Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Linagliptin Improves Cadmium-Evoked Retention and Recognition Memory Impairment in Rats
2.2. Linagliptin Attenuates the Degenerative Histological Changes Induced by Cadmium in the Hippocampi of Rats
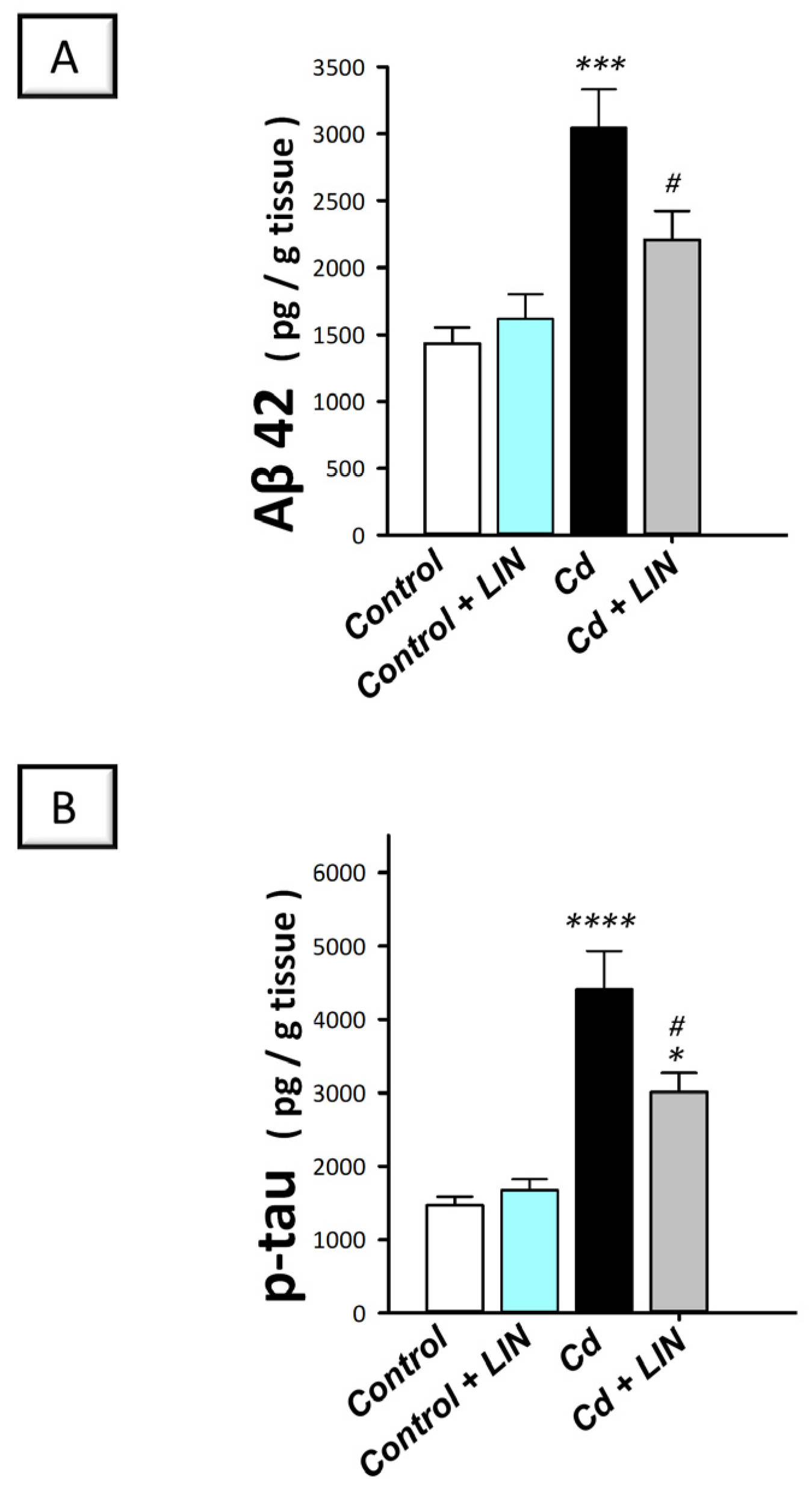
2.3. Linagliptin Lowers the Expression of the Neurotoxic Aβ42 and p-Tau in the Hippocampi of Cadmium-Intoxicated Rats
2.4. Linagliptin Augments Acetylcholine and GABA Neurotransmitters and Suppresses Glutamate and Acetylcholine Esterase in the Hippocampi of Cadmium-Intoxicated Rats
2.5. Linagliptin Attenuates the Pro-Oxidant Insult and Stimulates Hippocampal SIRT1/Nrf2/HO-1 Axis in Cadmium-Intoxicated Rats
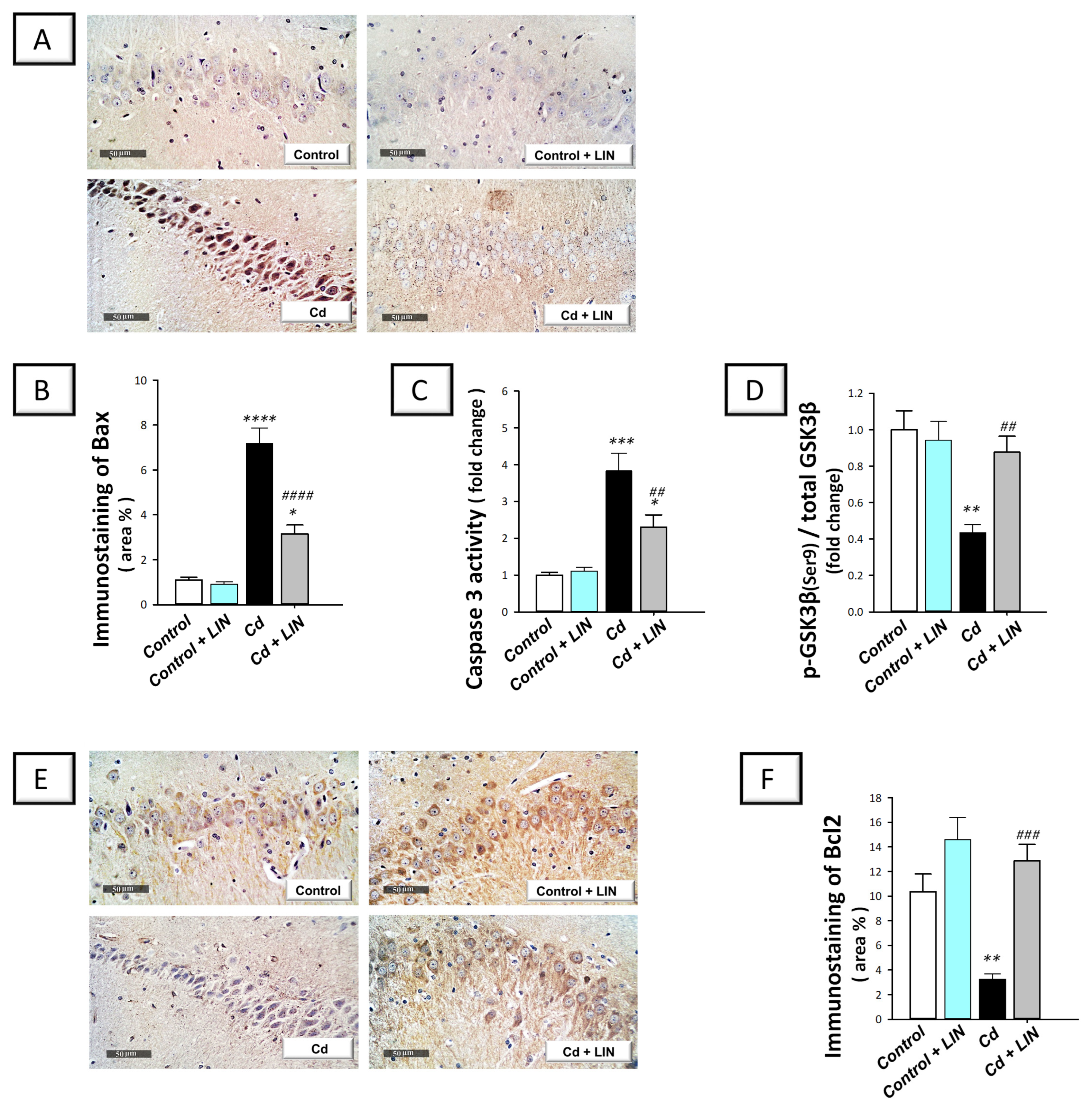
2.6. Linagliptin Counteracts Neuronal Apoptosis in the Hippocampi of Cadmium-Intoxicated Rats
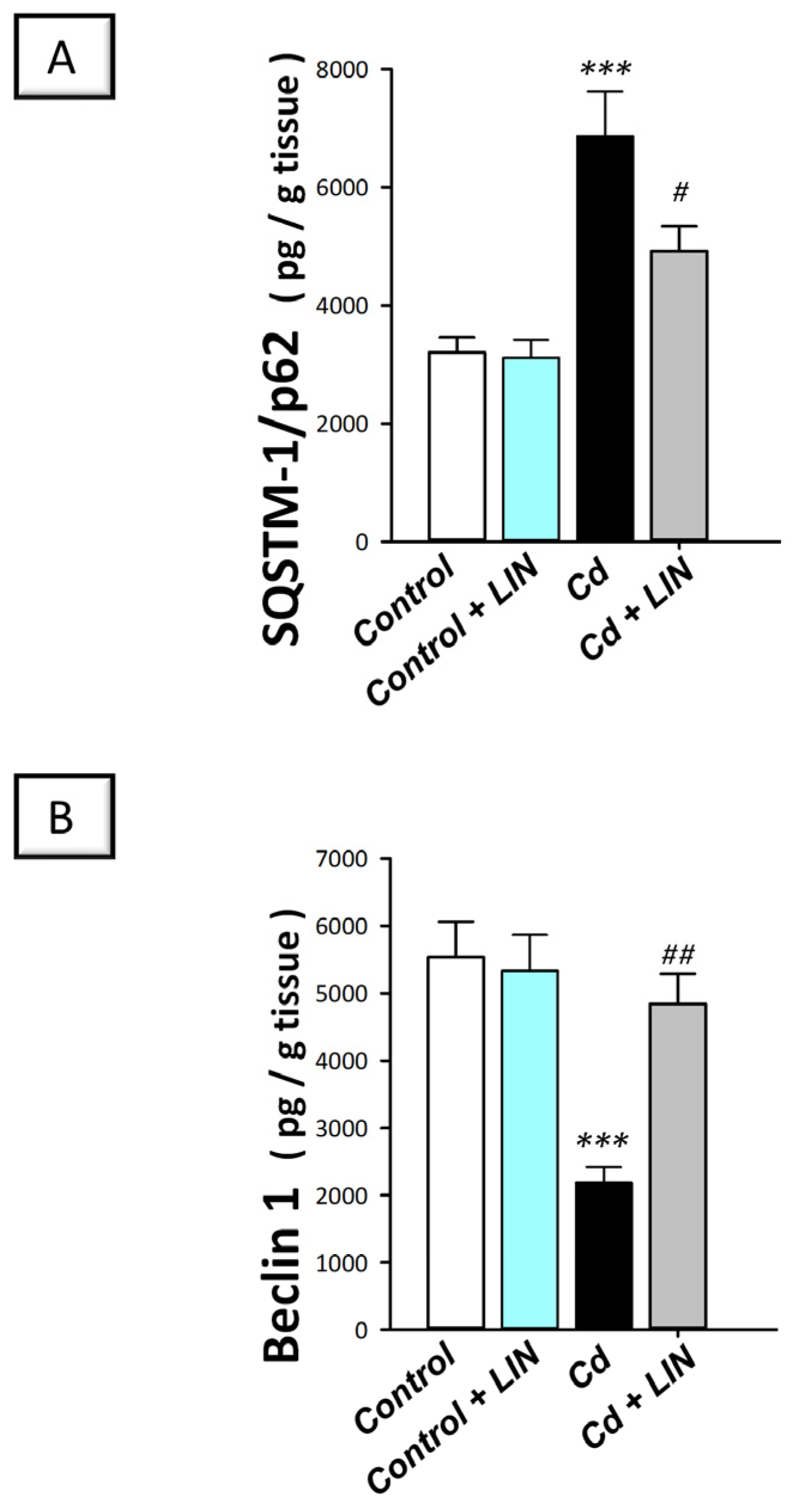
2.7. Linagliptin Enhances the Pro-Autophagy Events in the Hippocampi of Cadmium-Intoxicated Rats
2.8. Linagliptin Stimulates Hippocampal AMPK/mTOR Pathway in Cadmium-Intoxicated Rats
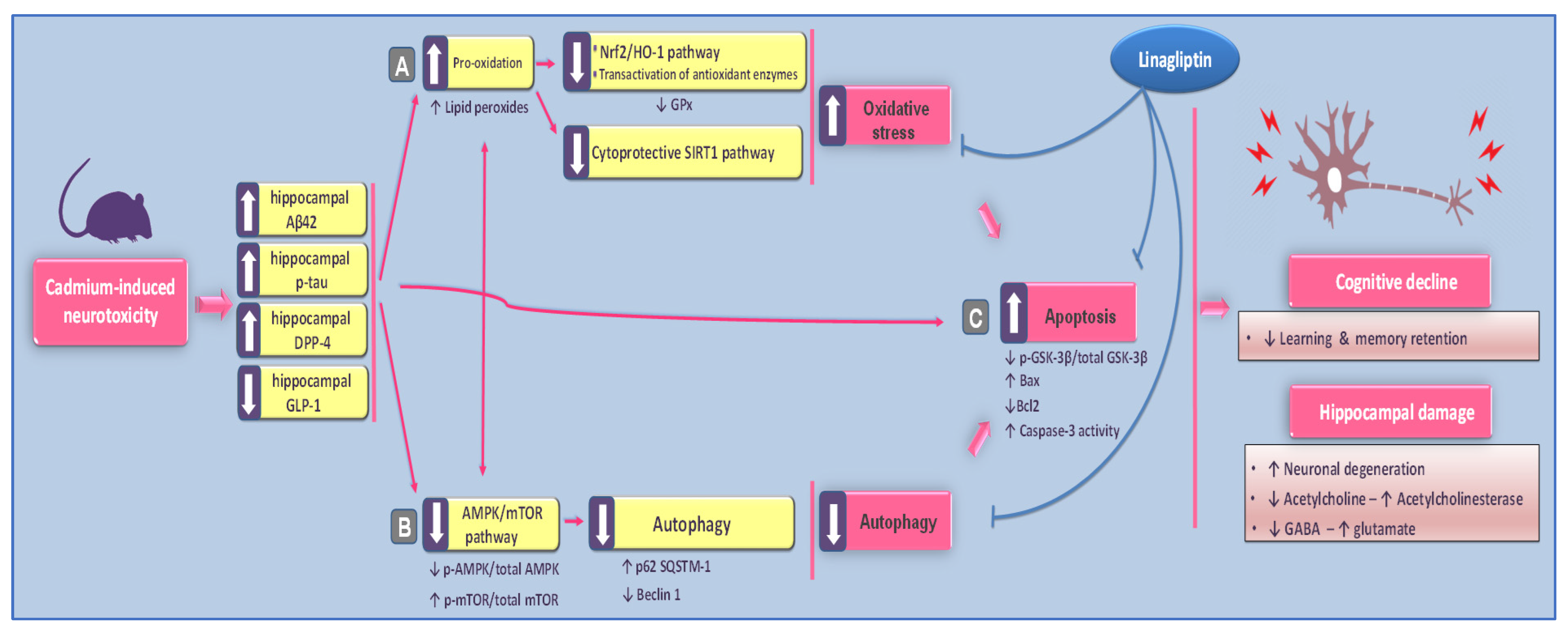
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Rats, Housing, and Ethical Statement
4.3. Establishing Preclinical Animal Model and Experimental Design
4.4. Evaluation of the Cognitive Function (Learning and Memory Acquisition)
4.4.1. Morris Water Maze (MWM)
4.4.2. Y-Maze Test
4.4.3. Novel Object Recognition Test (NORT)
4.5. Collecting the Brain Tissue
4.6. Serum Glucose and Hippocampal DPP-4 and GLP-1
4.7. Histopathology
4.8. Immunohistochemistry
4.9. Measurement of Hippocampal Aβ42, p-Tau, and Neurotransmitters
4.10. Markers of Autophagy and Apoptosis
4.11. Redox Milieu
4.12. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimers Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Khan, A.; Alam, S.I.; Ahmad, S.; Ikram, M.; Park, J.S.; Lee, H.J.; Kim, M.O. Cadmium, an Environmental Contaminant, Exacerbates Alzheimer’s Pathology in the Aged Mice’s Brain. Front. Aging Neurosci. 2021, 13, 650930. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Song, X.; Li, F.; Tan, X.; Sun-Waterhouse, D.; Li, D. Caffeic acid phenethyl ester reversed cadmium-induced cell death in hippocampus and cortex and subsequent cognitive disorders in mice: Involvements of AMPK/SIRT1 pathway and amyloid-tau-neuroinflammation axis. Food Chem. Toxicol. 2020, 144, 111636. [Google Scholar] [CrossRef]
- Tang, K.K.; Liu, X.Y.; Wang, Z.Y.; Qu, K.C.; Fan, R.F. Trehalose alleviates cadmium-induced brain damage by ameliorating oxidative stress, autophagy inhibition, and apoptosis. Metallomics 2019, 11, 2043–2051. [Google Scholar] [CrossRef]
- Ye, F.; Wu, A. The Protective Mechanism of SIRT1 in the Regulation of Mitochondrial Biogenesis and Mitochondrial Autophagy in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 149–157. [Google Scholar] [CrossRef]
- Rana, S.V. Metals and apoptosis: Recent developments. J. Trace Elem. Med. Biol. 2008, 22, 262–284. [Google Scholar] [CrossRef]
- Pi, H.; Li, M.; Tian, L.; Yang, Z.; Yu, Z.; Zhou, Z. Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci. Rep. 2017, 7, 43466. [Google Scholar] [CrossRef]
- Zhang, F.; Xing, S.; Li, Z. Antagonistic effects of lycopene on cadmium-induced hippocampal dysfunctions in autophagy, calcium homeostatis and redox. Oncotarget 2017, 8, 44720–44731. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Q.; Song, R.; Zhang, Y.; Yang, J.; Wang, Y.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; et al. Cadmium induced inhibition of autophagy is associated with microtubule disruption and mitochondrial dysfunction in primary rat cerebral cortical neurons. Neurotoxicol. Teratol. 2016, 53, 11–18. [Google Scholar] [CrossRef]
- Xu, C.; Chen, S.; Xu, M.; Chen, X.; Wang, X.; Zhang, H.; Dong, X.; Zhang, R.; Chen, X.; Gao, W.; et al. Cadmium Impairs Autophagy Leading to Apoptosis by Ca(2+)-Dependent Activation of JNK Signaling Pathway in Neuronal Cells. Neurochem. Res. 2021, 46, 2033–2045. [Google Scholar] [CrossRef]
- Querfurth, H.; Lee, H.K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 2021, 16, 44. [Google Scholar] [CrossRef]
- Ide, M.; Sonoda, N.; Inoue, T.; Kimura, S.; Minami, Y.; Makimura, H.; Hayashida, E.; Hyodo, F.; Yamato, M.; Takayanagi, R.; et al. The dipeptidyl peptidase-4 inhibitor, linagliptin, improves cognitive impairment in streptozotocin-induced diabetic mice by inhibiting oxidative stress and microglial activation. PLoS ONE 2020, 15, e0228750. [Google Scholar] [CrossRef]
- Siddiqui, N.; Ali, J.; Parvez, S.; Zameer, S.; Najmi, A.K.; Akhtar, M. Linagliptin, a DPP-4 inhibitor, ameliorates Abeta (1-42) peptides induced neurodegeneration and brain insulin resistance (BIR) via insulin receptor substrate-1 (IRS-1) in rat model of Alzheimer’s disease. Neuropharmacology 2021, 195, 108662. [Google Scholar] [CrossRef]
- Arab, H.H.; Eid, A.H.; Mahmoud, A.M.; Senousy, M.A. Linagliptin mitigates experimental inflammatory bowel disease in rats by targeting inflammatory and redox signaling. Life Sci. 2021, 273, 119295. [Google Scholar] [CrossRef]
- Tsuprykov, O.; Ando, R.; Reichetzeder, C.; von Websky, K.; Antonenko, V.; Sharkovska, Y.; Chaykovska, L.; Rahnenführer, J.; Hasan, A.A.; Tammen, H. The dipeptidyl peptidase inhibitor linagliptin and the angiotensin II receptor blocker telmisartan show renal benefit by different pathways in rats with 5/6 nephrectomy. Kidney Int. 2016, 89, 1049–1061. [Google Scholar] [CrossRef]
- Arab, H.H.; Elhemiely, A.A.; El-Sheikh, A.A.K.; Khabbaz, H.J.A.; Arafa, E.A.; Ashour, A.M.; Kabel, A.M.; Eid, A.H. Repositioning Linagliptin for the Mitigation of Cadmium-Induced Testicular Dysfunction in Rats: Targeting HMGB1/TLR4/NLRP3 Axis and Autophagy. Pharmaceuticals 2022, 15, 852. [Google Scholar] [CrossRef]
- Ma, M.; Hasegawa, Y.; Koibuchi, N.; Toyama, K.; Uekawa, K.; Nakagawa, T.; Lin, B.; Kim-Mitsuyama, S. DPP-4 inhibition with linagliptin ameliorates cognitive impairment and brain atrophy induced by transient cerebral ischemia in type 2 diabetic mice. Cardiovasc. Diabetol. 2015, 14, 54. [Google Scholar] [CrossRef]
- Nakaoku, Y.; Saito, S.; Yamamoto, Y.; Maki, T.; Takahashi, R.; Ihara, M. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice. Int. J. Mol. Sci. 2019, 20, 2539. [Google Scholar] [CrossRef]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef]
- Shannon, R.P. DPP-4 inhibition and neuroprotection: Do mechanisms matter? Diabetes 2013, 62, 1029–1031. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.L.; Peng, C.H.; Huang, C.N. DPP-4 Inhibitor Linagliptin Attenuates Abeta-induced Cytotoxicity through Activation of AMPK in Neuronal Cells. CNS Neurosci. Ther. 2015, 21, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Górski, K.; Walczak, M.; Wódkiewicz, E.; Słupski, M.; Pawlak-Osińska, K.; Malinowski, B. Neuroprotective Properties of Linagliptin: Focus on Biochemical Mechanisms in Cerebral Ischemia, Vascular Dysfunction and Certain Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4052. [Google Scholar] [CrossRef] [PubMed]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef] [PubMed]
- Mima, A.; Yasuzawa, T.; Nakamura, T.; Ueshima, S. Linagliptin affects IRS1/Akt signaling and prevents high glucose-induced apoptosis in podocytes. Sci. Rep. 2020, 10, 5775. [Google Scholar] [CrossRef]
- Kumar, P.; Raman, T.; Swain, M.M.; Mishra, R.; Pal, A. Hyperglycemia-Induced Oxidative-Nitrosative Stress Induces Inflammation and Neurodegeneration via Augmented Tuberous Sclerosis Complex-2 (TSC-2) Activation in Neuronal Cells. Mol. Neurobiol. 2017, 54, 238–254. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Hale, G.; Good, M. Impaired visuospatial recognition memory but normal object novelty detection and relative familiarity judgments in adult mice expressing the APPswe Alzheimer’s disease mutation. Behav. Neurosci. 2005, 119, 884–891. [Google Scholar] [CrossRef]
- Hussien, H.M.; Abd-Elmegied, A.; Ghareeb, D.A.; Hafez, H.S.; Ahmed, H.E.A.; El-Moneam, N.A. Neuroprotective effect of berberine against environmental heavy metals-induced neurotoxicity and Alzheimer’s-like disease in rats. Food Chem. Toxicol. 2018, 111, 432–444. [Google Scholar] [CrossRef]
- Vereecken, T.H.; Vogels, O.J.; Nieuwenhuys, R. Neuron loss and shrinkage in the amygdala in Alzheimer’s disease. Neurobiol. Aging 1994, 15, 45–54. [Google Scholar] [CrossRef]
- Zhang, Z.; Miah, M.; Culbreth, M.; Aschner, M. Autophagy in Neurodegenerative Diseases and Metal Neurotoxicity. Neurochem. Res. 2016, 41, 409–422. [Google Scholar] [CrossRef]
- Moyano, P.; de Frias, M.; Lobo, M.; Anadon, M.J.; Sola, E.; Pelayo, A.; Diaz, M.J.; Frejo, M.T.; Del Pino, J. Cadmium induced ROS alters M1 and M3 receptors, leading to SN56 cholinergic neuronal loss, through AChE variants disruption. Toxicology 2018, 394, 54–62. [Google Scholar] [CrossRef]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid ss peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef]
- Safar, M.M.; Arab, H.H.; Rizk, S.M.; El-Maraghy, S.A. Bone Marrow-Derived Endothelial Progenitor Cells Protect Against Scopolamine-Induced Alzheimer-Like Pathological Aberrations. Mol. Neurobiol. 2016, 53, 1403–1418. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Ma, L.; Dong, W.; Wang, R.; Li, Y.; Xu, B.; Zhang, J.; Zhao, Z.; Wang, Y. Effect of caloric restriction on the SIRT1/mTOR signaling pathways in senile mice. Brain Res. Bull. 2015, 116, 67–72. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Back to Nucleus: Combating with Cadmium Toxicity Using Nrf2 Signaling Pathway as a Promising Therapeutic Target. Biol. Trace Elem. Res. 2020, 197, 52–62. [Google Scholar] [CrossRef]
- Majd, S.; Power, J.H.; Grantham, H.J. Neuronal response in Alzheimer’s and Parkinson’s disease: The effect of toxic proteins on intracellular pathways. BMC Neurosci. 2015, 16, 69. [Google Scholar] [CrossRef]
- Del Pino, J.; Zeballos, G.; Anadon, M.J.; Moyano, P.; Diaz, M.J.; Garcia, J.M.; Frejo, M.T. Cadmium-induced cell death of basal forebrain cholinergic neurons mediated by muscarinic M1 receptor blockade, increase in GSK-3beta enzyme, beta-amyloid and tau protein levels. Arch. Toxicol. 2016, 90, 1081–1092. [Google Scholar] [CrossRef]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, X.; Zhao, R.; Zhang, R.; Xu, C.; Wang, X.; Liu, C.; Hu, X.; Huang, S.; Chen, L. Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell Signal. 2019, 55, 26–39. [Google Scholar] [CrossRef]
- Charan, J.; Kantharia, N. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 2013, 4, 303–306. [Google Scholar] [CrossRef]
- El-Kott, A.F.; Bin-Meferij, M.M.; Eleawa, S.M.; Alshehri, M.M. Kaempferol Protects Against Cadmium Chloride-Induced Memory Loss and Hippocampal Apoptosis by Increased Intracellular Glutathione Stores and Activation of PTEN/AMPK Induced Inhibition of Akt/mTOR Signaling. Neurochem. Res. 2020, 45, 295–309. [Google Scholar] [CrossRef]
- Arab, H.H.; Gad, A.M.; Reda, E.; Yahia, R.; Eid, A.H. Activation of autophagy by sitagliptin attenuates cadmium-induced testicular impairment in rats: Targeting AMPK/mTOR and Nrf2/HO-1 pathways. Life Sci. 2021, 269, 119031. [Google Scholar] [CrossRef]
- Weitzner, D.S.; Engler-Chiurazzi, E.B.; Kotilinek, L.A.; Ashe, K.H.; Reed, M.N. Morris Water Maze Test: Optimization for Mouse Strain and Testing Environment. J. Vis. Exp. 2015, 100, e52706. [Google Scholar] [CrossRef]
- Kraeuter, A.-K.; Guest, P.C.; Sarnyai, Z. The Y-maze for assessment of spatial working and reference memory in mice. In Pre-clinical Models; Springer: Berlin/Heidelberg, Germany, 2019; pp. 105–111. [Google Scholar]
- Arab, H.H.; Khames, A.; Mohammad, M.K.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Gad, A.M. Meloxicam Targets COX-2/NOX1/NOX4/Nrf2 Axis to Ameliorate the Depression-like Neuropathology Induced by Chronic Restraint Stress in Rats. Pharmaceuticals 2023, 16, 848. [Google Scholar] [CrossRef]
- Arab, H.H.; Al-Shorbagy, M.Y.; Saad, M.A. Activation of autophagy and suppression of apoptosis by dapagliflozin attenuates experimental inflammatory bowel disease in rats: Targeting AMPK/mTOR, HMGB1/RAGE and Nrf2/HO-1 pathways. Chem. Biol. Interact. 2021, 335, 109368. [Google Scholar] [CrossRef]
- Thoresen, M.; Bagenholm, R.; Loberg, E.M.; Apricena, F.; Kjellmer, I. Posthypoxic cooling of neonatal rats provides protection against brain injury. Arch. Dis. Child. Fetal Neonatal Ed. 1996, 74, F3–F9. [Google Scholar] [CrossRef]
- Muhammad, R.N.; Ahmed, L.A.; Abdul Salam, R.M.; Ahmed, K.A.; Attia, A.S. Crosstalk Among NLRP3 Inflammasome, ET(B)R Signaling, and miRNAs in Stress-Induced Depression-Like Behavior: A Modulatory Role for SGLT2 Inhibitors. Neurotherapeutics 2021, 18, 2664–2681. [Google Scholar] [CrossRef]
- Nassar, N.N.; Al-Shorbagy, M.Y.; Arab, H.H.; Abdallah, D.M. Saxagliptin: A novel antiparkinsonian approach. Neuropharmacology 2015, 89, 308–317. [Google Scholar] [CrossRef]
- Van de Berg, W.D.; Blokland, A.; Cuello, A.C.; Schmitz, C.; Vreuls, W.; Steinbusch, H.W.; Blanco, C.E. Perinatal asphyxia results in changes in presynaptic bouton number in striatum and cerebral cortex-a stereological and behavioral analysis. J. Chem. Neuroanat. 2000, 20, 71–82. [Google Scholar] [CrossRef]
- Arab, H.H.; Abd El Aal, H.A.; Alsufyani, S.E.; El-Sheikh, A.A.K.; Arafa, E.A.; Ashour, A.M.; Kabel, A.M.; Eid, A.H. Topiramate Reprofiling for the Attenuation of Cadmium-Induced Testicular Impairment in Rats: Role of NLRP3 Inflammasome and AMPK/mTOR-Linked Autophagy. Pharmaceuticals 2022, 15, 1402. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | Received |
|---|---|---|
| Control | 10 | Animals were given normal saline as the vehicle for cadmium chloride by gavage (10 mL/kg/day). Likewise, carboxymethyl cellulose (CMC; 0.5%) was given as linagliptin vehicle by gavage to animals. Each day, the 2 doses were separated by 2 h to avoid potential interaction. The treatments were received for 8 weeks. |
| Control + LIN | 10 | Animals were given saline (10 mL/kg/day). In addition, linagliptin (5 mg/kg/day; 10 mL/kg/day) was given by gavage. Each day, the 2 doses were separated by 2 h to avoid potential interaction. The treatments were received for 8 weeks. |
| Cd | 10 | Animals were given cadmium chloride solution by gavage (5 mg/kg/day; 10 mL/kg/day). In addition, animals received an oral gavage of CMC (10 mL/kg/day). Each day, the 2 doses were separated by 2 h to avoid potential interaction. The treatments were received for 8 weeks. The chosen regimen is in accordance with published studies [29,43,44]. |
| Cd + LIN | 10 | Animals were given cadmium chloride solution by gavage (5 mg/kg/day; 10 mL/kg/day). In addition, linagliptin (5 mg/kg/day; 10 mL/kg/day) was given by gavage. Each day, the 2 doses were separated by 2 h to avoid potential interaction. The treatments were received for 8 weeks. The selected linagliptin dose was based on literature that demonstrated such dose as effective for amelioration of streptozotocin-induced diabetic dementia [13] and high-fat-evoked cognitive deficit in PS19 transgenic mice [19]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Georgy, G.S. Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals 2023, 16, 1065. https://doi.org/10.3390/ph16081065
Arab HH, Eid AH, Alsufyani SE, Ashour AM, El-Sheikh AAK, Darwish HW, Georgy GS. Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals. 2023; 16(8):1065. https://doi.org/10.3390/ph16081065
Chicago/Turabian StyleArab, Hany H., Ahmed H. Eid, Shuruq E. Alsufyani, Ahmed M. Ashour, Azza A. K. El-Sheikh, Hany W. Darwish, and Gehan S. Georgy. 2023. "Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy" Pharmaceuticals 16, no. 8: 1065. https://doi.org/10.3390/ph16081065
APA StyleArab, H. H., Eid, A. H., Alsufyani, S. E., Ashour, A. M., El-Sheikh, A. A. K., Darwish, H. W., & Georgy, G. S. (2023). Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals, 16(8), 1065. https://doi.org/10.3390/ph16081065