

In Vitro Evaluation of Pharmacokinetic Properties of Selected Dual COX-2 and 5-LOX Inhibitors


 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Evaluation of Gastrointestinal Absorption
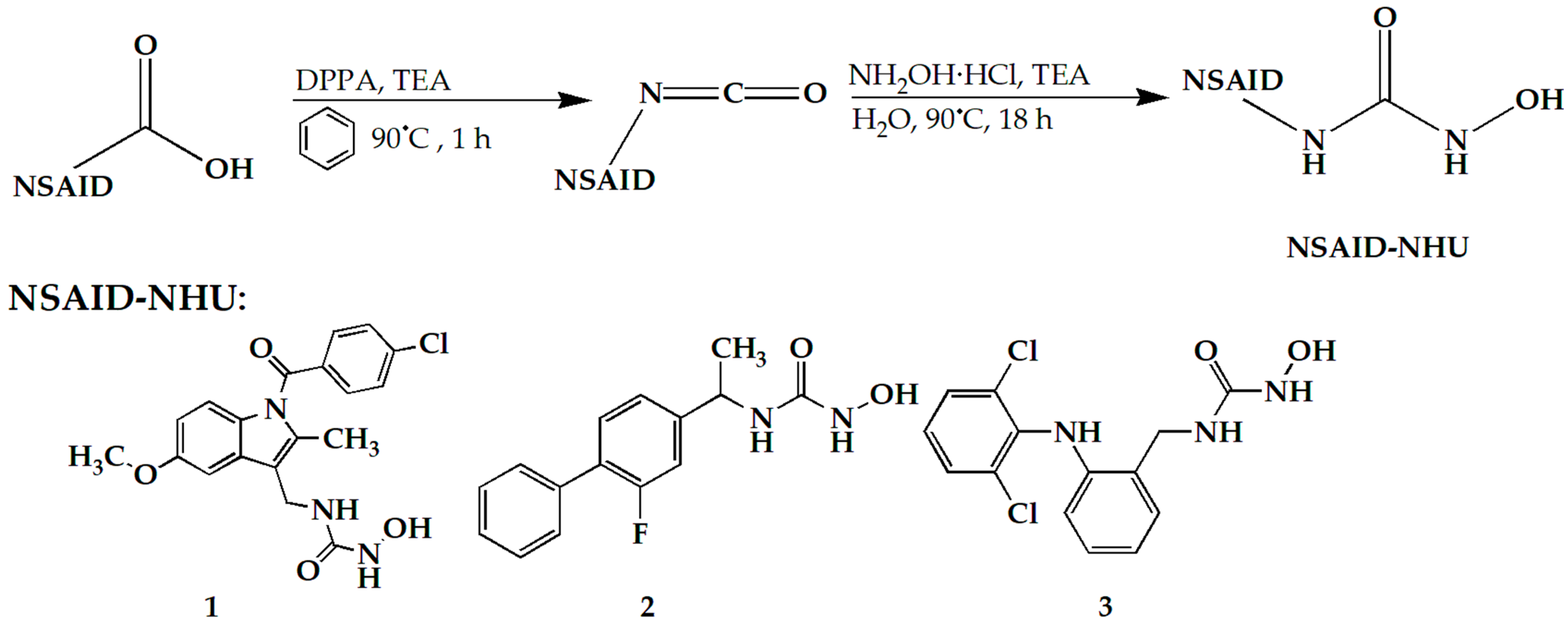
2.1.1. BMC
2.1.2. PAMPA Test
2.1.3. Comparison of the Results of Two Methods and Discussion on Passive Gastrointestinal Absorption Potential
2.2. Evaluation of HSA Binding
2.2.1. High-Performance Affinity Chromatography (HPAC)
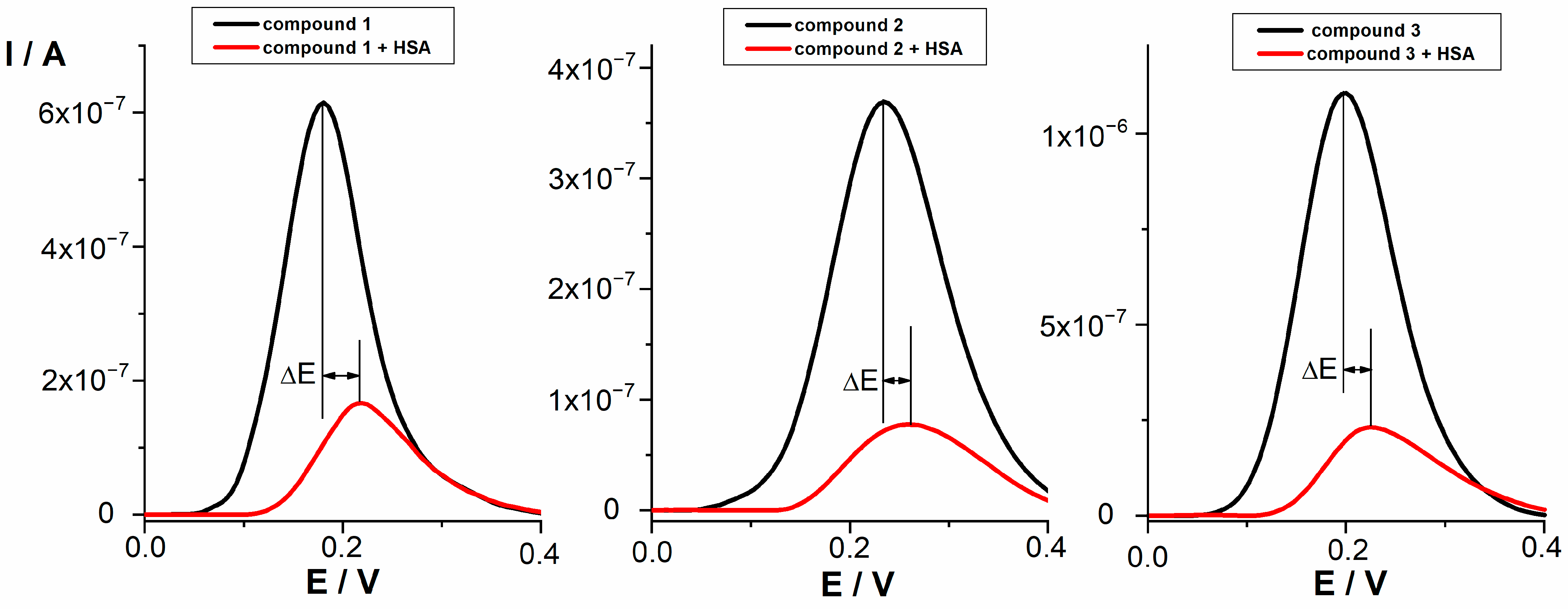
2.2.2. Differential Pulse Voltammetry (DPV)
2.2.3. Comparison of the Results of Two Methods and Discussion on Distribution Properties
2.3. Stability in Acidic Medium and Solubility
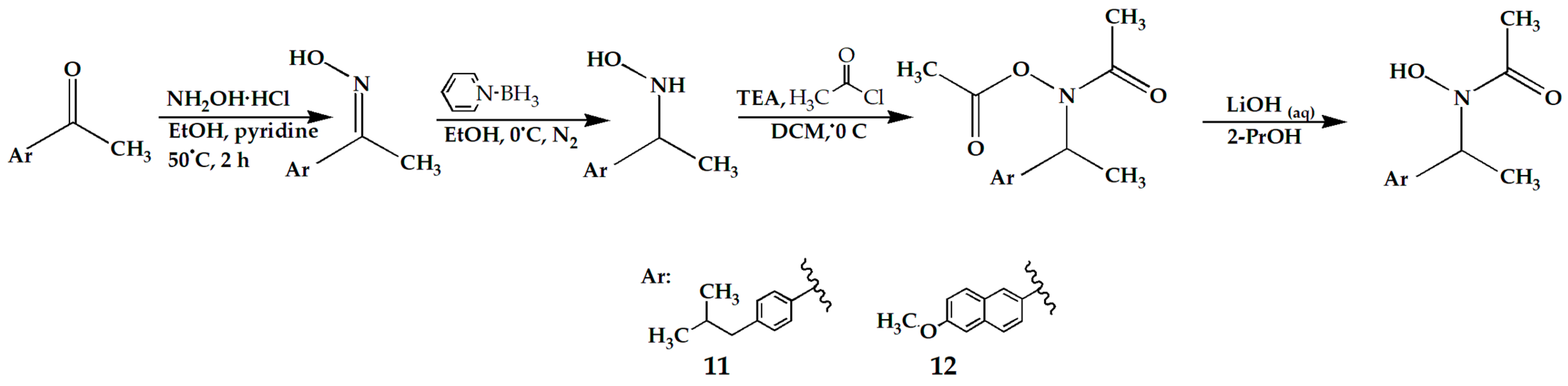
2.4. Microsomal Stability
3. Materials and Methods
3.1. Chemicals
3.2. Biopartitioning Micellar Chromatography (BMC)
3.3. PAMPA Test (Parallel Artificial Membrane Permeability Assay)
3.3.1. HPLC Method
3.3.2. Calculation of PAMPA Parameters
3.4. HPAC—Chromatographic Conditions
3.5. Differential Pulse Voltammetry (DPV)
3.6. Stability in Acidic Medium and Solubility
3.7. Microsomal Stability
3.7.1. HPLC-MS/MS Method
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- P, J.J.; Manju, S.L.; Ethiraj, K.R.; Elias, G. Safer Anti-Inflammatory Therapy through Dual COX-2/5-LOX Inhibitors: A Structure-Based Approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Cashman, J.R.; Kantner, S.S.; Wright, S.W. Rationally Designed, Potent Competitive Inhibitors of Leukotriene Biosynthesis. J. Am. Chem. Soc. 1984, 106, 1503–1504. [Google Scholar] [CrossRef]
- Flynn, D.L.; Capiris, T.; Cetenko, W.J.; Connor, D.T.; Dyer, R.D.; Kostlan, C.R.; Nies, D.E.; Schrier, D.J.; Sircar, J.C. Nonsteroidal anti-inflammatory drug hydroxamic acids. Dual inhibitors of both cyclooxygenase and 5-lipoxygenase. J. Med. Chem. 1990, 33, 2070–2072. [Google Scholar] [CrossRef] [PubMed]
- Kolasa, T.; Brooks, C.D.; Rodriques, K.E.; Summers, J.B.; Dellaria, J.F.; Hulkower, K.I.; Bouska, J.; Bell, R.L.; Carter, G.W. Nonsteroidal Anti-Inflammatory Drugs as Scaffolds for the Design of 5-Lipoxygenase Inhibitors. J. Med. Chem. 1997, 40, 819–824. [Google Scholar] [CrossRef]
- Argentieri, D.C.; Ritchie, D.M.; Ferro, M.P.; Kirchner, T.; Wachter, M.P.; Anderson, D.W.; Rosenthale, M.E.; Capetola, R.J. Tepoxalin: A Dual Cyclooxygenase/5-Lipoxygenase Inhibitor of Arachidonic Acid Metabolism with Potent Anti-Inflammatory Activity and a Favorable Gastrointestinal Profile. J. Pharmacol. Exp. Ther. 1994, 271, 1399–1408. [Google Scholar]
- Connolly, P.J.; Wetter, S.K.; Beers, K.N.; Hamel, S.C.; Chen, R.H.; Wachter, M.P.; Ansell, J.; Singer, M.M.; Steber, M.; Ritchie, D.M.; et al. N-Hydroxyurea and Hydroxamic Acid Inhibitors of Cyclooxygenase and 5-Lipoxygenase. Bioorg. Med. Chem. Lett. 1999, 9, 979–984. [Google Scholar] [CrossRef]
- Barbey, S.; Goossens, L.; Taverne, T.; Cornet, J.; Choesmel, V.; Rouaud, C.; Gimeno, G.; Yannic-Arnoult, S.; Michaux, C.; Charlier, C.; et al. Synthesis and Activity of a New Methoxytetrahydropyran Derivative as Dual Cyclooxygenase-2/5-Lipoxygenase Inhibitor. Bioorg. Med. Chem. Lett. 2002, 779–782. [Google Scholar] [CrossRef]
- Weisman, S.M.; Doyle, M.J.; Wehmeyer, K.R.; Hynd, B.A.; Eichhold, T.H.; Clear, R.M.; Coggeshall, C.W.; Kuhlenbeck, D.L. Effects of Tebufelone (NE-11740), a New Anti-Inflammatory Drug, on Arachidonic Acid Metabolism. Agents Actions 1994, 41, 156–163. [Google Scholar] [CrossRef]
- Unangst, P.C.; Connor, D.T.; Cetenko, W.A.; Sorenson, R.J.; Kostlan, C.R.; Sircar, J.C.; Wright, C.D.; Schrier, D.J.; Dyer, R.D. Synthesis and Biological Evaluation of 5-[[3,5-Bis(1,1-Dimethylethyl)-4-Hydroxyphenyl]Methylene]Oxazoles, -Thiazoles, and -Imidazoles: Novel Dual 5-Lipoxygenase and Cyclooxygenase Inhibitors with Antiinflammatory Activity. J. Med. Chem. 1994, 37, 322–328. [Google Scholar] [CrossRef]
- Ramanan, M.; Sinha, S.; Sudarshan, K.; Aidhen, I.S.; Doble, M. Inhibition of the Enzymes in the Leukotriene and Prostaglandin Pathways in Inflammation by 3-Aryl Isocoumarins. Eur. J. Med. Chem. 2016, 124, 428–434. [Google Scholar] [CrossRef]
- Bošković, J.; Dobričić, V.; Mihajlović, M.; Kotur-Stevuljević, J.; Čudina, O. Synthesis, Evaluation of Enzyme Inhibition and Redox Properties of Potential Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals 2023, 16, 549. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhu, C.; Qiang, H.; Zhou, X.; Zhou, G. Enhancing Antitumor Effects in Pancreatic Cancer Cells by Combined Use of COX-2 and 5-LOX Inhibitors. Biomed. Pharmacother. 2011, 65, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-X.; Ding, X.-L.; Wu, S.-B.; Zhang, H.-F.; Cao, W.; Qu, L.-S.; Zhang, H. Inhibition of 5-Lipoxygenase Triggers Apoptosis in Pancreatic Cancer Cells. Oncol. Rep. 2015, 33, 661–668. [Google Scholar] [CrossRef]
- Che, X.-H.; Chen, C.-L.; Ye, X.-L.; Weng, G.-B.; Guo, X.-Z.; Yu, W.-Y.; Tao, J.; Chen, Y.-C.; Chen, X. Dual Inhibition of COX-2/5-LOX Blocks Colon Cancer Proliferation, Migration and Invasion in Vitro. Oncol. Rep. 2016, 35, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Bošković, J.; Dobričić, V.; Keta, O.; Korićanac, L.; Žakula, J.; Dinić, J.; Jovanović Stojanov, S.; Pavić, A.; Čudina, O. Unveiling Anticancer Potential of COX-2 and 5-LOX Inhibitors: Cytotoxicity, Radiosensitization Potential and Antimigratory Activity against Colorectal and Pancreatic Carcinoma. Pharmaceutics 2024, 16, 826. [Google Scholar] [CrossRef]
- Valkó, K.L.; Nunhuck, S.B.; Hill, A.P. Estimating Unbound Volume of Distribution and Tissue Binding by in Vitro HPLC-Based Human Serum Albumin and Immobilised Artificial Membrane-Binding Measurements. J. Pharm. Sci. 2011, 100, 849–862. [Google Scholar] [CrossRef]
- Byers, J.P.; Sarver, J.G. Pharmacokinetic Modeling. In Pharmacology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 201–277. [Google Scholar] [CrossRef]
- Barthe, L.; Woodley, J.; Houin, G. Gastrointestinal Absorption of Drugs: Methods and Studies. Fundam. Clin. Pharmacol. 1999, 13, 154–168. [Google Scholar] [CrossRef]
- Pagliara, A.; Reist, M.; Geinoz, S.; Carrupt, P.-A.; Testa, B. Evaluation and Prediction of Drug Permeation. J. Pharm. Pharmacol. 1999, 51, 1339–1357. [Google Scholar] [CrossRef]
- Tsopelas, F.; Danias, P.; Pappa, A.; Tsantili-Kakoulidou, A. Biopartitioning Micellar Chromatography under Different Conditions: Insight into the Retention Mechanism and the Potential to Model Biological Processes. J. Chromatogr. A 2020, 1621, 461027. [Google Scholar] [CrossRef]
- Stępnik, K.E.; Malinowska, I. The Use of Biopartitioning Micellar Chromatography and Immobilized Artificial Membrane Column for in Silico and in Vitro Determination of Blood-Brain Barrier Penetration of Phenols. J. Chromatogr. A 2013, 1286, 127–136. [Google Scholar] [CrossRef]
- Escuder-Gilabert, L.; Martínez-Pla, J.J.; Sagrado, S.; Villanueva-Camañas, R.M.; Medina-Hernández, M.J. Biopartitioning Micellar Separation Methods: Modelling Drug Absorption. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 797, 21–35. [Google Scholar] [CrossRef]
- Avdeef, A. The Rise of PAMPA. Expert Opin. Drug Metab. Toxicol. 2005, 1, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Bujard, A.; Voirol, H.; Carrupt, P.-A.; Schappler, J. Modification of a PAMPA Model to Predict Passive Gastrointestinal Absorption and Plasma Protein Binding. Eur. J. Pharm. Sci. 2015, 77, 273–278. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Bezar, I.F.; Petusky, S.L.; Huang, Y. Comparison of Blood-Brain Barrier Permeability Assays: In Situ Brain Perfusion, MDR1-MDCKII and PAMPA-BBB. J. Pharm. Sci. 2009, 98, 1980–1991. [Google Scholar] [CrossRef]
- Ottaviani, G.; Martel, S.; Carrupt, P.-A. Parallel Artificial Membrane Permeability Assay: A New Membrane for the Fast Prediction of Passive Human Skin Permeability. J. Med. Chem. 2006, 49, 3948–3954. [Google Scholar] [CrossRef]
- Kragh-Hansen, U. Human Serum Albumin: A Multifunctional Protein. In Albumin in Medicine; Otagiri, M., Giam Chuang, V.T., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–24. [Google Scholar]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human Serum Albumin: From Bench to Bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin-Drug Interaction and Its Clinical Implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Otagiri, M. A Molecular Functional Study on the Interactions of Drugs with Plasma Proteins. Drug Metab. Pharmacokinet. 2005, 20, 309–323. [Google Scholar] [CrossRef]
- Vuignier, K.; Guillarme, D.; Veuthey, J.-L.; Carrupt, P.-A.; Schappler, J. High Performance Affinity Chromatography (HPAC) as a High-Throughput Screening Tool in Drug Discovery to Study Drug-Plasma Protein Interactions. J. Pharm. Biomed. Anal. 2013, 74, 205–212. [Google Scholar] [CrossRef]
- Ravelli, D.; Isernia, P.; Acquarulo, A.; Profumo, A.; Merli, D. Voltammetric Determination of Binding Constant and Stoichiometry of Albumin (Human, Bovine, Ovine)-Drug Complexes. Anal. Chem. 2019, 91, 10110–10115. [Google Scholar] [CrossRef] [PubMed]
- Heli, H.; Sattarahmady, N.; Jabbari, A.; Moosavi-Movahedi, A.A.; Hakimelahi, G.H.; Tsai, F.-Y. Adsorption of Human Serum Albumin onto Glassy Carbon Surface—Applied to Albumin-Modified Electrode: Mode of Protein–Ligand Interactions. J. Electroanal. Chem. 2007, 610, 67–74. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Hao, J.; Wang, L.; Tong, Y.-J.; Fu, Z.-Q.; Zhang, A.P. Investigation of Interaction Between Ozagrel and Human Serum Albumin by Spectroscopic and Electrochemical Methods. J. Appl. Spectrosc. 2017, 83, 1076–1083. [Google Scholar] [CrossRef]
- Rezaeinasab, M.; Benvidi, A.; Gharaghani, S.; Zare, H.R. Chemometrics approaches based on electrochemical methods for the investigation of interaction between bovine serum albumin and carvacrol with the aim of its applicaion to protein sensing. J. Electroanal. Chem. 2019, 845, 48–56. [Google Scholar] [CrossRef]
- Naggar, A.H.; El Kaoutit, M.; Naranjo-Rodriguez, I.; El-Sayed, A.Y.; Hidalgo-Hidalgo de Cisneros, J.L. Voltammetric and Spectroscopic Investigation of the Interaction Between 1,4-Benzodiazepines and Bovine Serum Albumin. J. Solut. Chem. 2016, 45, 1659–1678. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef]
- Molero-Monfort, M.; Escuder-Gilabert, L.; Villanueva-Camañas, R.M.; Sagrado, S.; Medina-Hernández, M.J. Biopartitioning Micellar Chromatography: An in Vitro Technique for Predicting Human Drug Absorption. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2001, 753, 225–236. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Nome, F. Partitioning Behavior of Solutes Eluted with Micellar Mobile Phases in Liquid Chromatography. Anal. Chem. 1981, 53, 1662–1666. [Google Scholar] [CrossRef]
- Arunyanart, M.; Love, L.J.C. Model for Micellar Effects on Liquid Chromatography Capacity Factors and for Determination of Micelle-Solute Equilibrium Constants. Anal. Chem. 1984, 56, 1557–1561. [Google Scholar] [CrossRef]
- Ruiz-Ángel, M.J.; Carda-Broch, S.; Torres-Lapasió, J.R.; García-Álvarez-Coque, M.C. Retention Mechanisms in Micellar Liquid Chromatography. J. Chromatogr. A 2009, 1216, 1798–1814. [Google Scholar] [CrossRef]
- CH Harmonised Guideline. Biopharmaceutics Classification System-Based Biowaivers M9. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. 2019. Available online: https://database.ich.org/sites/default/files/M9_Guideline_Step4_2019_1116.pdf (accessed on 17 September 2024).
- Czub, M.P.; Handing, K.B.; Venkataramany, B.S.; Cooper, D.R.; Shabalin, I.G.; Minor, W. Albumin-Based Transport of Nonsteroidal Anti-Inflammatory Drugs in Mammalian Blood Plasma. J. Med. Chem. 2020, 63, 6847–6862. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.Y. Adverse Interactions Between Warfarin and Nonsteroidal Antiinflammatory Drugs: Mechanisms, Clinical Significance, and Avoidance. Ann. Pharmacother. 1995, 29, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Woolfork, A.G.; Suh, K.; Weigand, M.; Hage, D.S. Studies of Binding by 2-Imidazolines to Human Serum Albumin and Alpha1-Acid Glycoprotein by High-Performance Affinity Chromatography. J. Pharm. Biomed. Anal. 2021, 202, 114135. [Google Scholar] [CrossRef] [PubMed]
- Turković, N.; Anđelković, N.; Obradović, D.; Vujić, Z.; Ivković, B. Application of Liquid Chromatography in Defining the Interaction of Newly Synthesized Chalcones and Related Compounds with Human Serum Albumin. J. Serbian Chem. Soc. 2023, 88, 765–776. [Google Scholar] [CrossRef]
- Sofia Almeida, A.; Cardoso, T.; Cravo, S.; Elizabeth Tiritan, M.; Remião, F.; Fernandes, C. Binding Studies of Synthetic Cathinones to Human Serum Albumin by High-Performance Affinity Chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2023, 1227, 123836. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, W.; Wu, D.; Chen, L.; Liu, X. Investigation of the Interaction of Batatasin Derivatives with Human Serum Albumin Using Voltammetric and Spectroscopic Methods. RSC Adv. 2016, 6, 36281–36292. [Google Scholar] [CrossRef]
- Evoli, S.; Mobley, D.L.; Guzzi, R.; Rizzuti, B. Multiple binding modes of ibuprofen in human serum albumin identified by absolute binding free energy calculations. Phys. Chem. Chem. Phys. 2016, 18, 32358–32368. [Google Scholar] [CrossRef]
- MarvinSketch 23.14.0; ChemAxon: Budapest, Hungary, 2023; Available online: http://www.chemaxon.com (accessed on 30 September 2024).
- Hervé, F.; Urien, S.; Albengres, E.; Duché, J.C.; Tillement, J.P. Drug binding in plasma: A summary of recent trends in the study of drug and hormone binding. Clin. Pharmacokinet. 1994, 26, 44–58. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef]
- The United States Pharmacopoeia 34/National Formulary 29; United States Pharmacopoeia Convention; United States Pharmacopoeia: Rockville, MD, USA, 2011.
- Petit, C.; Bujard, A.; Skalicka-Woźniak, K.; Cretton, S.; Houriet, J.; Christen, P.; Carrupt, P.-A.; Wolfender, J.-L. Prediction of the Passive Intestinal Absorption of Medicinal Plant Extract Constituents with the Parallel Artificial Membrane Permeability Assay (PAMPA). Planta Med. 2016, 82, 424–431. [Google Scholar] [CrossRef]
- Wohnsland, F.; Faller, B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J. Med. Chem. 2001, 44, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Bujard, A.; Sol, M.; Carrupt, P.A.; Martel, S. Predicting both passive intestinal absorption and the dissociation constant toward albumin using the PAMPA technique. Eur. J. Pharm. Sci. 2014, 63, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In vitro drug metabolism using liver microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7–8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | k |
|---|---|
| 1 | 9.50 ± 0.01 (p = 0.0156) a |
| 2 | 5.55 ± 0.01 (p = 0.0383) |
| 3 | 9.33 ± 0.03 (p = 0.0056) |
| 11 | 3.03 ± 0.01 (p = 0.0003) |
| 12 | 3.02 ± 0.00 (p = 0.0270) |
| Indomethacin | 10.20 ± 0.03 |
| Flurbiprofen | 7.14 ± 0.02 |
| Diclofenac | 8.25 ± 0.00 |
| Ibuprofen | 13.36 ± 0.00 |
| Naproxen | 5.29 ± 0.27 |
| Compound | Pe | –logPe |
|---|---|---|
| 1 | 4.29 × 10−5 ± 7.85 × 10−6 | 4.37 ± 0.08 |
| 2 | 1.76 × 10−5 ± 0.96 × 10−6 | 4.76 ± 0.02 |
| 3 | 7.73 × 10−5 ± 4.19 × 10−6 | 4.11 ± 0.02 |
| 11 | 14.59 × 10−5 ± 15.67 × 10−6 | 3.84 ± 0.05 |
| 12 | 15.70 × 10−5 ± 13.41 × 10−6 | 3.81 ± 0.04 |
| Indomethacin | 38.63 × 10−5 ± 45.00 × 10−66 | 3.41 ± 0.05 |
| Flurbiprofen a | >40.00 × 10−5 | <3.40 |
| Diclofenac a | >40.00 × 10−5 | <3.40 |
| Ibuprofen a | >40.00 × 10−5 | <3.40 |
| Naproxen | 34.67 × 10−5 ± 50.83 × 10−6 | 3.46 ± 0.06 |
| Compound | Retention Factors (logk) |
|---|---|
| 1 | 9.54 ± 0.29 |
| 2 | 7.45 ± 0.02 |
| 3 | 8.24 ± 0.01 |
| 11 | 5.81 ± 0.04 a |
| 12 | 2.03 ± 0.02 |
| Indomethacin | >70 |
| Flurbiprofen | 11.03 ± 0.02 |
| Diclofenac | >70 |
| Ibuprofen | >70 |
| Naproxen | >70 |
| Compound | ΔE (mV) | % Bound Drug |
|---|---|---|
| 1 | 34 | 73.3 |
| 2 | 30 | 80.2 |
| 3 | 29 | 80.2 |
| 11 | 7 | 70.8 |
| 12 | 13 | 70.4 |
| Indomethacin | 70 | 94.3 |
| Flurbiprofen | Electrochemically inactive | Electrochemically inactive |
| Diclofenac | 54 | 80.5 |
| Ibuprofen | Electrochemically inactive | Electrochemically inactive |
| Naproxen | 168 | 88.4 |
| Compound | Peak Area (t0) | Peak Area (t) | The Decrease in Peak Area (%) |
|---|---|---|---|
| 1 | 8.28 ± 0.01 | 8.27 ± 0.00 | 0.07 (p = 0.2839) a |
| 2 | 6.57 ± 0.12 | 6.55 ± 0.07 | 0.34 (p = 0.4501) |
| 3 | 6.42 ± 0.11 | 6.27 ± 0.10 | 2.33 (p = 0.0159) |
| 11 | 11.01 ± 0.02 | 10.83 ± 0.03 | 1.63 (p = 0.073) |
| 12 | 48.66 ± 0.49 | 48.54 ± 0.39 | 0.24 (p = 0.4399) |
| Compound | Peak Area, t0 (×103) | Peak Area, t (×103) | The Decrease in Peak Area (%) |
|---|---|---|---|
| 1 | 131.5 ± 16.8 | 60.5 ± 6.4 | 53.98 (p = 0.0328) a |
| 2 | 16.5 ± 3.1 | 9.6 ± 3.0 | 41.65 (p = 0.0034) |
| 3 | 1033.6 ± 42.5 | 455.5 ± 8.9 | 55.93 (p = 0.0200) |
| 11 | 60.0 ± 1.4 | 44.6 ± 1.9 | 25.07 (p = 0.0499) |
| 12 | 43.9 ± 3.2 | 18.2 ± 0.1 | 58.44 (p = 0.0273) |
| Indomethacin | 241.9 ± 2.5 | 206.9 ± 9.0 | 14.47 (p = 0.0419) |
| Diclofenac | 126.2 ± 2.8 | 99.7 ± 5.3 | 20.99 (p = 0. 0213) |
| Propranolol | 268.5 ± 17.0 | 155.7 ± 17.9 | 42.03 (p = 0.0017) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bošković, J.; Dobričić, V.; Savić, J.; Rupar, J.; Aleksić, M.; Marković, B.; Čudina, O. In Vitro Evaluation of Pharmacokinetic Properties of Selected Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals 2024, 17, 1329. https://doi.org/10.3390/ph17101329
Bošković J, Dobričić V, Savić J, Rupar J, Aleksić M, Marković B, Čudina O. In Vitro Evaluation of Pharmacokinetic Properties of Selected Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals. 2024; 17(10):1329. https://doi.org/10.3390/ph17101329
Chicago/Turabian StyleBošković, Jelena, Vladimir Dobričić, Jelena Savić, Jelena Rupar, Mara Aleksić, Bojan Marković, and Olivera Čudina. 2024. "In Vitro Evaluation of Pharmacokinetic Properties of Selected Dual COX-2 and 5-LOX Inhibitors" Pharmaceuticals 17, no. 10: 1329. https://doi.org/10.3390/ph17101329
APA StyleBošković, J., Dobričić, V., Savić, J., Rupar, J., Aleksić, M., Marković, B., & Čudina, O. (2024). In Vitro Evaluation of Pharmacokinetic Properties of Selected Dual COX-2 and 5-LOX Inhibitors. Pharmaceuticals, 17(10), 1329. https://doi.org/10.3390/ph17101329