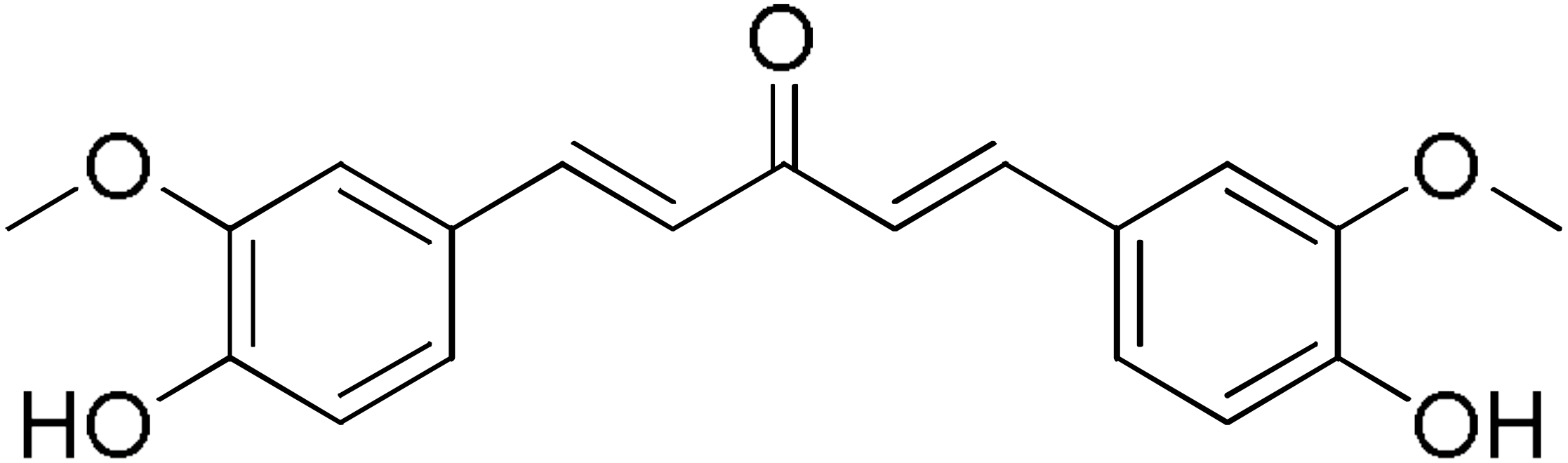 | 94.1 |
DM109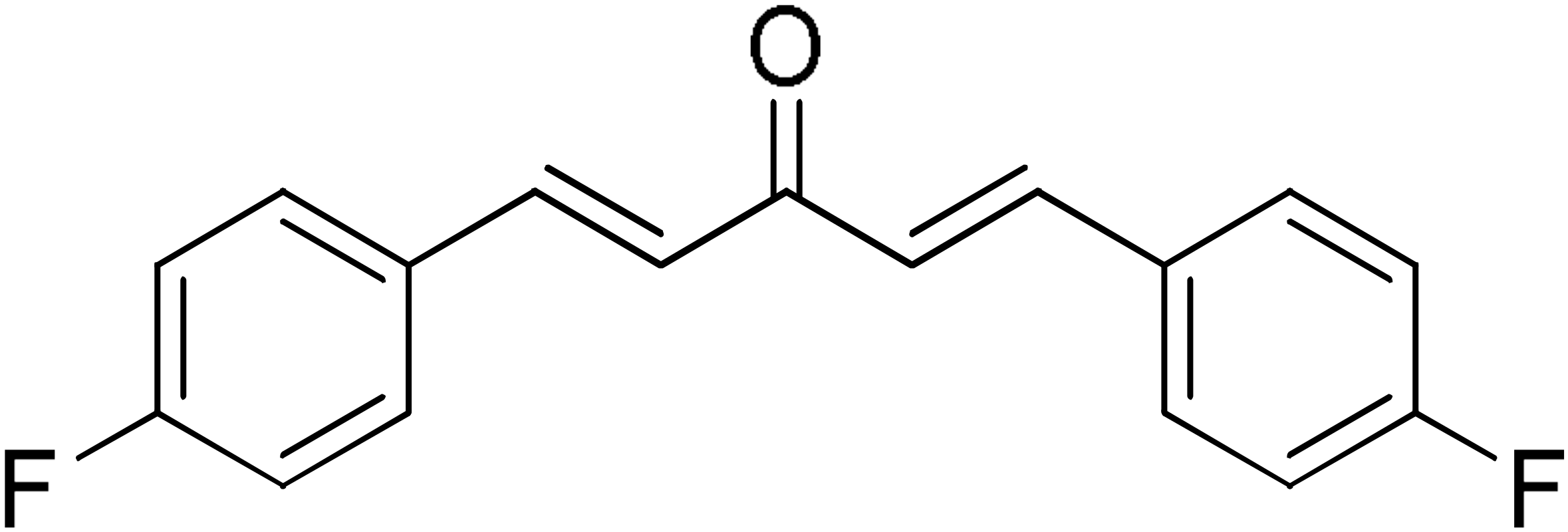 | 73.9 | DM100 | 95.1 |
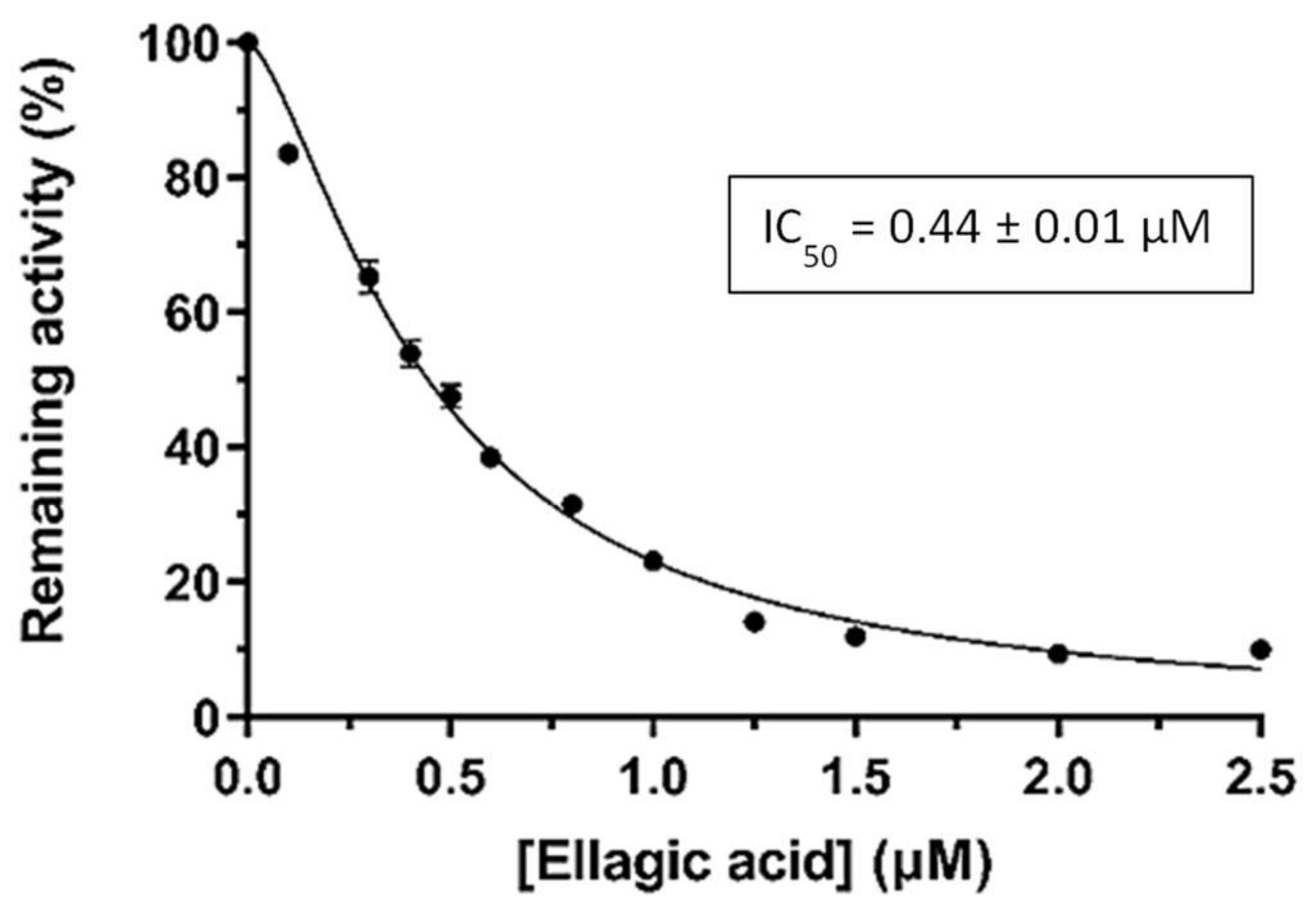
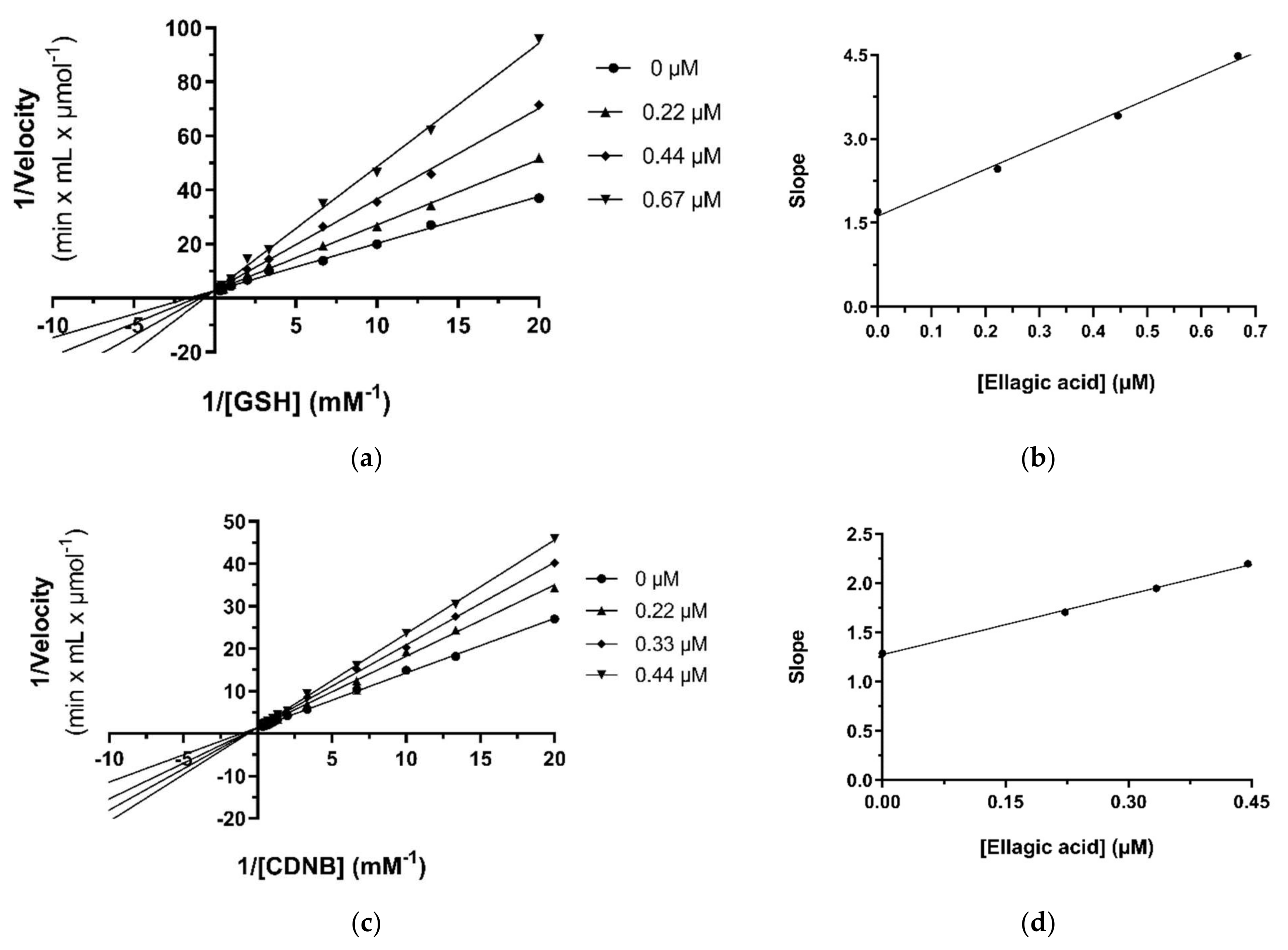
2.4. Kinetic Inhibition Studies of hGSTA4-4 with Ellagic Acid and DM151
2.4.1. Kinetic Inhibition Studies of hGSTA4-4 with Ellagic Acid
2.4.2. Kinetic Inhibition Studies of hGSTA4-4 with DM151
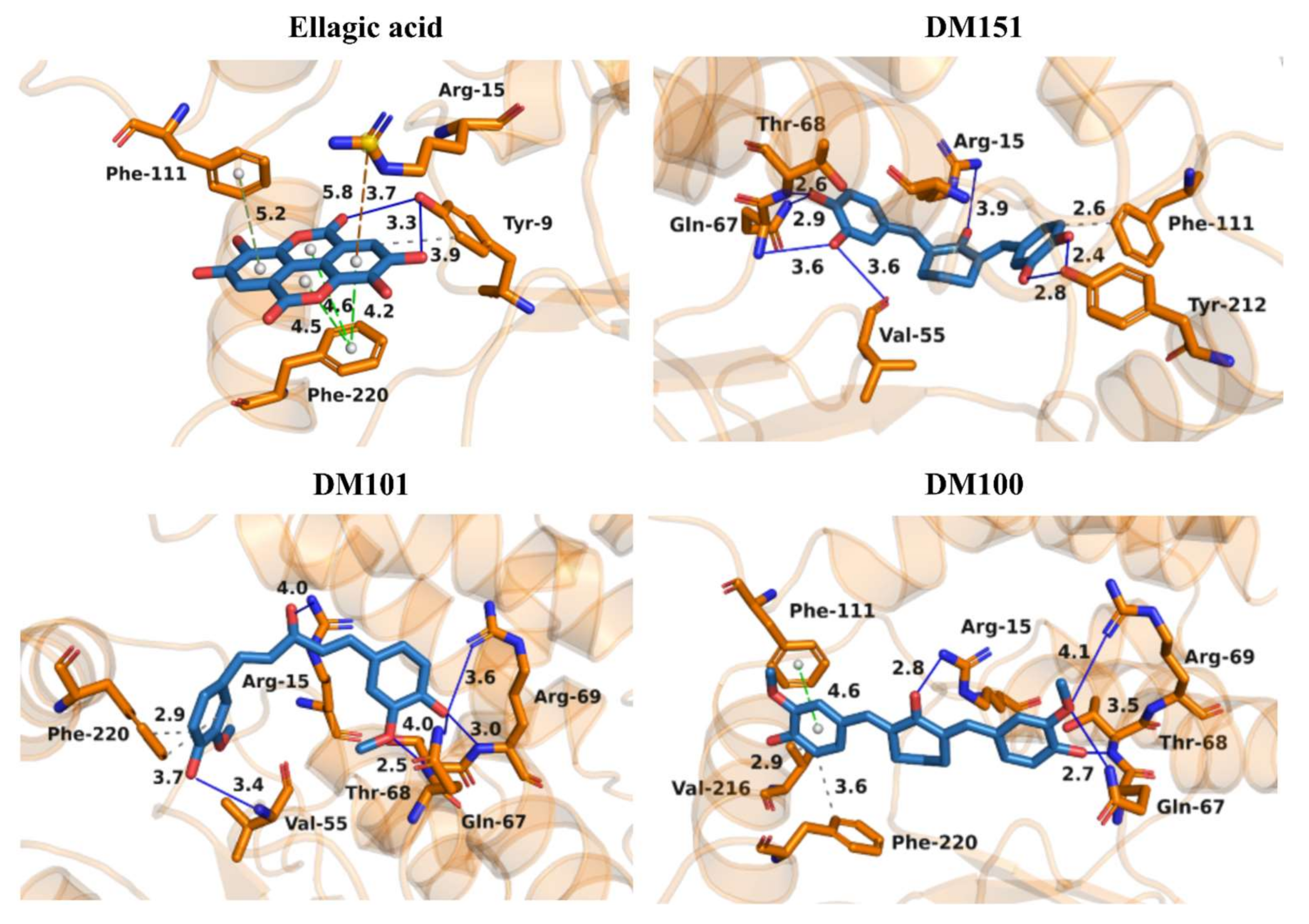
2.5. Molecular Docking of Ellagic Acid, DM151, DM101, and DM100 to hGSTA4-4
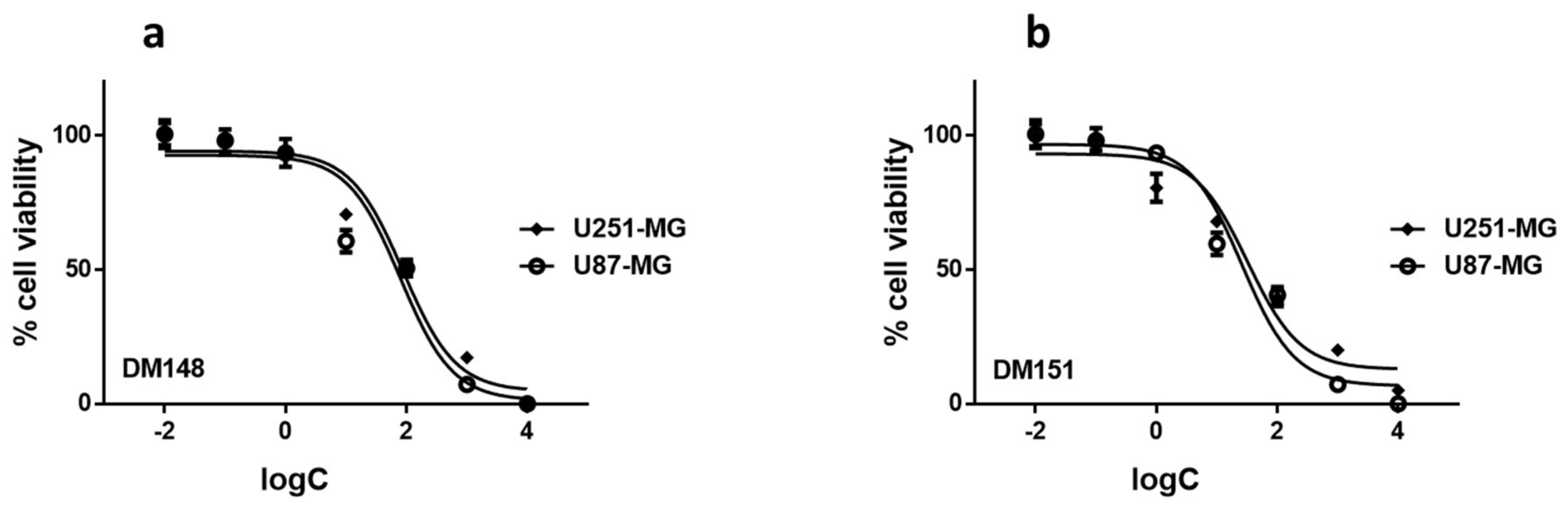
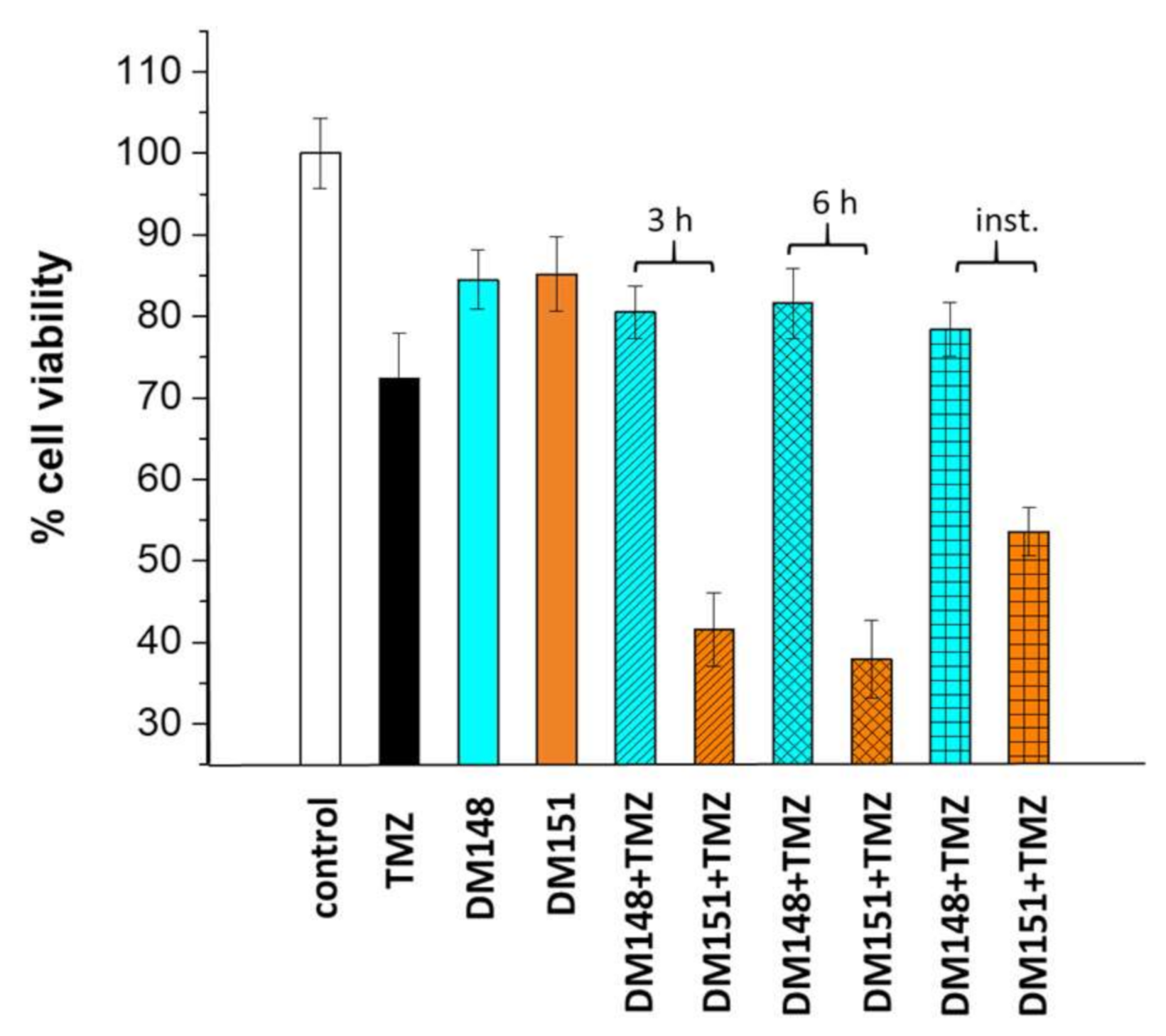
2.6. Cytotoxicity Studies of DM148 and DM151 against Glioblastoma Cells
3. Materials and Methods
3.1. Materials
3.1.1. Chemicals
3.1.2. Bacterial Strains and Plasmids
3.1.3. Curcumin Derivatives
3.2. Methods
3.2.1. Cloning, Expression, and Purification of hGSTA4-4 from Recombinant E. coli Cells
3.2.2. Enzyme Assays and Inhibition Studies
3.2.3. Kinetic Inhibition Analysis
3.2.4. Molecular Docking
3.2.5. In Vitro Cytotoxicity with MTT Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Chen, Y.; Wan, Y.; Zhao, Y.; Wen, Q.; Tang, X.; Shen, J.; Wu, X.; Li, M.; Li, X.; et al. Circular RNAs in the Regulation of Oxidative Stress. Front. Pharmacol. 2021, 12, 697903. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hou, L.C.; Guo, Z.; Wang, G.C.; Xu, J.T.; Zheng, Z.H.; Sun, K.; Guo, F.J. Lipid peroxidation in osteoarthritis: Focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 2023, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Aspects Med. 2003, 24, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361–370. [Google Scholar] [CrossRef]
- Parola, M.; Bellomo, G.; Robino, G.; Barrera, G.; Dianzani, M.U. 4-Hydroxynonenal as a biological signal: Molecular basis and pathophysiological implications. Antioxid. Redox Signal. 1999, 1, 255–284. [Google Scholar] [CrossRef]
- Mazari, A.M.A.; Zhang, L.; Ye, Z.W.; Zhang, J.; Tew, K.D.; Townsend, D.M. The Multifaceted Role of Glutathione S-Transferases in Health and Disease. Biomolecules 2023, 13, 688. [Google Scholar] [CrossRef]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta 2015, 1852, 826–838. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Dwivedi, S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal self-limits Fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry 2008, 47, 143–156. [Google Scholar] [CrossRef] [PubMed]
- McElhanon, K.E.; Bose, C.; Sharma, R.; Wu, L.; Awasthi, Y.C.; Singh, S.P. Gsta4 null mouse embryonic fibroblasts exhibit enhanced sensitivity to oxidants: Role of 4-Hydroxynonenal in oxidant toxicity. Open J. Apoptosis 2013, 2, 1–22. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ridderström, M.; Mannervik, B. Human glutathione transferase A4-4: An alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem. J. 1998, 330, 175–179. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, Z.; Li, T.; Tao, Y.; Zhu, G.; Xu, L.; Ju, Y.; Huang, X.; Zhai, J.; Wang, X. Colonic expression of glutathione S-transferase alpha 4 and 4-hydroxynonenal adducts is correlated with the pathology of murine colitis-associated cancer. Heliyon 2023, 9, e19815. [Google Scholar] [CrossRef]
- Ng, K.T.; Yeung, O.W.; Lam, Y.F.; Liu, J.; Liu, H.; Pang, L.; Yang, X.X.; Zhu, J.; Zhang, W.; Lau, M.Y.H.; et al. Glutathione S-transferase A2 promotes hepatocellular carcinoma recurrence after liver transplantation through modulating reactive oxygen species metabolism. Cell Death Discov. 2021, 7, 188. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Marusic, Z.; Zarkovic, K.; Jaganjac, M. The 4-Hydroxynonenal-Protein Adducts and Their Biological Relevance: Are Some Proteins Preferred Targets? Antioxidants 2023, 12, 856. [Google Scholar] [CrossRef]
- Singh, R.R.; Mohammad, J.; Orr, M.; Reindl, K.M. Glutathione S-Transferase Pi-1 knockdown reduces pancreatic ductal adenocarcinoma growth by activating oxidative stress response pathways. Cancers 2020, 12, 1501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, L.; Huang, L.; Li, T.; Wang, J.Y.; Ma, C.; Bian, X.; Ren, X.; Li, H.; Wang, X. Glutathione S-Transferase alpha 4 promotes proliferation and chemoresistance in colorectal cancer cells. Front. Oncol. 2022, 12, 887127. [Google Scholar] [CrossRef] [PubMed]
- Drozd, E.; Krzysztoń-Russjan, J.; Marczewska, J.; Drozd, J.; Bubko, I.; Bielak, M.; Lubelska, K.; Wiktorska, K.; Chilmonczyk, Z.; Anuszewska, E.; et al. Up-regulation of glutathione-related genes, enzyme activities and transport proteins in human cervical cancer cells treated with doxorubicin. Biomed. Pharmacother. 2016, 83, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Singh, S. Cytoprotective and regulatory functions of glutathione S-transferases in cancer cell proliferation and cell death. Cancer Chemother. Pharmacol. 2015, 75, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Ellis, B.; Sharma, A. Role of alpha class glutathione transferases (GSTs) in chemoprevention: GSTA1 and A4 overexpressing human leukemia (HL60) cells resist sulforaphane and curcumin induced toxicity. Phytother. Res. 2011, 25, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Phytother. Res. 2010, 17, 1373–1380. [Google Scholar] [CrossRef]
- Pan, D.; Tang, Y.; Tong, J.; Xie, C.; Chen, J.; Feng, C.; Hwu, P.; Huang, W.; Zhou, D. An antibody-drug conjugate targeting a GSTA glycosite-signature epitope of MUC1 expressed by non-small cell lung cancer. Cancer Med. 2020, 9, 9529–9540. [Google Scholar] [CrossRef]
- Liu, C.J.; Yang, J.H.; Huang, F.Z.; Nie, W.P.; Liu, C.P.; Mao, X.H.; Yin, X.M.; Shen, X.B.; Peng, C.; Chen, M.F.; et al. Glutathione-s- transferase A 4 (GSTA4) suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting AKT pathway. Am. J. Transl. Res. 2017, 9, 301–315. [Google Scholar] [PubMed]
- Lv, N.; Huang, C.; Huang, H.; Dong, Z.; Chen, X.; Lu, C.; Zhang, Y. Overexpression of glutathione S-transferases in human diseases: Drug targets and therapeutic implications. Antioxidants 2023, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Jani, J.P.; Jacobs, S.; Levitt, M.; Fields, L.; Awasthi, S.; Xu, B.H.; Sreevardhan, M.; Awasthi, Y.C.; Singh, S.V. Activity of melphalan in combination with the glutathione transferase inhibitor sulfasalazine. Cancer Chemother. Pharmacol. 1995, 36, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Hu, X.; Xu, B.; Tong, T.; Jing, Y.; Xi, L.; Zhou, W.; Lu, J.; Wang, X.; Yang, X.; et al. Glutathione S-transferase isozyme alpha 1 is predominantly involved in the cisplatin resistance of common types of solid cancer. Oncol. Rep. 2019, 41, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Perperopoulou, F.D.; Tsoungas, P.G.; Thireou, T.N.; Rinotas, V.E.; Douni, E.K.; Eliopoulos, E.E.; Labrou, N.E.; Clonis, Y.D. 2,2′-Dihydroxybenzophenones and their carbonyl N-analogues as inhibitor scaffolds for MDR-involved human glutathione transferase isoenzyme A1-1. Bioorganic Med. Chem. 2014, 22, 3957–3970. [Google Scholar] [CrossRef] [PubMed]
- Pouliou, F.M.; Thireou, T.N.; Eliopoulos, E.E.; Tsoungas, P.G.; Labrou, N.E.; Clonis, Y.D. Isoenzyme- and allozyme-specific inhibitors: 2,2′-dihydroxybenzophenones and their carbonyl N-analogues that discriminate between human glutathione transferase A1-1 and P1-1 allozymes. Chem. Biol. Drug Des. 2015, 86, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Labrou, N.E. The Interaction of the Flavonoid Fisetin with Human Glutathione Transferase A1-1. Metabolites 2021, 11, 190. [Google Scholar] [CrossRef]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Alam, A.; Labrou, N.E. Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1-1. Biomolecules 2022, 12, 1364. [Google Scholar] [CrossRef]
- Appiah-Opong, R.; Commandeur, J.N.M.; Istyastono, E.; Bogaards, J.J.; Vermeulen, N.P.E. Inhibition of human glutathione S-transferases by curcumin and analogues. Xenobiotica 2009, 39, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Van Iersel, M.L.; Ploemen, J.P.; Lo Bello, M.; Federici, G.; Van Bladeren, P.J. Interactions of alpha, beta-unsaturated aldehydes and ketones with human glutathione S-transferase P1-1. Chem. Biol. Interact. 1997, 108, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Guo, J.; Wang, Q.; Zhu, S.; Gao, S.; Yang, C.; Wei, M.; Pan, X.; Zhu, W.; et al. Cytotoxic and antitumor effects of curzerene from Curcuma longa. Planta Med. 2017, 83, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Hong, X.; Wang, L.; Cao, Y.; Qin, D.; Zhou, H.; Gao, D. Curzerene suppresses progression of human glioblastoma through inhibition of glutathione S-transferase A4. CNS Neurosci. Ther. 2022, 28, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.M.; Hubatsch, I.; Ridderstrom, M.; Mannervik, B.; Tainer, J.A. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J. Mol. Biol. 1999, 288, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Groom, H.; Lee, M.; Patil, P.; Josephy, P.D. Inhibition of human glutathione transferases by dinitronaphthalene derivatives. Arch. Biochem. Biophys. 2014, 555, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Pantiora, P.; Furlan, V.; Matiadis, D.; Mavroidi, B.; Perperopoulou, F.; Papageorgiou, A.C.; Sagnou, M.; Bren, U.; Pelecanou, M.; Labrou, N.E. Monocarbonyl Curcumin Analogues as Potent Inhibitors against Human Glutathione Transferase P1-1. Antioxidants 2022, 12, 63. [Google Scholar] [CrossRef]
- Perperopoulou, F.; Pouliou, F.; Labrou, N.E. Recent advances in protein engineering and biotechnological applications of glutathione transferases. Crit. Rev. Biotechnol. 2018, 38, 511–528. [Google Scholar] [CrossRef]
- Alary, J.; Fernandez, Y.; Debrauwer, L.; Perdu, E.; Guéraud, F. Identification of intermediate pathways of 4-hydroxynonenal metabolism in the rat. Chem. Res. Toxicol. 2003, 16, 320–327. [Google Scholar] [CrossRef]
- Yuan, X.; Li, H.; Bai, H.; Su, Z.; Xiang, Q.; Wang, C.; Zhao, B.; Zhang, Y.; Zhang, Q.; Chu, Y.; et al. Synthesis of novel curcumin analogues for inhibition of 11-hydroxysteroid dehydrogenase type 1 with anti-diabetic properties. Eur. J. Med. Chem. 2014, 77, 223–230. [Google Scholar] [CrossRef]
- Wang, Z.S.; Chen, L.Z.; Zhou, H.P.; Liu, X.H.; Chen, F.H. Diarylpentadienone derivatives (curcumin analogues): Synthesis and anti-inflammatory activity. Bioorg. Med. Chem. Lett. 2017, 27, 1803–1807. [Google Scholar] [CrossRef] [PubMed]
- Hotsumi, M.; Tajiri, M.; Nikaido, Y.; Sato, T.; Makabe, K.; Konno, H. Design, synthesis, and evaluation of a water soluble C5-monoketone type curcumin analogue as a potent amyloid aggregation inhibitor. Bioorg. Med. Chem. Lett. 2019, 29, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.Y.; Jiang, Y.F.; Tang, Z.K.; Mo, R.Q.; Xue, G.H.; Lu, Y.J.; Zheng, X.; Dong, C.Z.; Zhang, K. Antioxidation and tyrosinase inhibition of polyphenolic curcumin analogs. Biosci. Biotechnol. Biochem. 2011, 75, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Du, Z.Y.; Huang, Z.S.; Lee, K.S.; Gu, L.Q. Inhibition of thioredoxin reductase by curcumin analogs. Biosci. Biotechnol. Biochem. 2008, 72, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.Y.; Bao, Y.D.; Liu, Z.; Qiao, W.; Ma, L.; Huang, Z.S.; Gu, L.Q.; Chan, A.S.C. Curcumin analogs as potent aldose reductase inhibitors. Arch. Pharm. 2006, 339, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Buolamwini, J.K.; Assefa, H. CoMFA and CoMSIA 3D QSAR and Docking studies on conformationally-restrained cinnamoyl HIV-1 integrase inhibitors: Exploration of a binding mode at the active site. J. Med. Chem. 2002, 45, 841–852. [Google Scholar] [CrossRef]
- Fine, J.; Konc, J.; Samudrala, R.; Chopra, G. CANDOCK: Chemical atomic network-based hierarchical flexible docking algorithm using generalized statistical potentials. J. Chem. Inf. Model. 2020, 60, 1509–1527. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef]
- Wang, D.; Chen, Q.; Tan, Y.; Liu, B.; Liu, C. Ellagic acid inhibits human glioblastoma growth in vitro and in vivo. Oncol. Rep. 2017, 37, 1084–1092. [Google Scholar] [CrossRef]
- Zuccari, G.; Baldassari, S.; Ailuno, G.; Turrini, F.; Alfei, S.; Caviglioli, G. Formulation strategies to improve oral bioavailability of ellagic acid. Appl. Sci. 2020, 10, 3353. [Google Scholar] [CrossRef]
- Poon, M.T.C.; Bruce, M.; Simpson, J.E.; Hannan, C.J.; Brennan, P.M. Temozolomide sensitivity of malignant glioma cell lines—A systematic review assessing consistencies between in vitro studies. BMC Cancer 2021, 21, 1240. [Google Scholar] [CrossRef]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An updated overview of resistance mechanisms, Nanotechnology Advances and Clinical Applications. Curr. Neuropharmacol. 2021, 19, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer. 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Guo, J.; Yu, L.; Liu, J.; Chen, J.; Xin, J.; Zhang, Y.; Luo, J.; Duan, C. Synergistic effect of cryptotanshinone and temozolomide treatment against human glioblastoma cells. Sci Rep. 2023, 13, 21835. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Chen, N.F.; Wen, Z.H.; Yao, Z.K.; Tsui, K.H.; Kuo, H.M.; Chen, W.F. Glutathione S-transferase M3 is associated with glycolysis in intrinsic temozolomide-resistant glioblastoma multiforme cells. Int. J. Mol. Sci. 2021, 22, 7080. [Google Scholar] [CrossRef] [PubMed]
- Mazarei, M.; Mohammadi Arvejeh, P.; Mozafari, M.R.; Khosravian, P.; Ghasemi, S. Anticancer Potential of Temozolomide-Loaded Eudragit-Chitosan Coated Selenium Nanoparticles: In Vitro Evaluation of Cytotoxicity, Apoptosis and Gene Regulation. Nanomaterials 2021, 11, 1704. [Google Scholar] [CrossRef]
- Georgakis, N.D.; Karagiannopoulos, D.A.; Thireou, T.N.; Eliopoulos, E.E.; Labrou, N.E.; Tsoungas, P.G.; Koutsilieris, M.N.; Clonis, Y.D. Concluding the trilogy: The interaction of 2, 20-dihydroxy-benzophenones and their carbonyl N-analogues with human glutathione transferase M1-1 face to face with the P1-1 and A1-1 isoenzymes involved in MDR. Chem. Biol. Drug Des. 2017, 90, 900–908. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4. Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
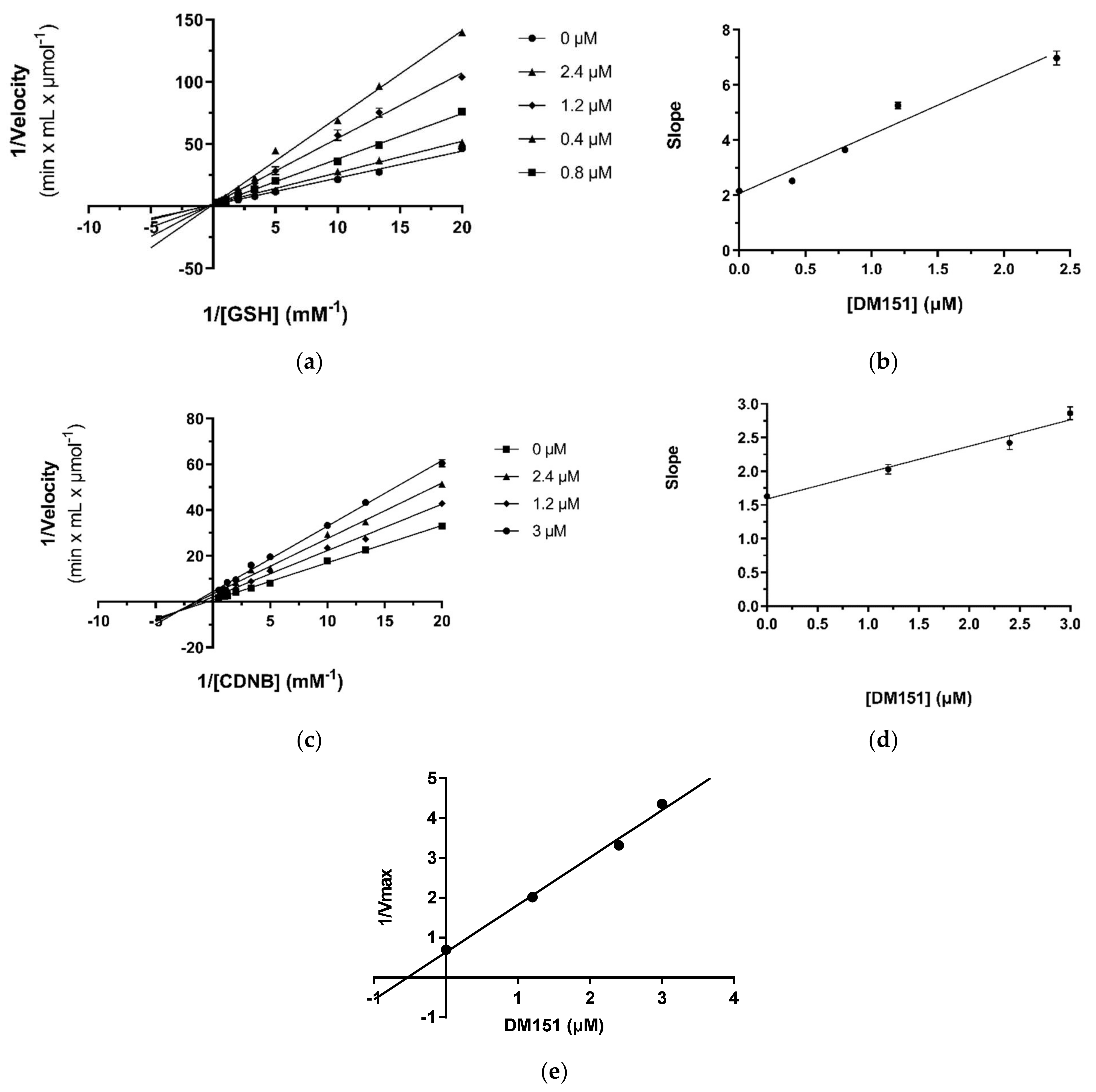
| Inhibitor | IC50 (μΜ) | Variable Substrate | Type of Inhibition | Inhibition Constant Ki (μΜ) | Inhibition Constant Ki′ (μΜ) |
|---|---|---|---|---|---|
| Ellagic acid | 0.44 ± 0.01 | CDNB | competitive | 0.63 ± 0.03 | - |
| GSH | competitive | 0.39 ± 0.02 | - | ||
| DM151 | 2.4 ± 0.1 | CDNB | mixed | 4.1 ± 0.5 | 0.53 ± 0.03 |
| GSH | competitive | 0.98 ± 0.11 | - |
| Ιnhibitors | Docking Score Values (Arbitrary Units) |
|---|---|
| Ellagic acid | −45.18 |
| DM151 | −44.59 |
| DM101 | −36.37 |
| DM100 | −33.37 |
| IC50 (μM) | ||
|---|---|---|
| U251-MG | U87-MG | |
| DM148 | 89.21 ± 3.41 | 91.09 ± 5.64 |
| DM151 | 25.57 ± 4.83 | 18.95 ± 5.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsouri, S.; Tselo, E.; Premetis, G.E.; Furlan, V.; Pantiora, P.D.; Mavroidi, B.; Matiadis, D.; Pelecanou, M.; Papageorgiou, A.C.; Bren, U.; et al. A Monocarbonyl Curcuminoid Derivative Inhibits the Activity of Human Glutathione Transferase A4-4 and Chemosensitizes Glioblastoma Cells to Temozolomide. Pharmaceuticals 2024, 17, 365. https://doi.org/10.3390/ph17030365
Tsouri S, Tselo E, Premetis GE, Furlan V, Pantiora PD, Mavroidi B, Matiadis D, Pelecanou M, Papageorgiou AC, Bren U, et al. A Monocarbonyl Curcuminoid Derivative Inhibits the Activity of Human Glutathione Transferase A4-4 and Chemosensitizes Glioblastoma Cells to Temozolomide. Pharmaceuticals. 2024; 17(3):365. https://doi.org/10.3390/ph17030365
Chicago/Turabian StyleTsouri, Steliana, Evanthia Tselo, Georgios E. Premetis, Veronika Furlan, Panagiota D. Pantiora, Barbara Mavroidi, Dimitris Matiadis, Maria Pelecanou, Anastassios C. Papageorgiou, Urban Bren, and et al. 2024. "A Monocarbonyl Curcuminoid Derivative Inhibits the Activity of Human Glutathione Transferase A4-4 and Chemosensitizes Glioblastoma Cells to Temozolomide" Pharmaceuticals 17, no. 3: 365. https://doi.org/10.3390/ph17030365
APA StyleTsouri, S., Tselo, E., Premetis, G. E., Furlan, V., Pantiora, P. D., Mavroidi, B., Matiadis, D., Pelecanou, M., Papageorgiou, A. C., Bren, U., Sagnou, M., & Labrou, N. E. (2024). A Monocarbonyl Curcuminoid Derivative Inhibits the Activity of Human Glutathione Transferase A4-4 and Chemosensitizes Glioblastoma Cells to Temozolomide. Pharmaceuticals, 17(3), 365. https://doi.org/10.3390/ph17030365