

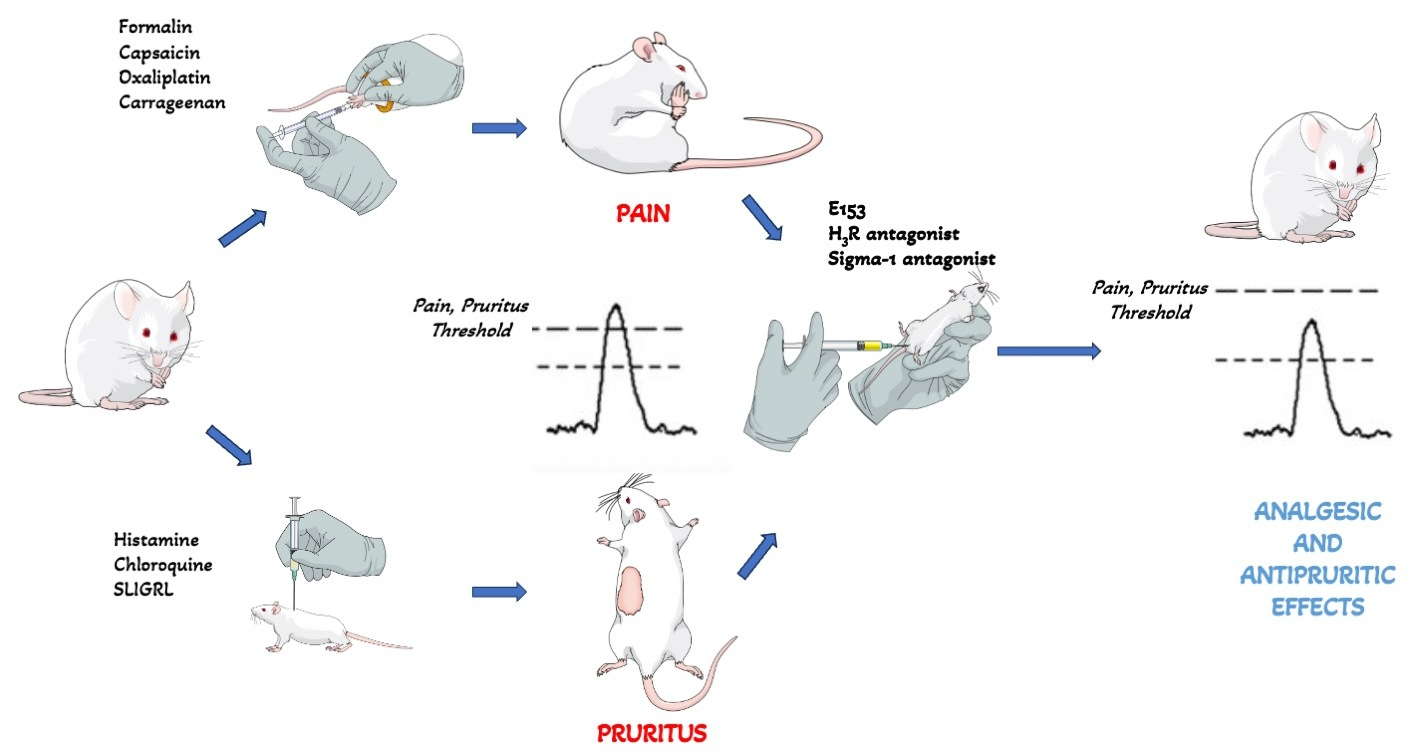
Efficacy of the Multi-Target Compound E153 in Relieving Pain and Pruritus of Different Origins
 , ,
, ,  ,
, 
Abstract
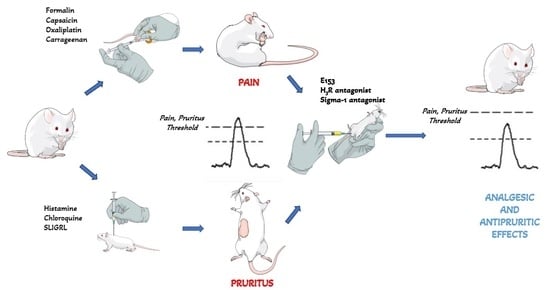
:
1. Introduction
2. Results
2.1. In Vitro Radioligand Binding Studies
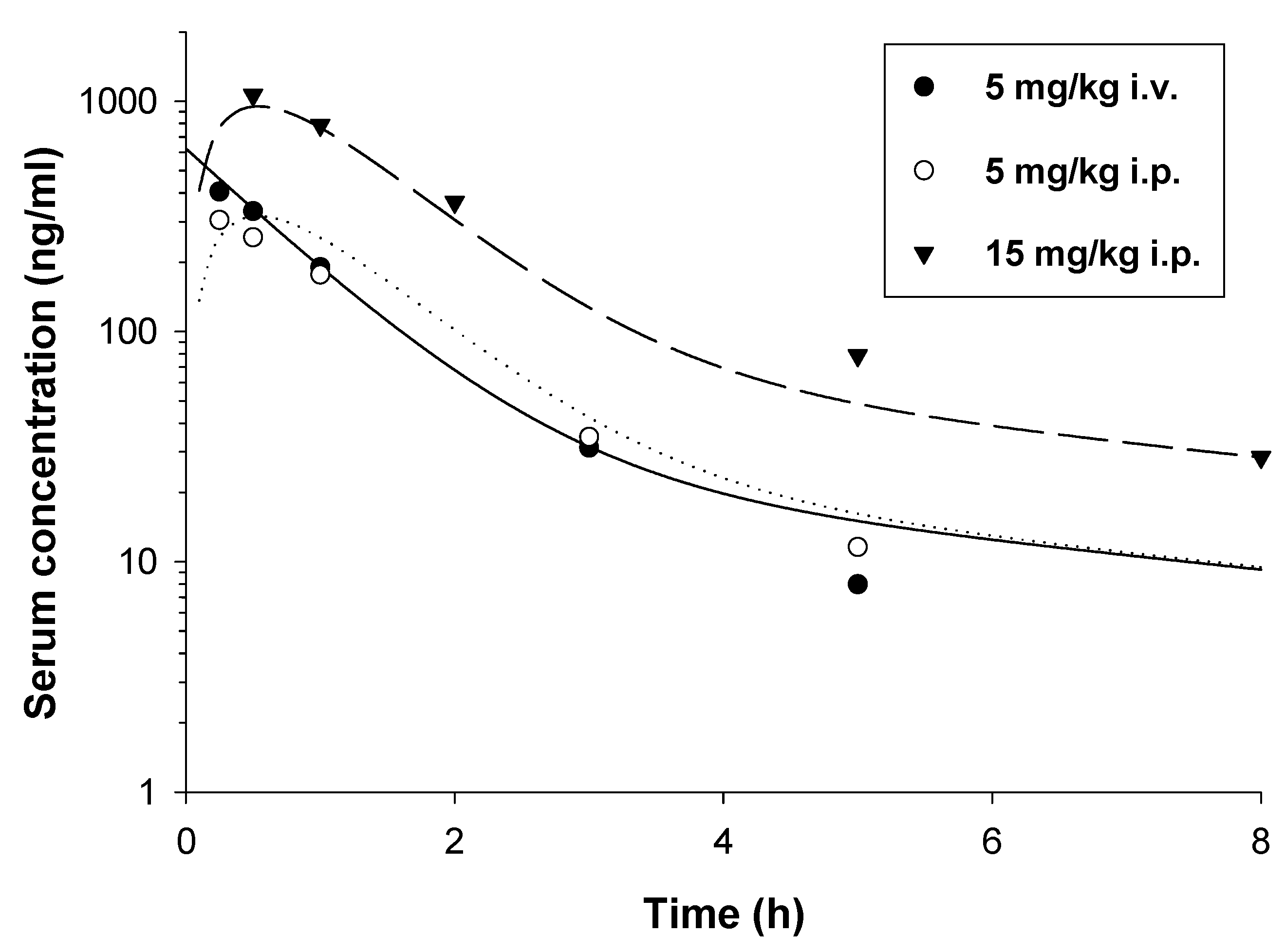
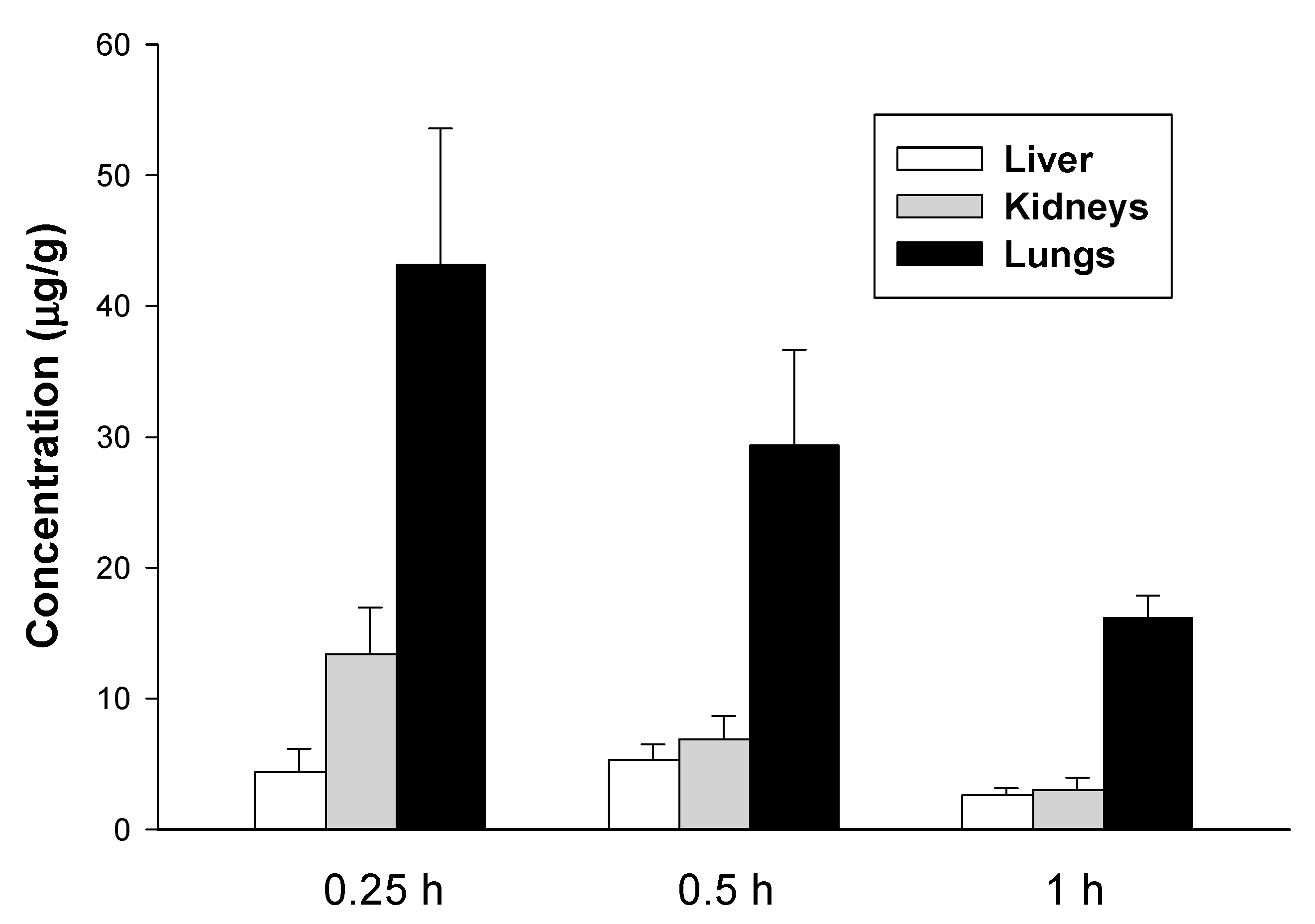
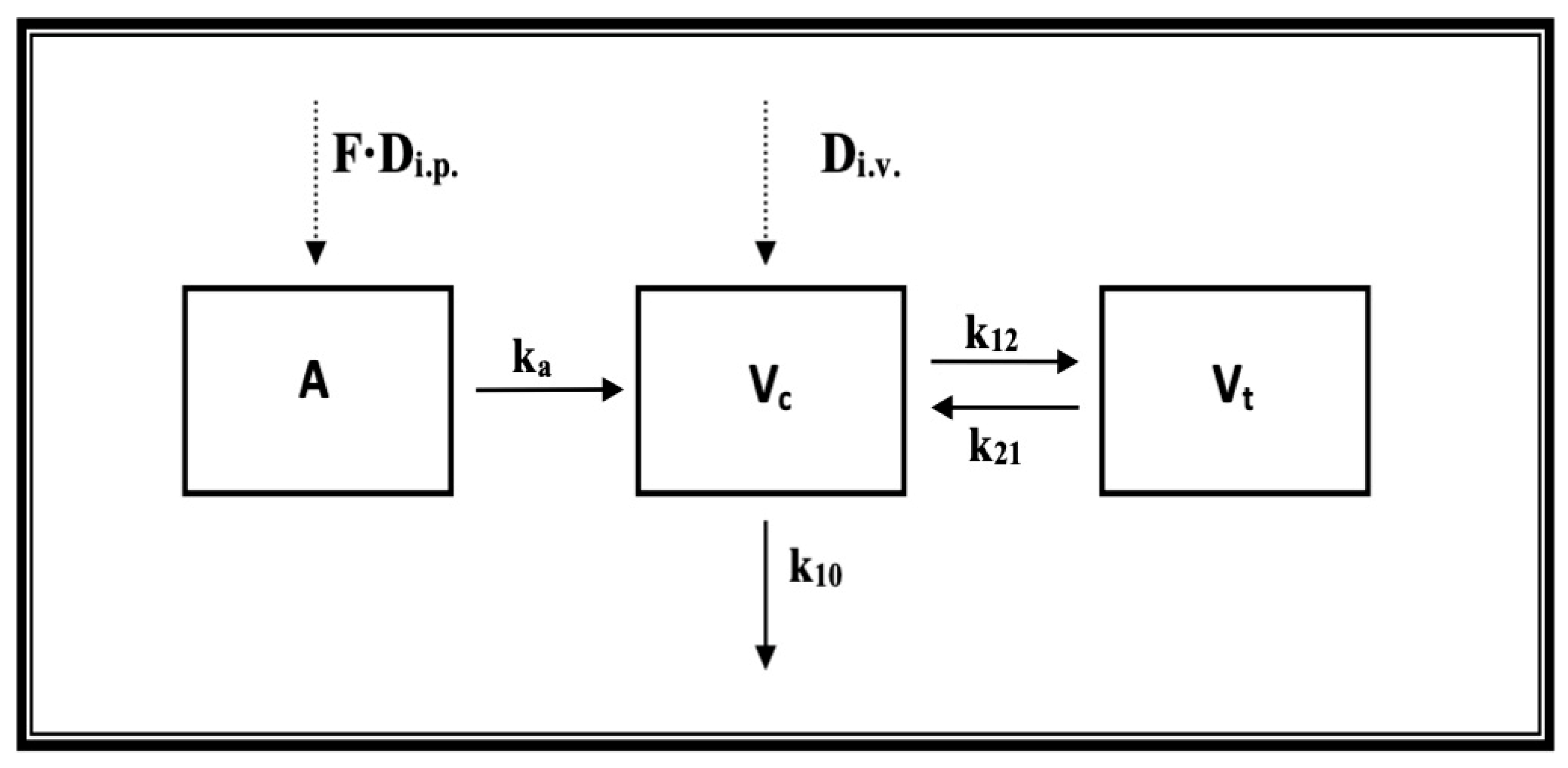
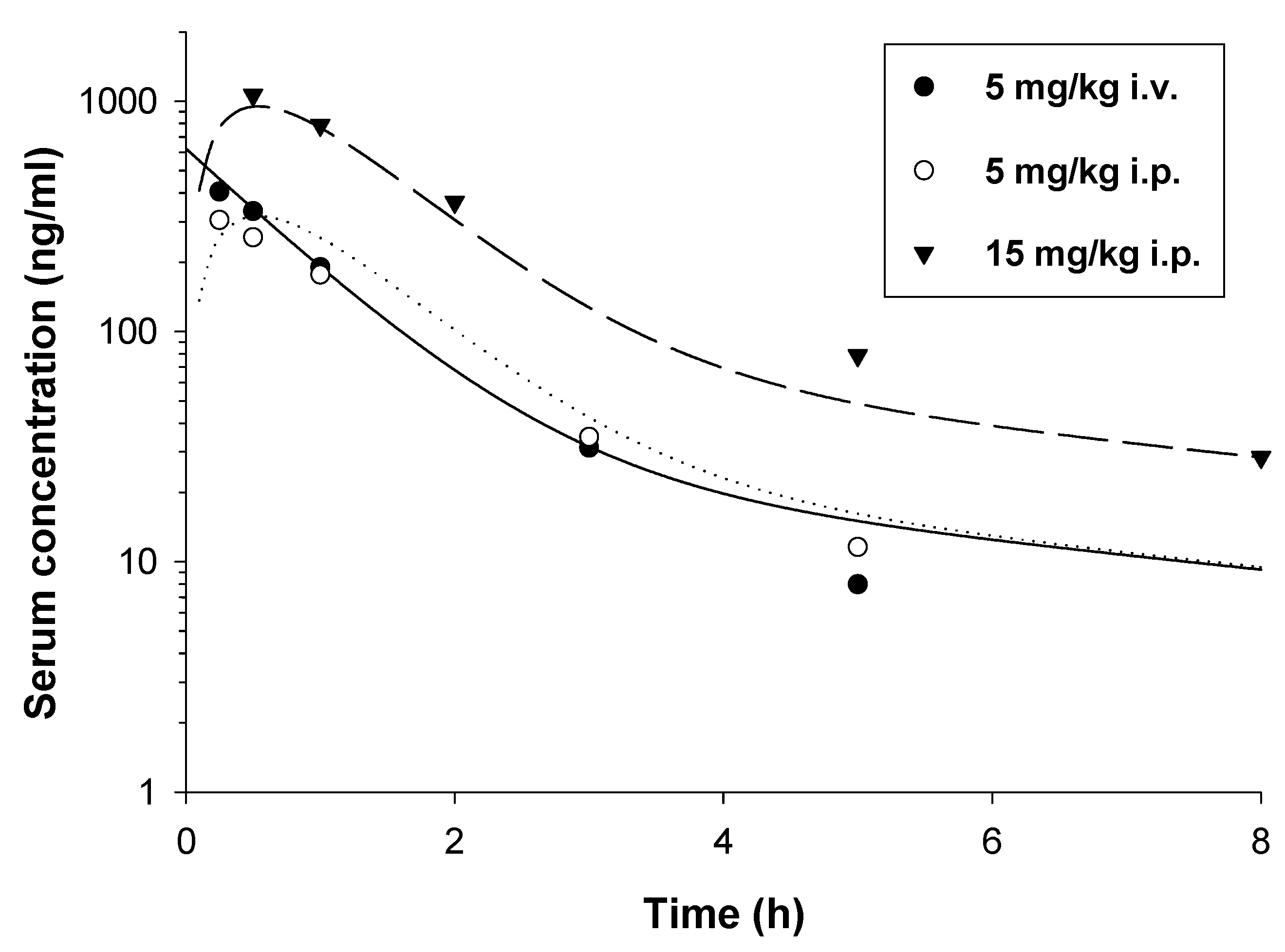
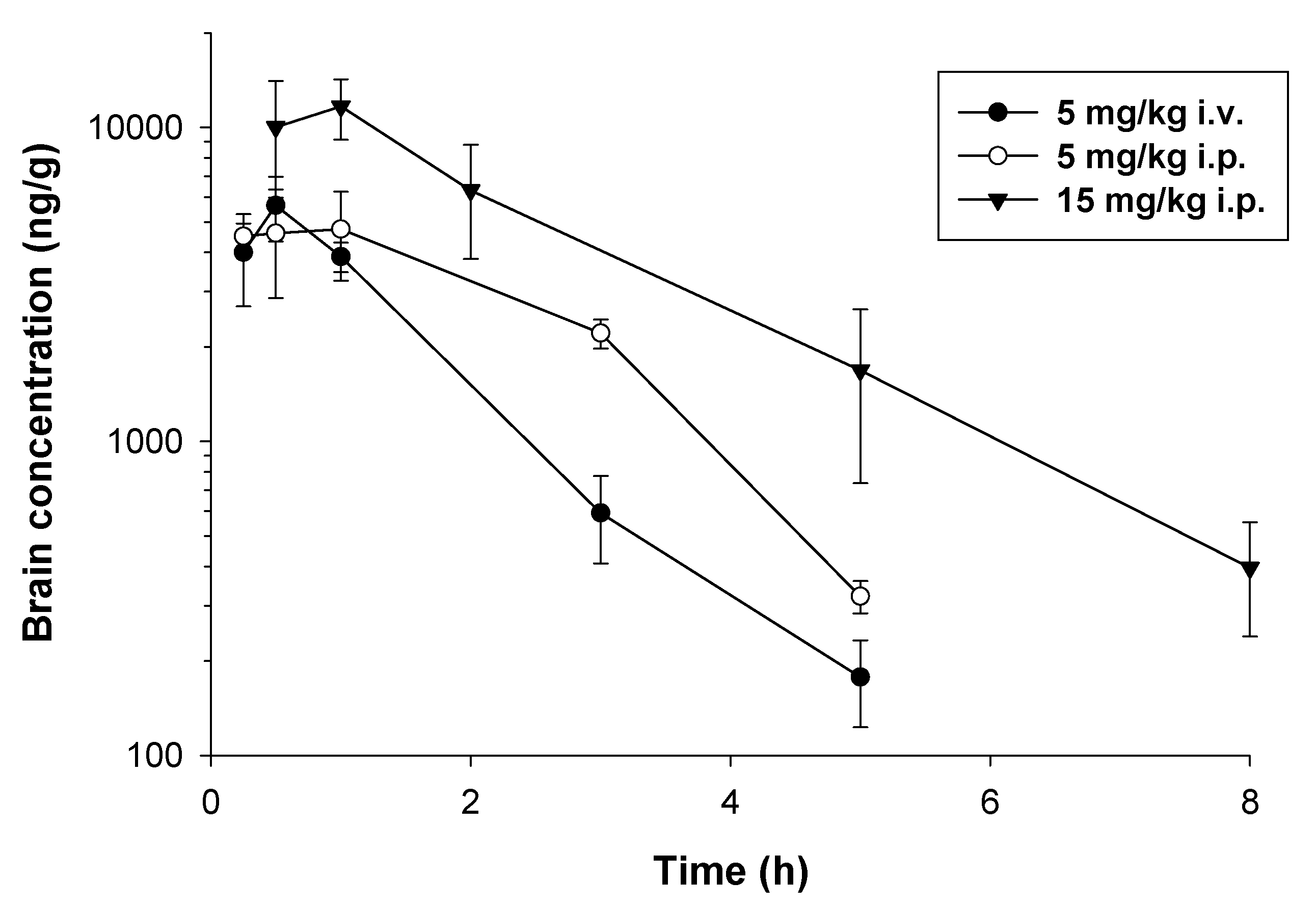
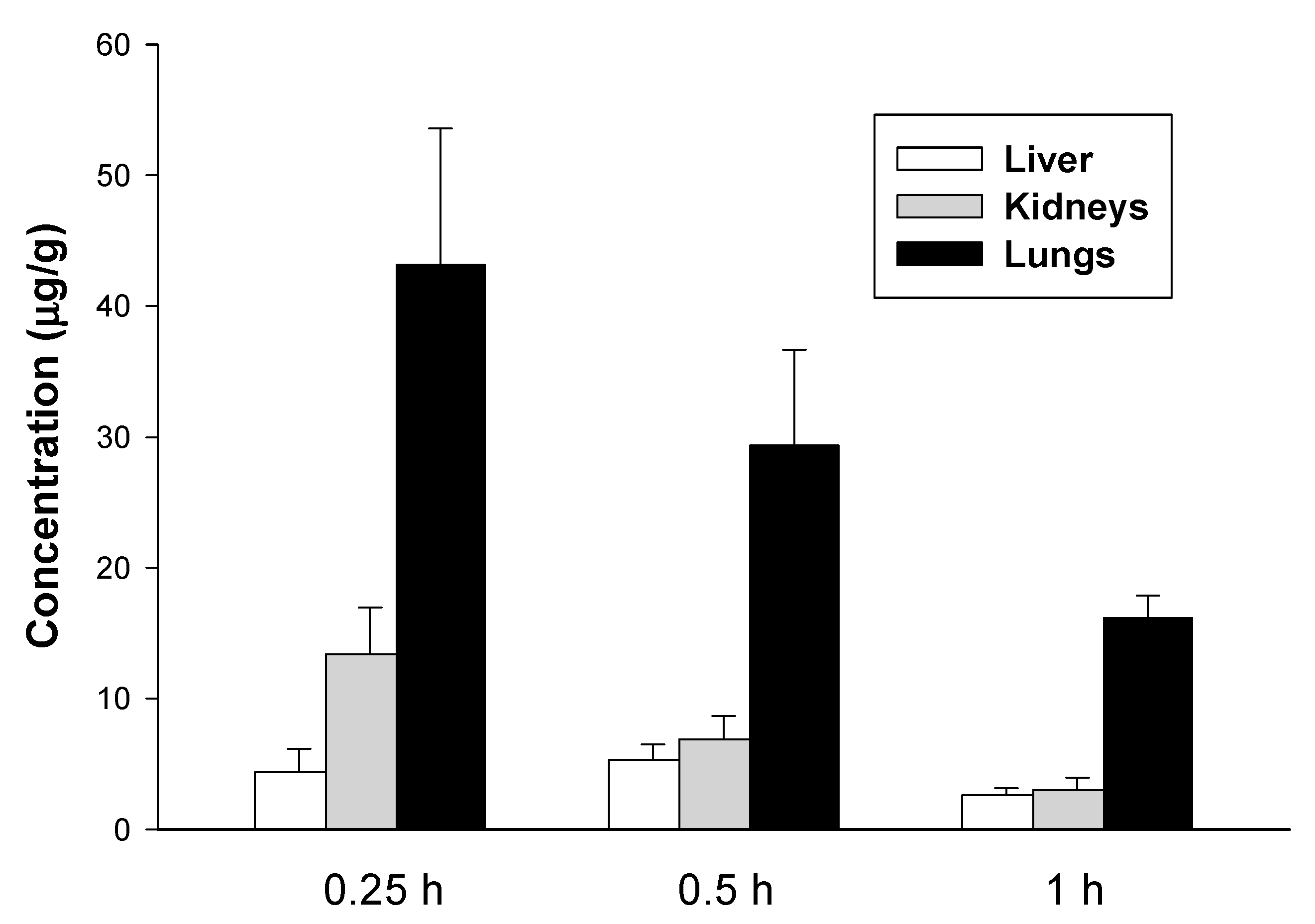
2.2. Pharmacokinetic Studies
2.3. In Vivo Pharmacological Studies
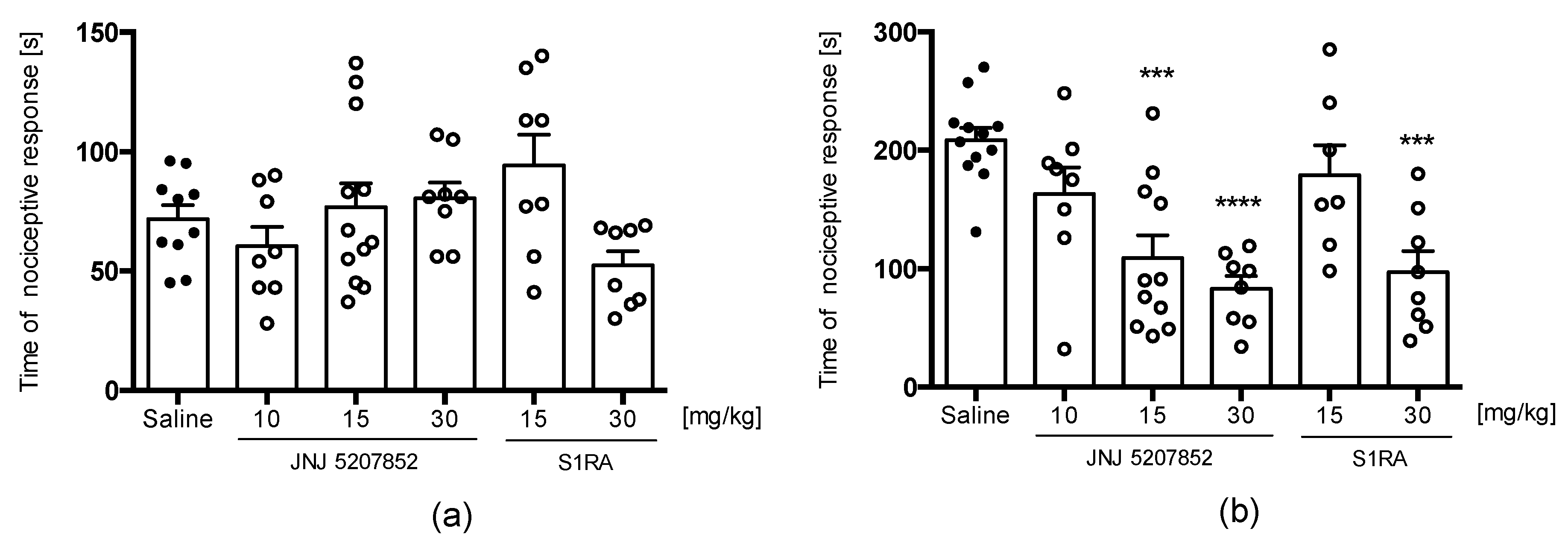
2.3.1. E153 Attenuates Acute, Inflammatory, and Neuropathic Pain
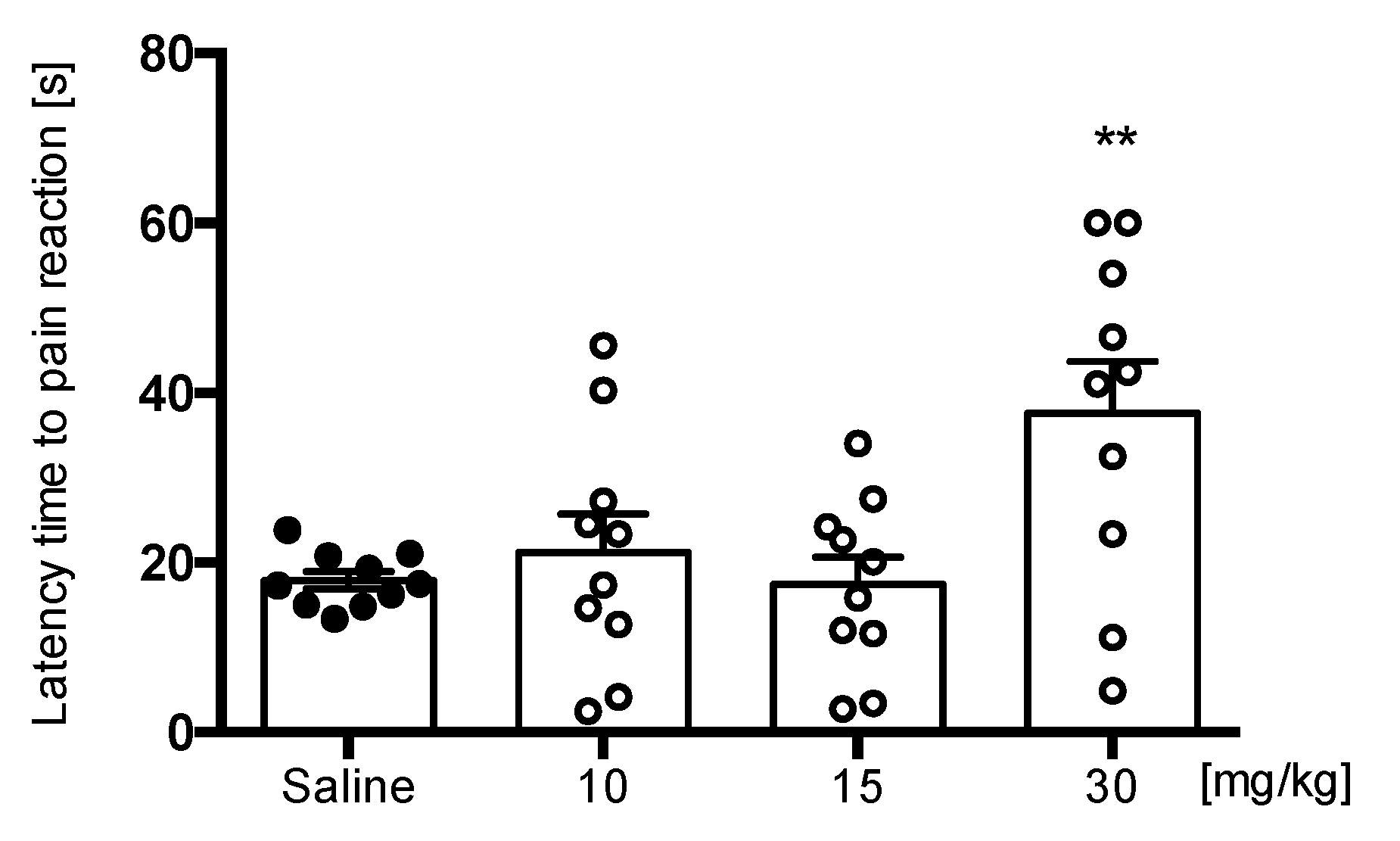
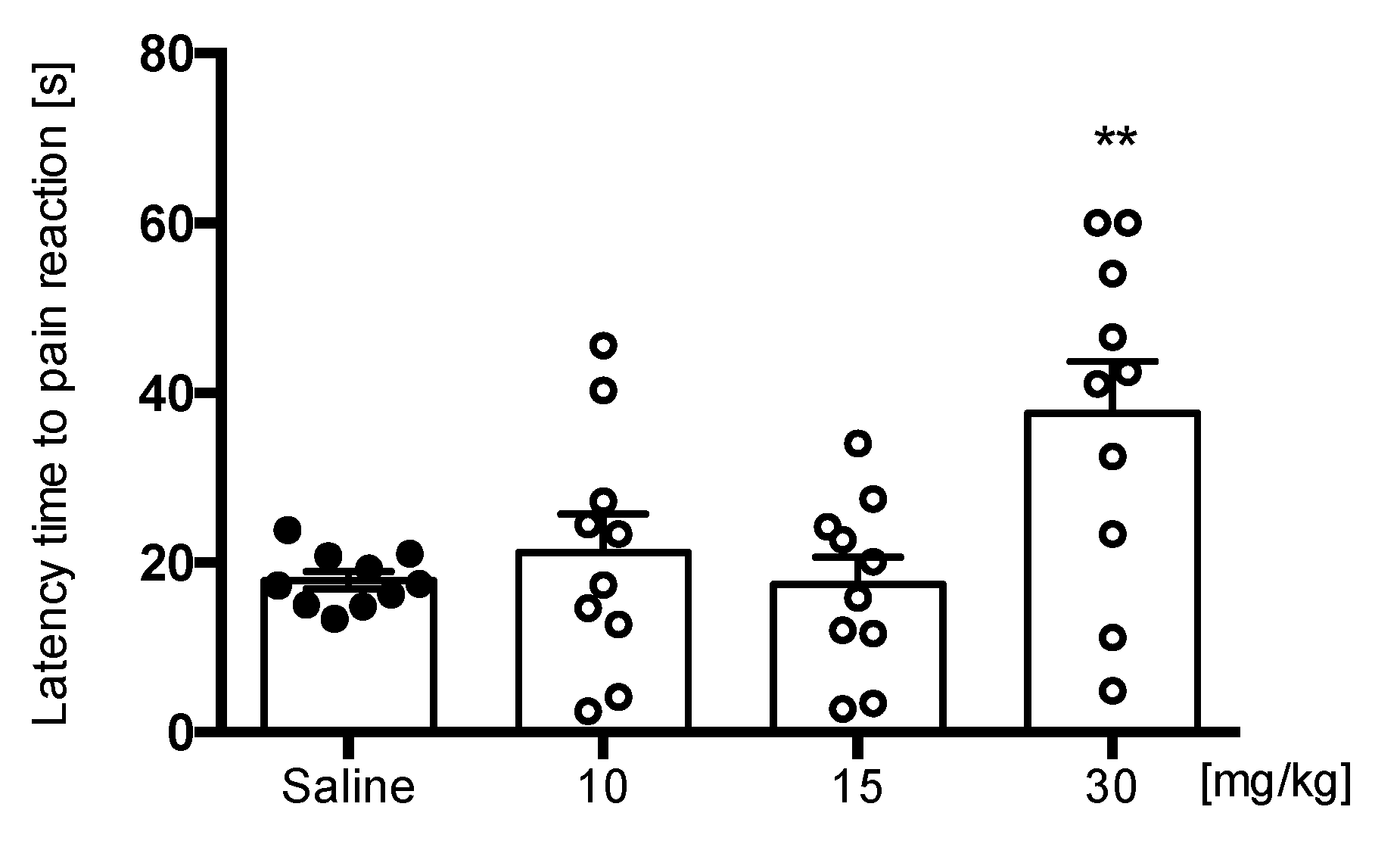
Hot Plate Test
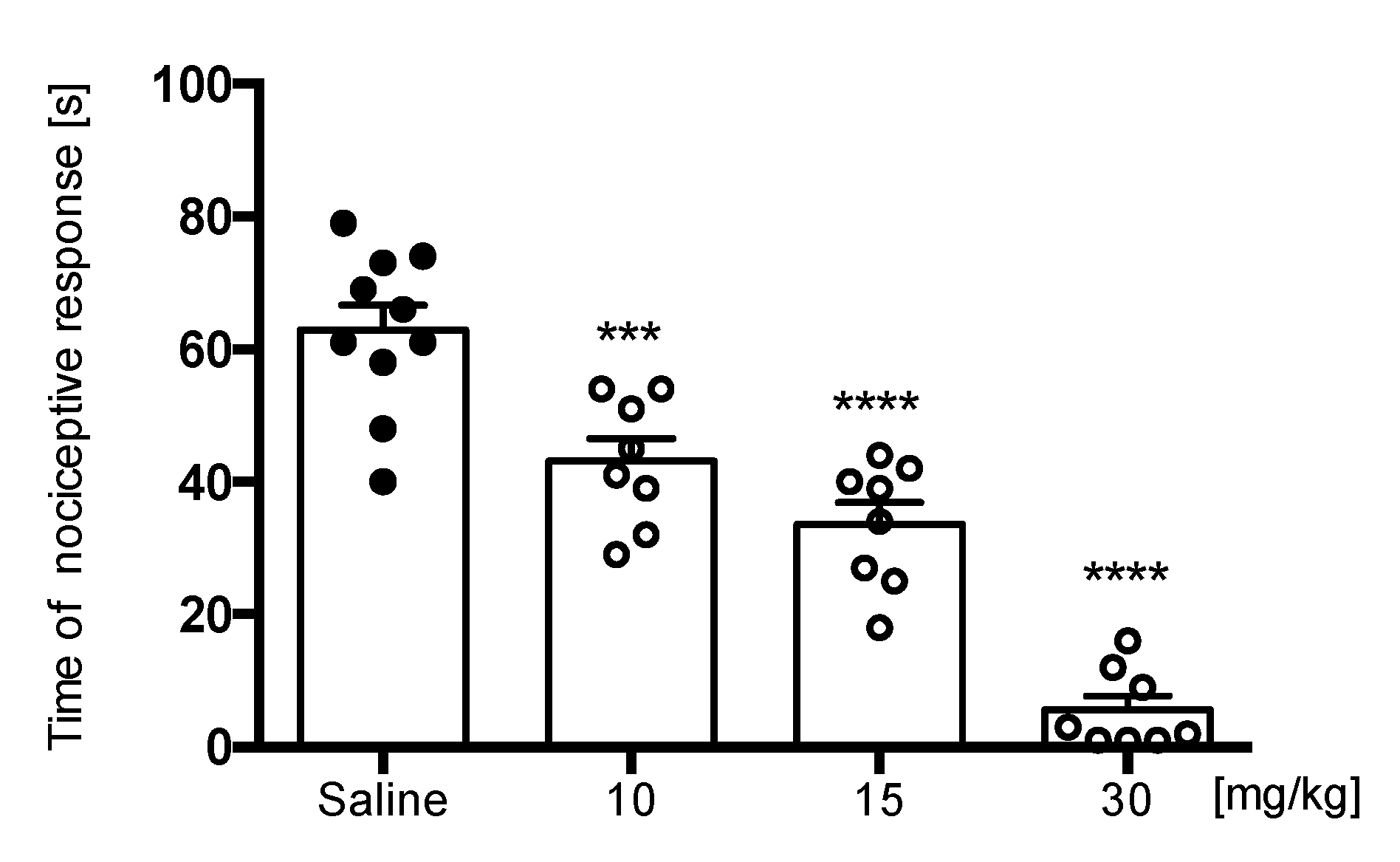
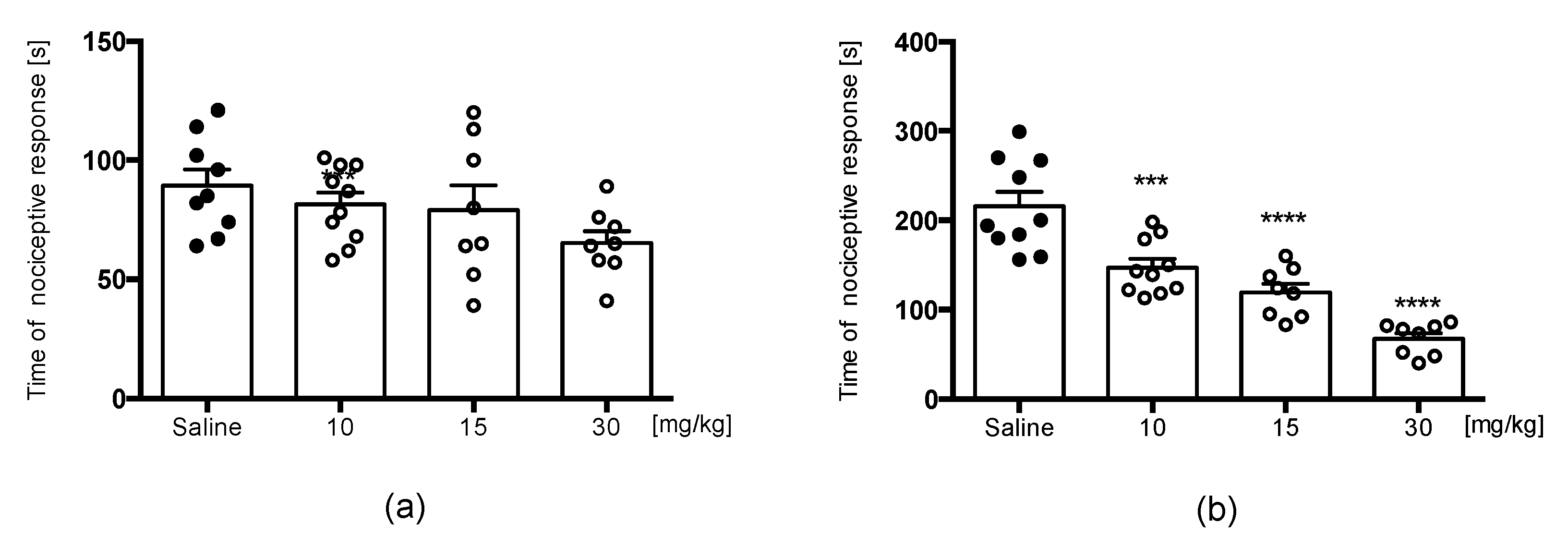
Formalin Test
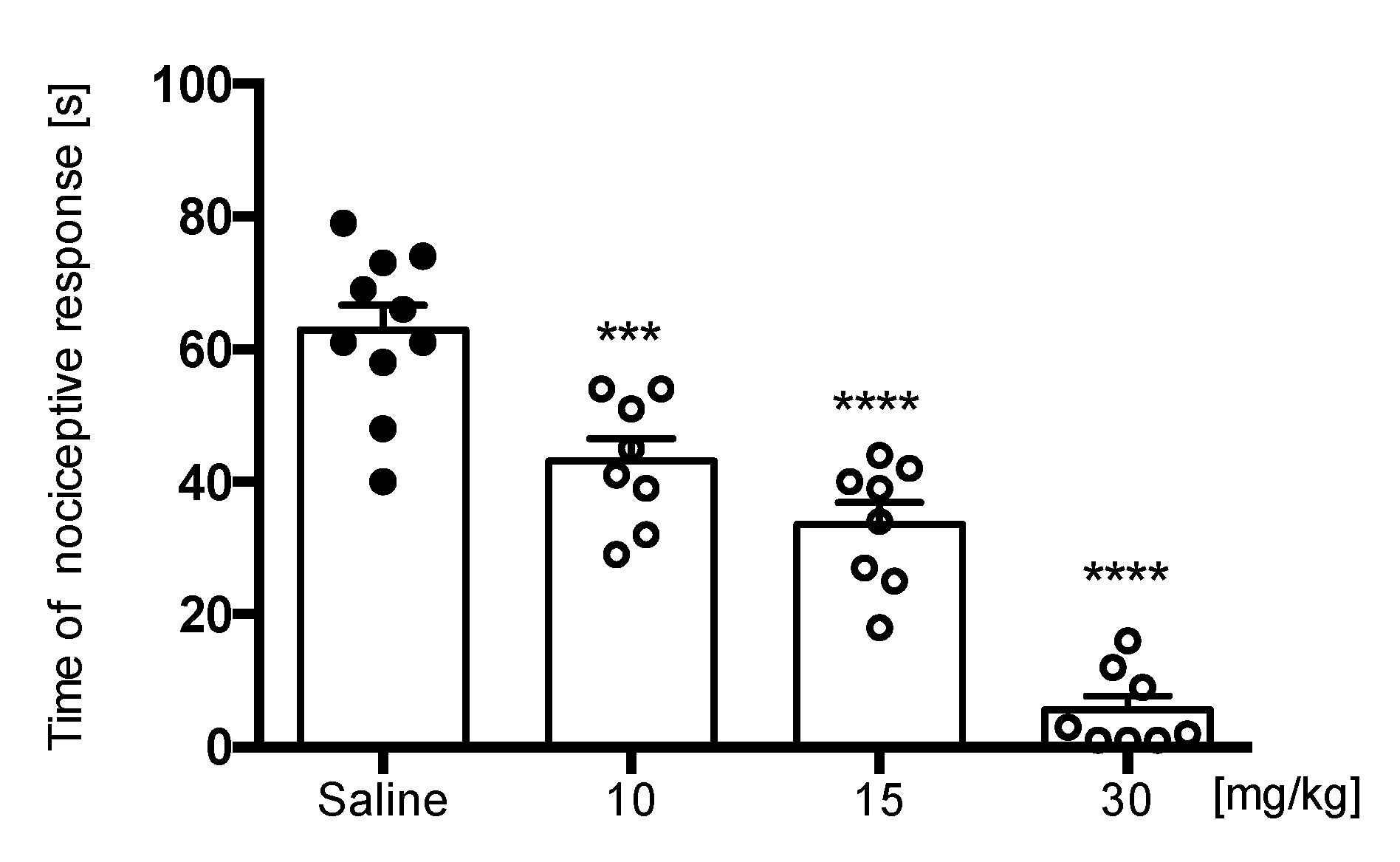
Capsaicin Test
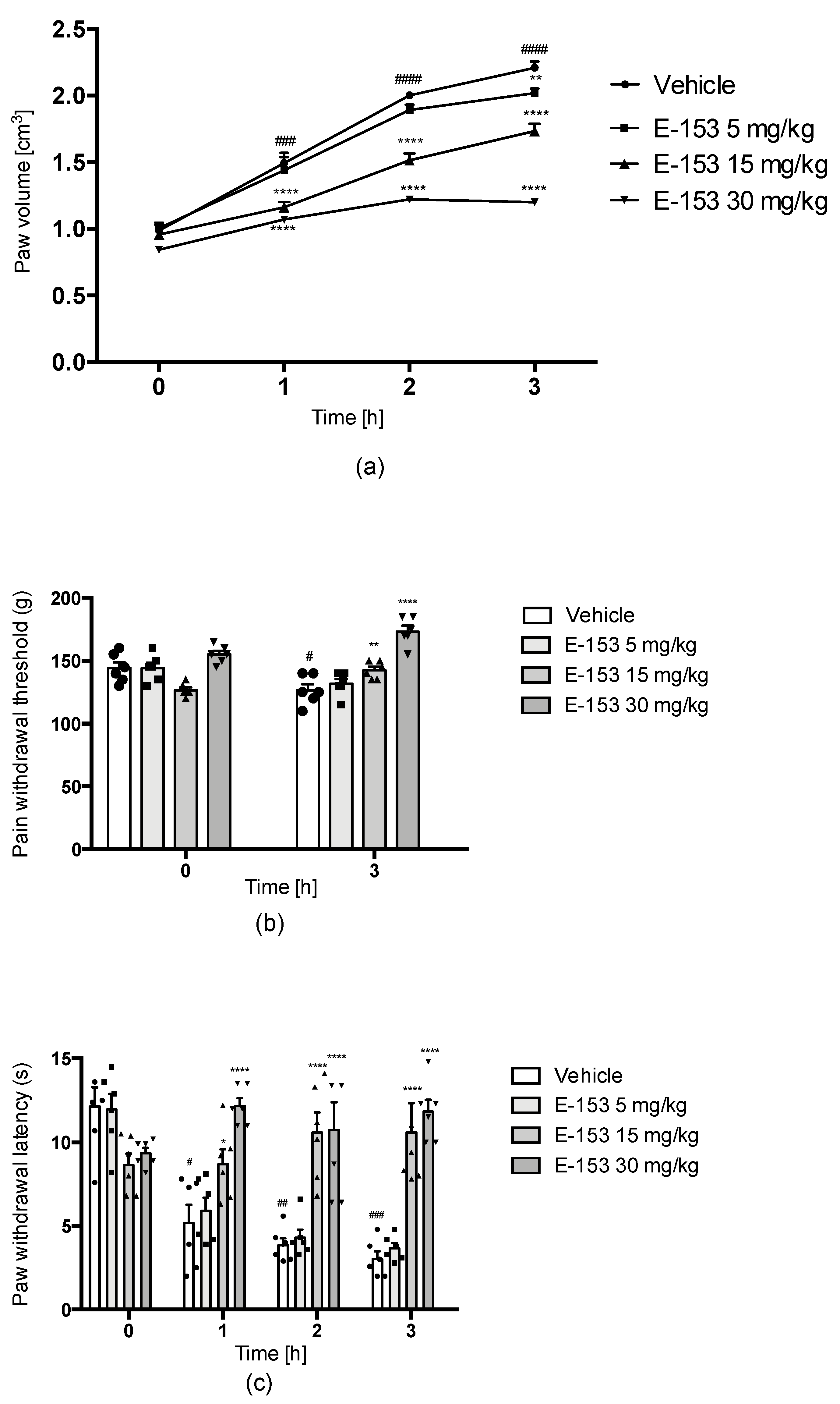
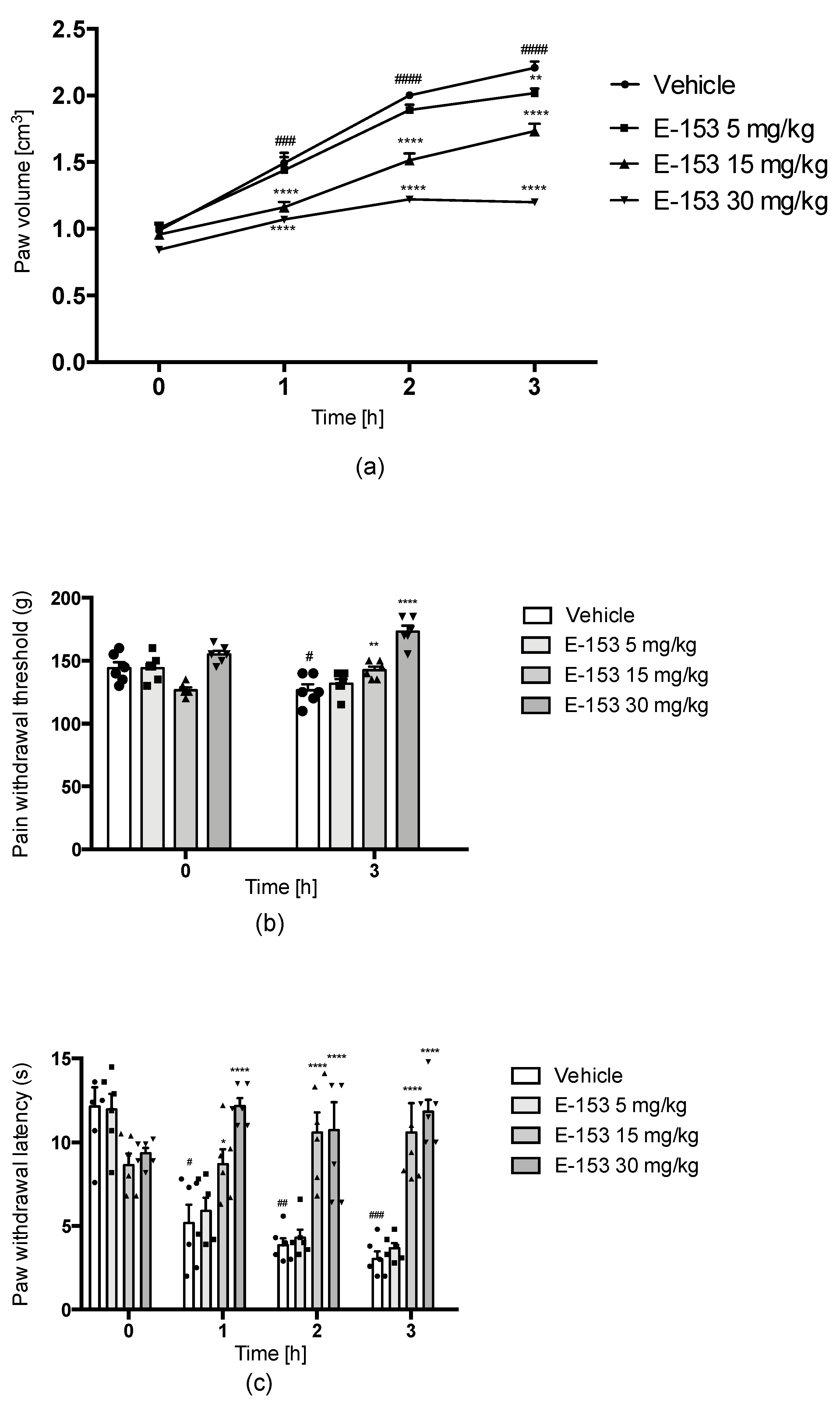
Carrageenan-Induced Inflammatory Hyperalgesia and Edema
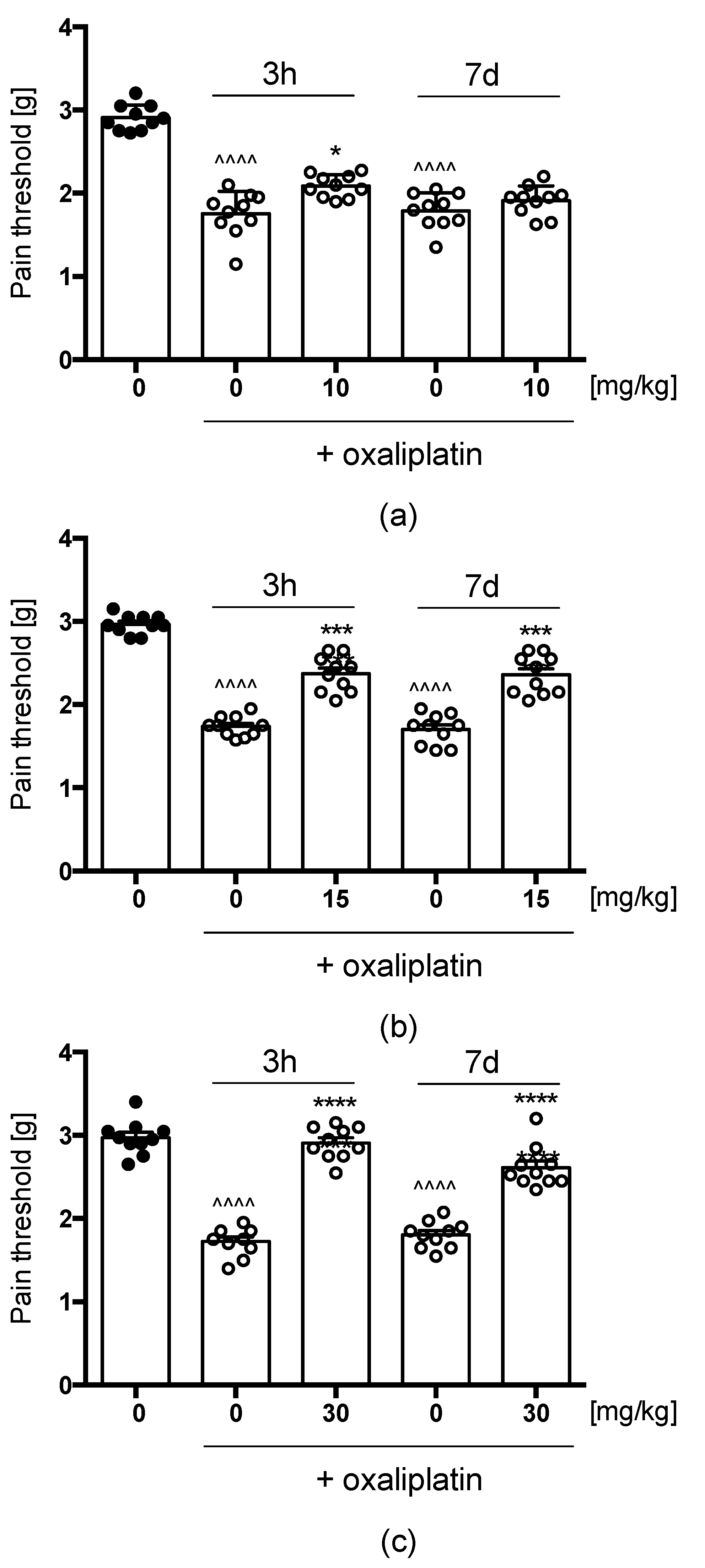
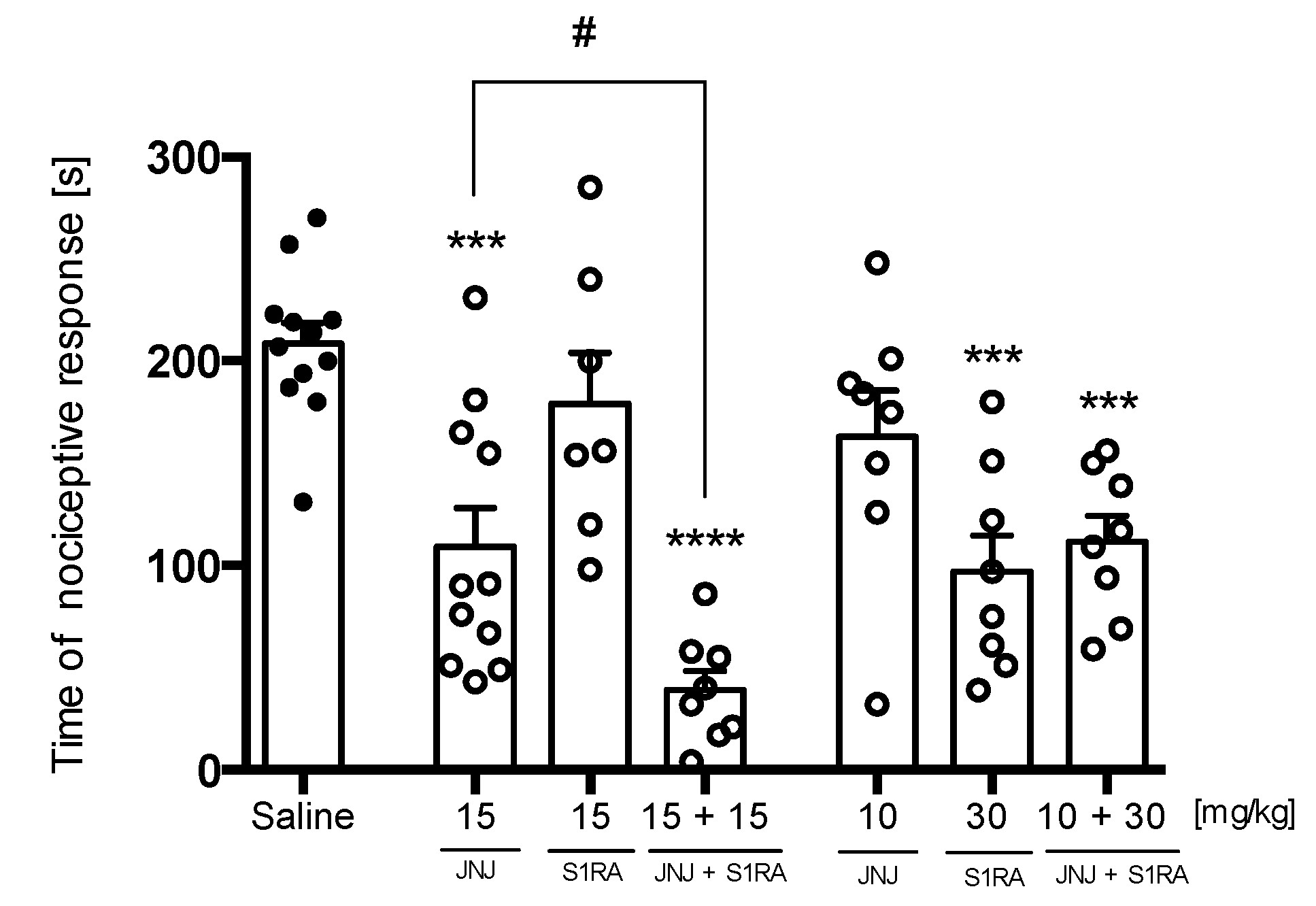
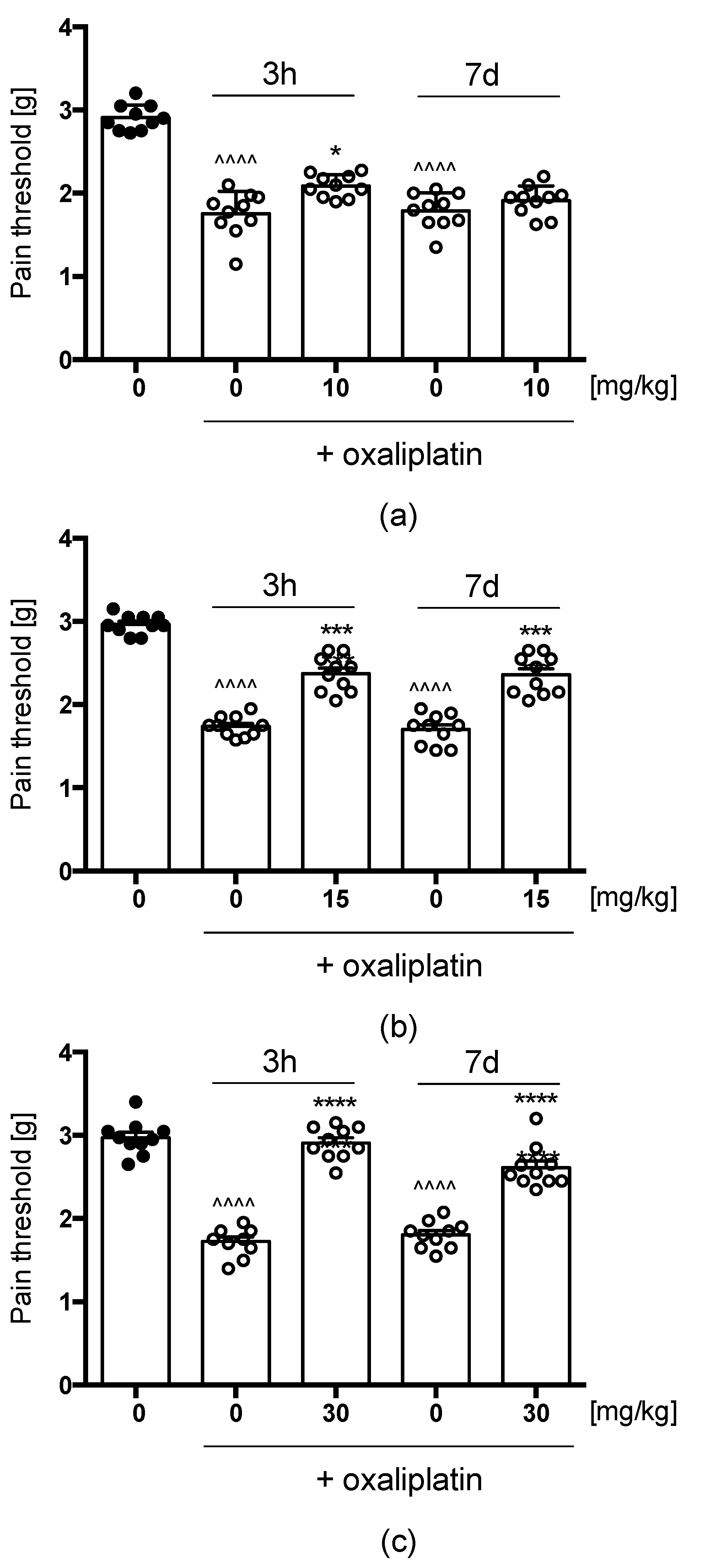
Oxaliplatin(OXPT)-Induced Neuropathic Pain
2.3.2. E153 Attenuates Pruritus of Different Origins
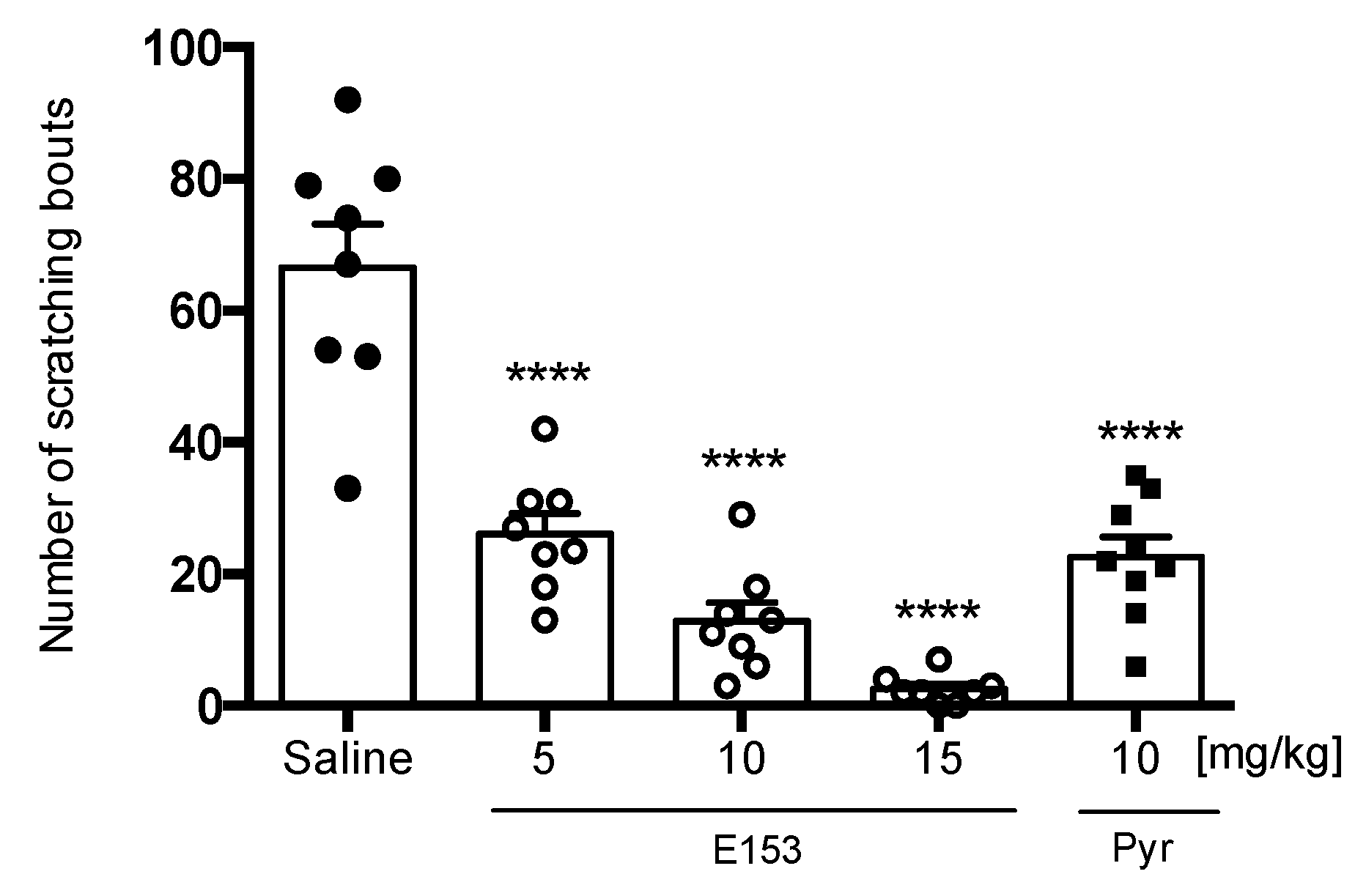
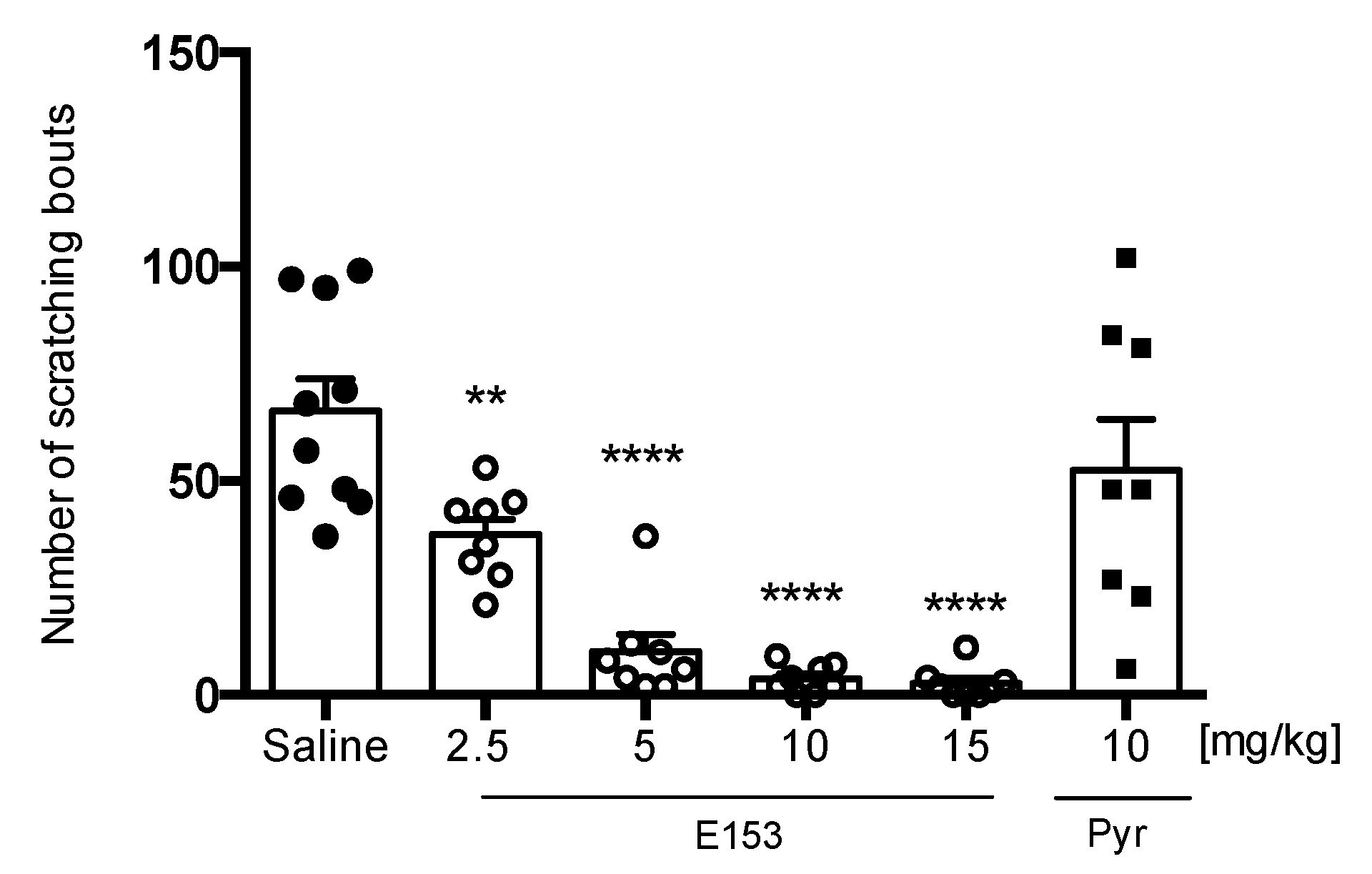
Histamine-Induced Pruritus
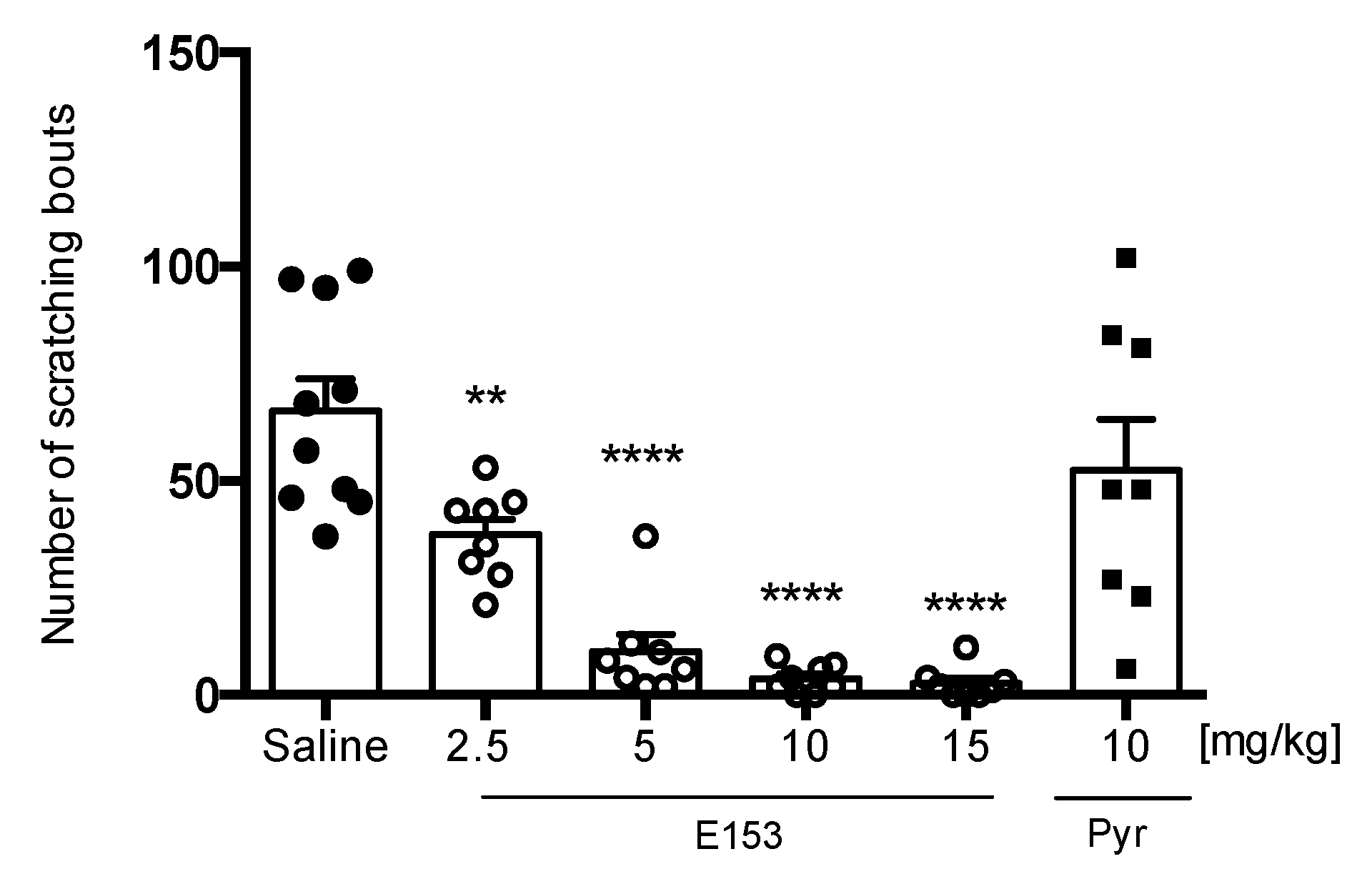
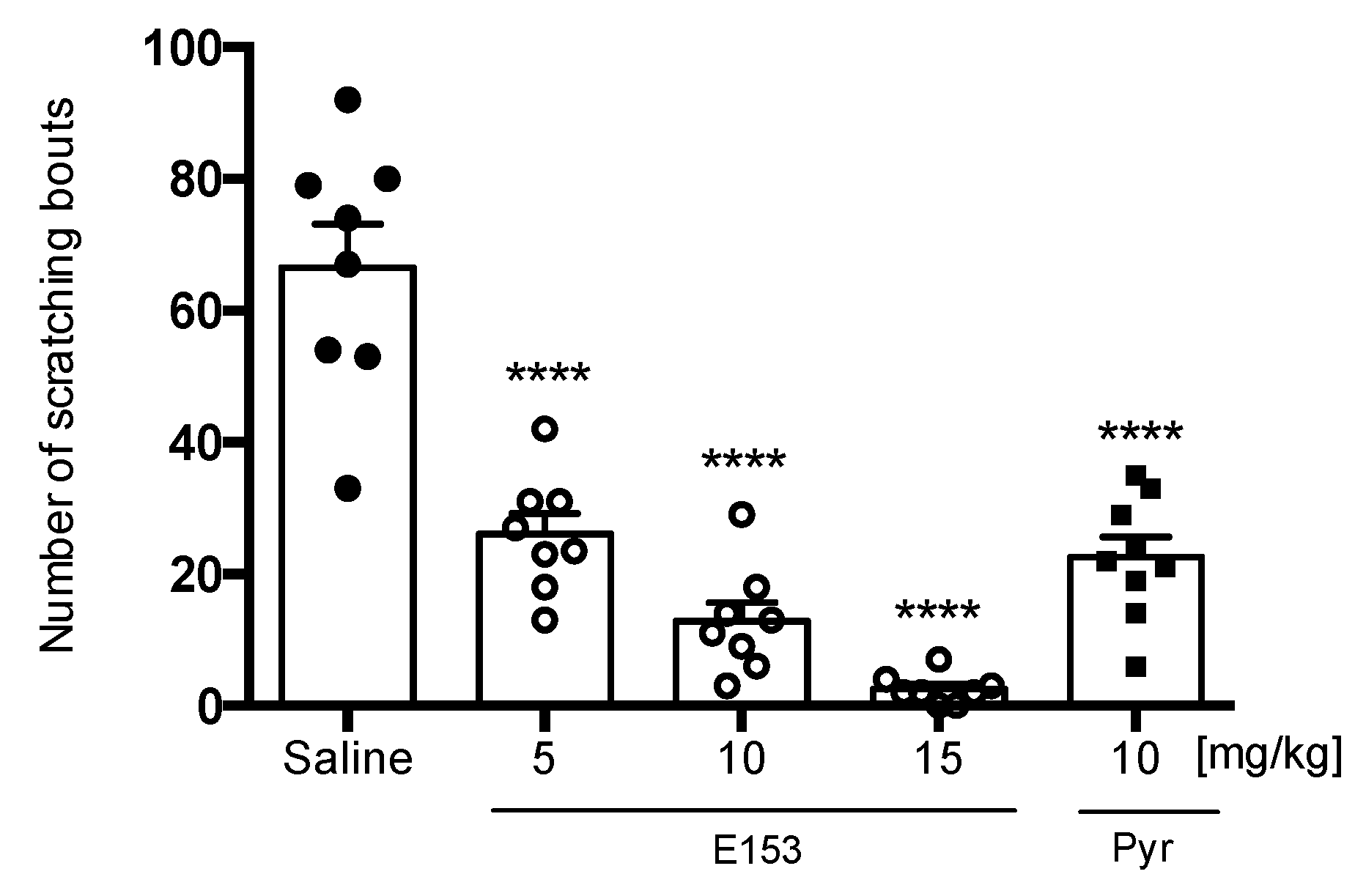
Chloroquine (CQ)-Induced Pruritus
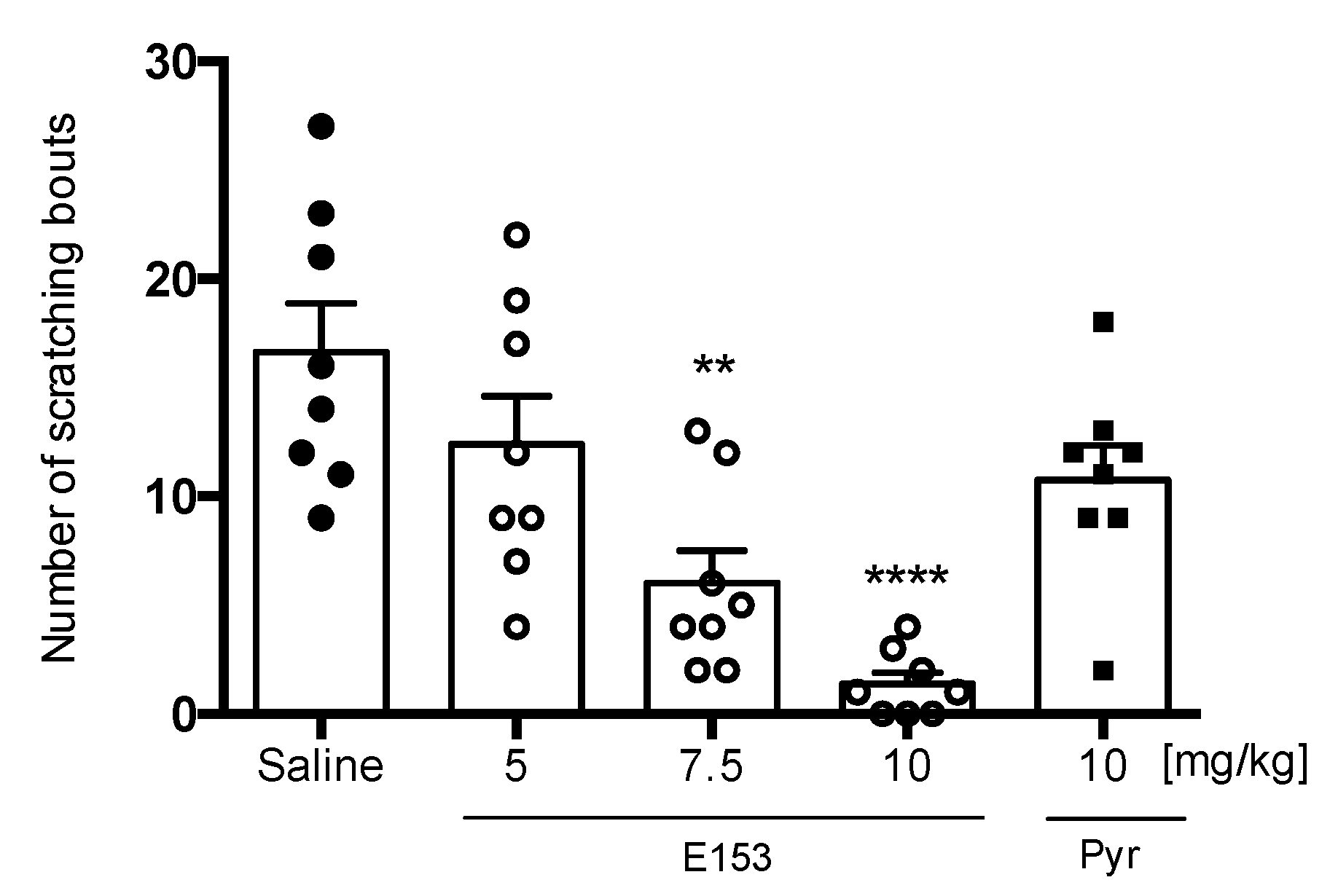
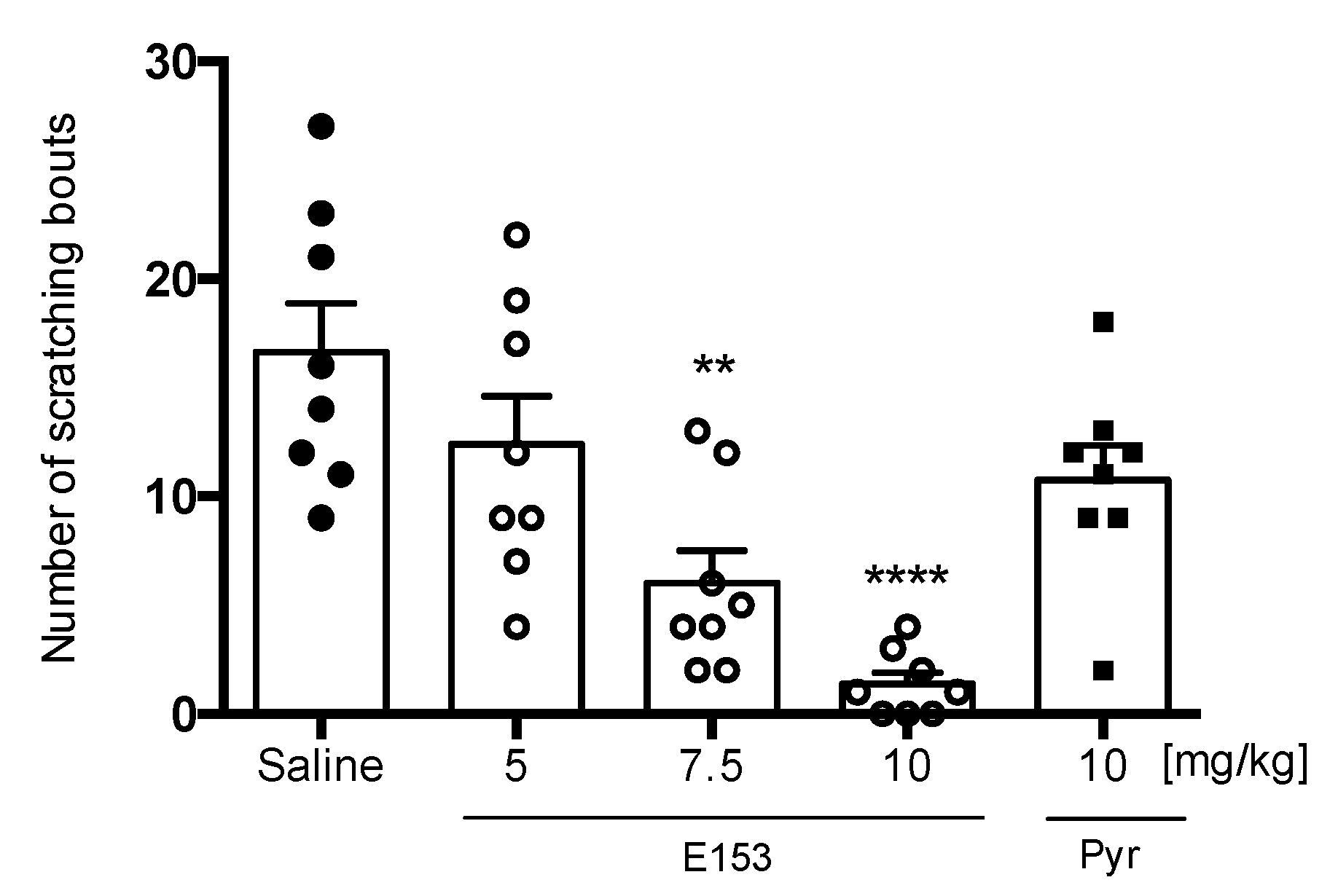
SLIGRL-Induced Pruritus
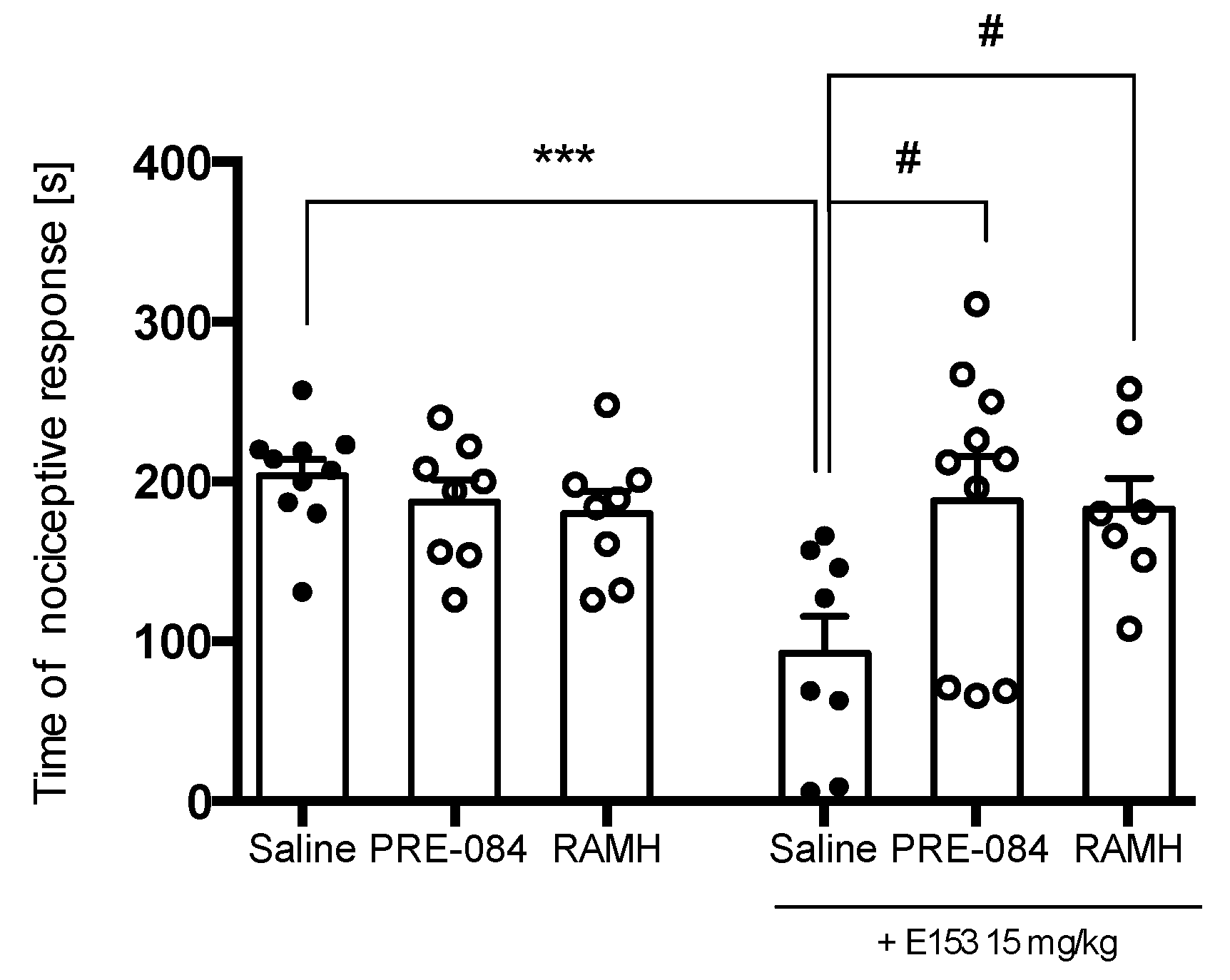
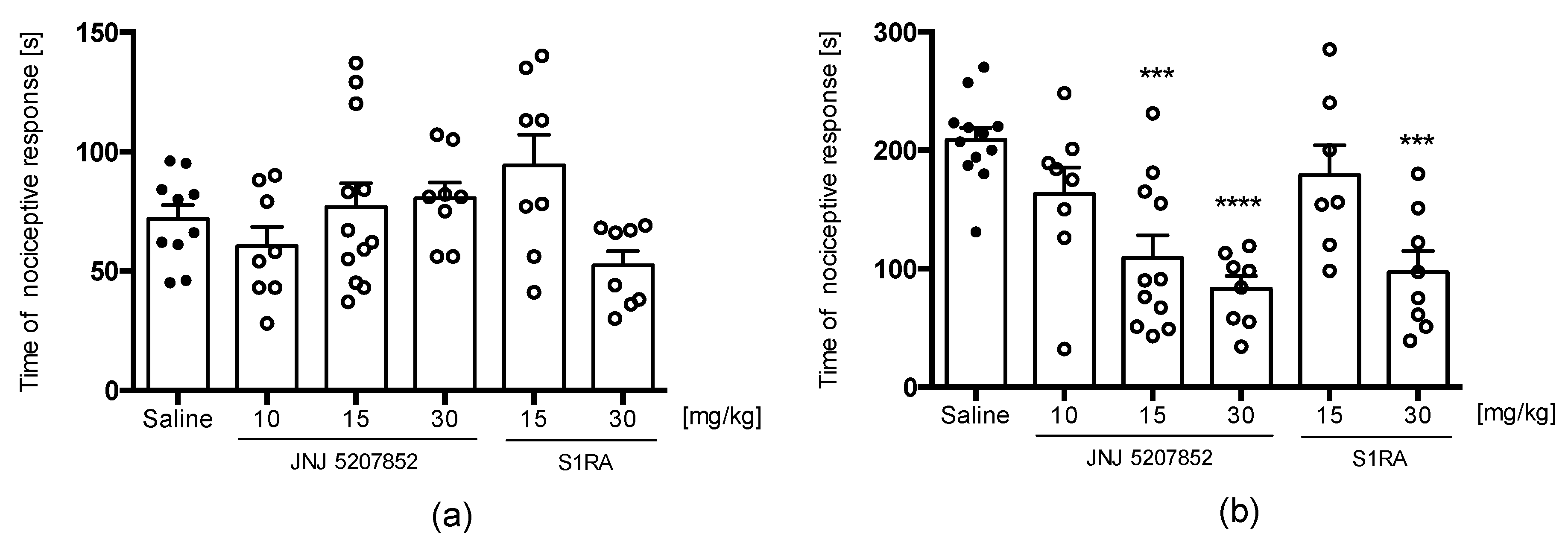
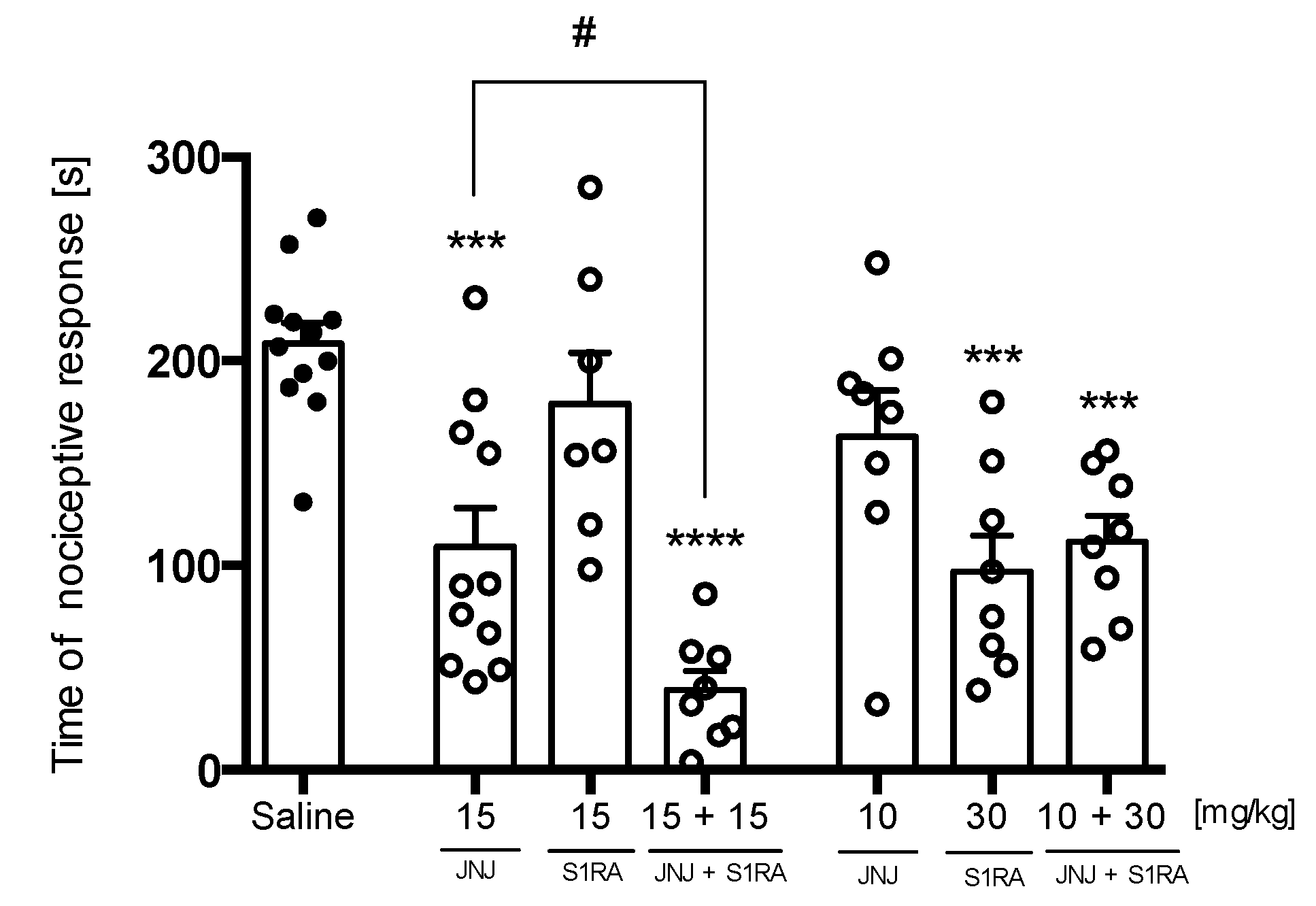
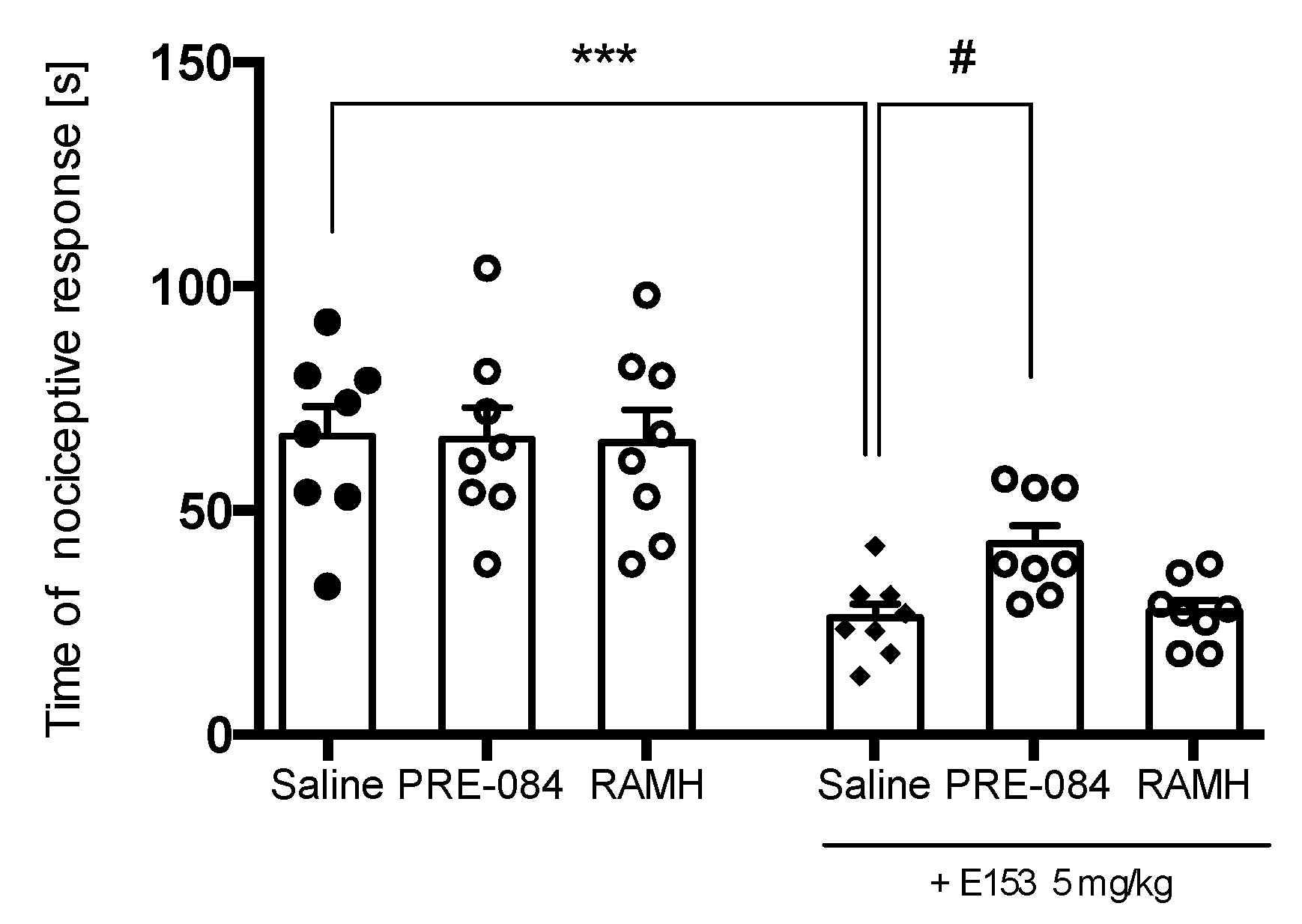
2.3.3. PRE-084 and RAMH Attenuate E153 Analgesic Activity
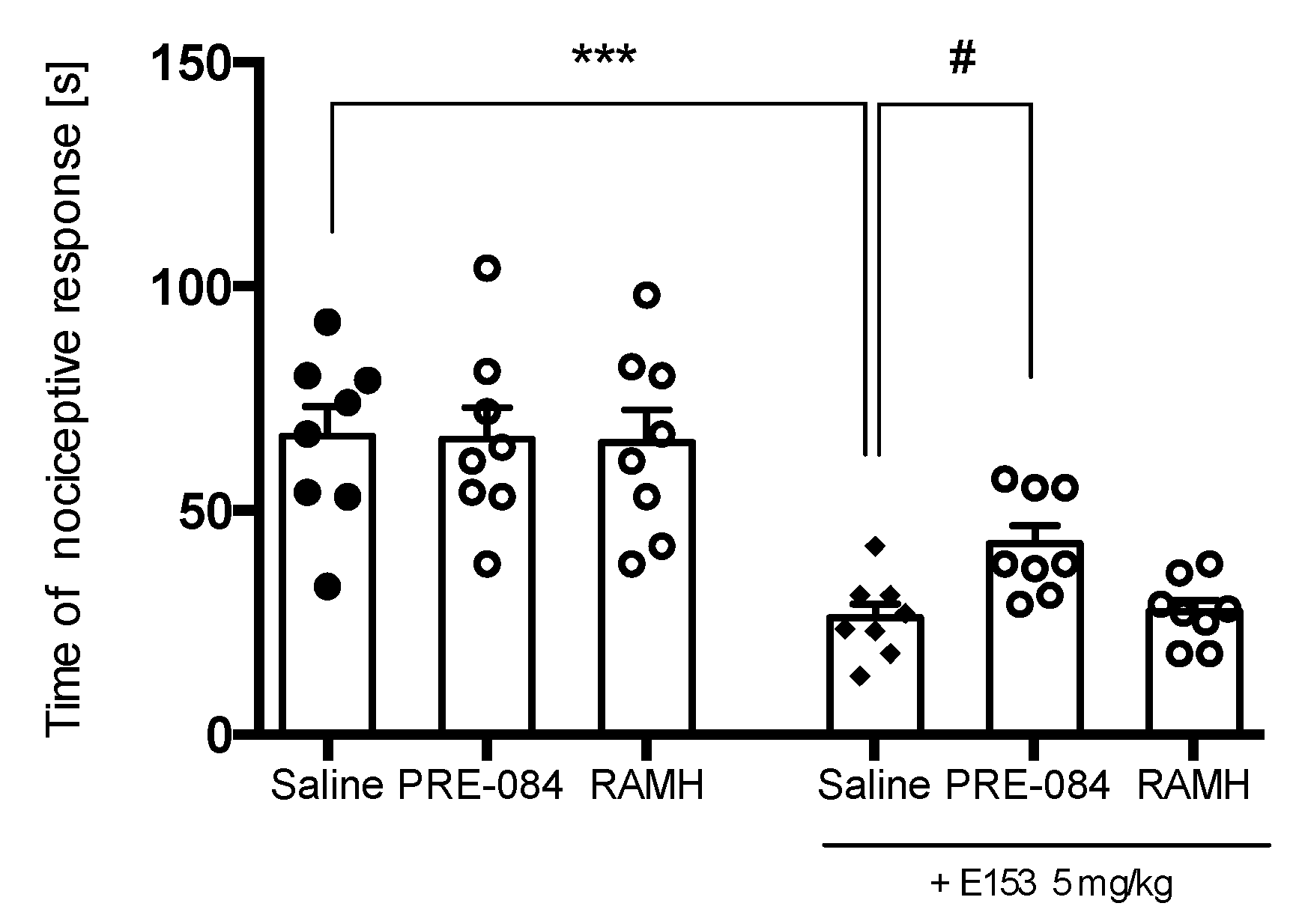
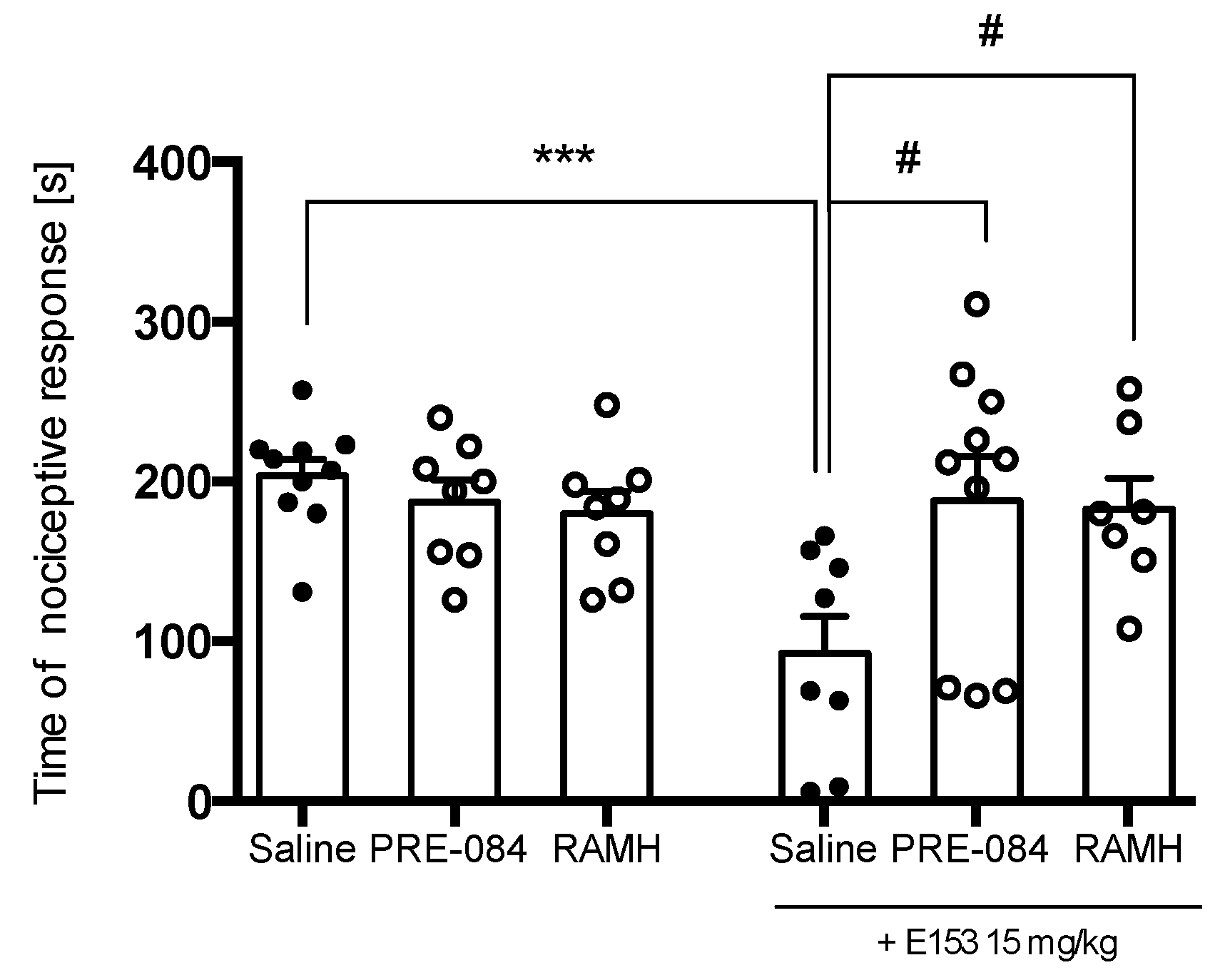
2.3.4. PRE-084 but Not RAMH Attenuates E153 Antipruritic Activity
3. Discussion
4. Materials and Methods
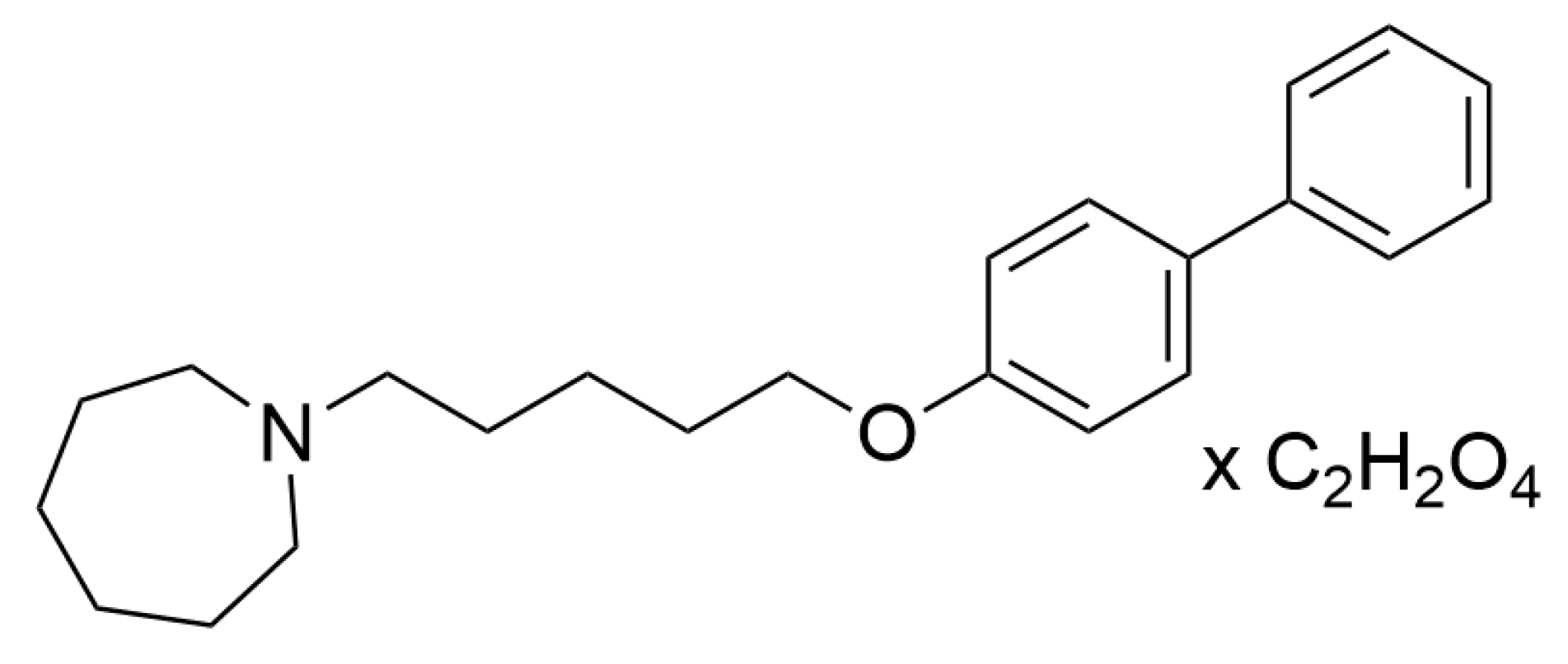
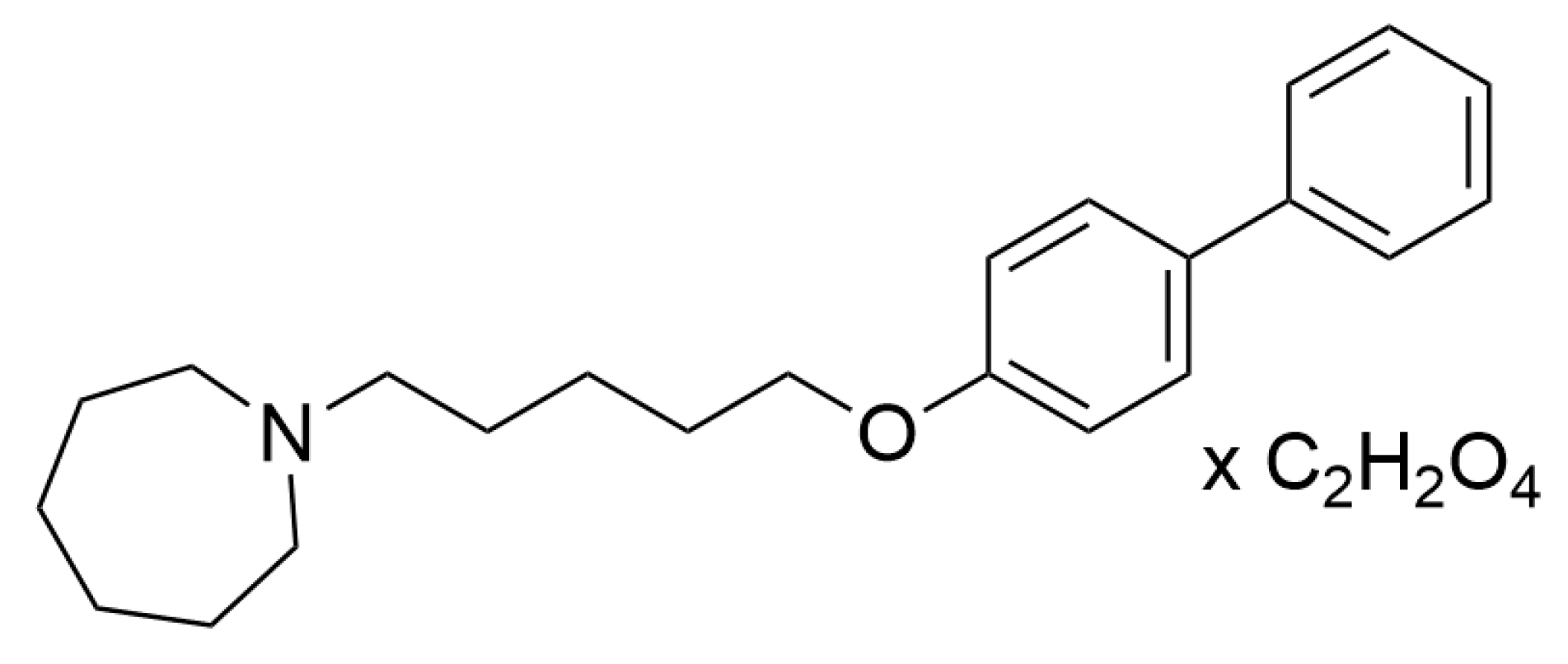
4.1. Drugs
4.2. Animals
4.3. In Vitro Radioligand Binding Studies
4.4. Pharmacokinetic Studies
4.4.1. Study Design
4.4.2. Analytical Method
4.4.3. Chromatographic Conditions
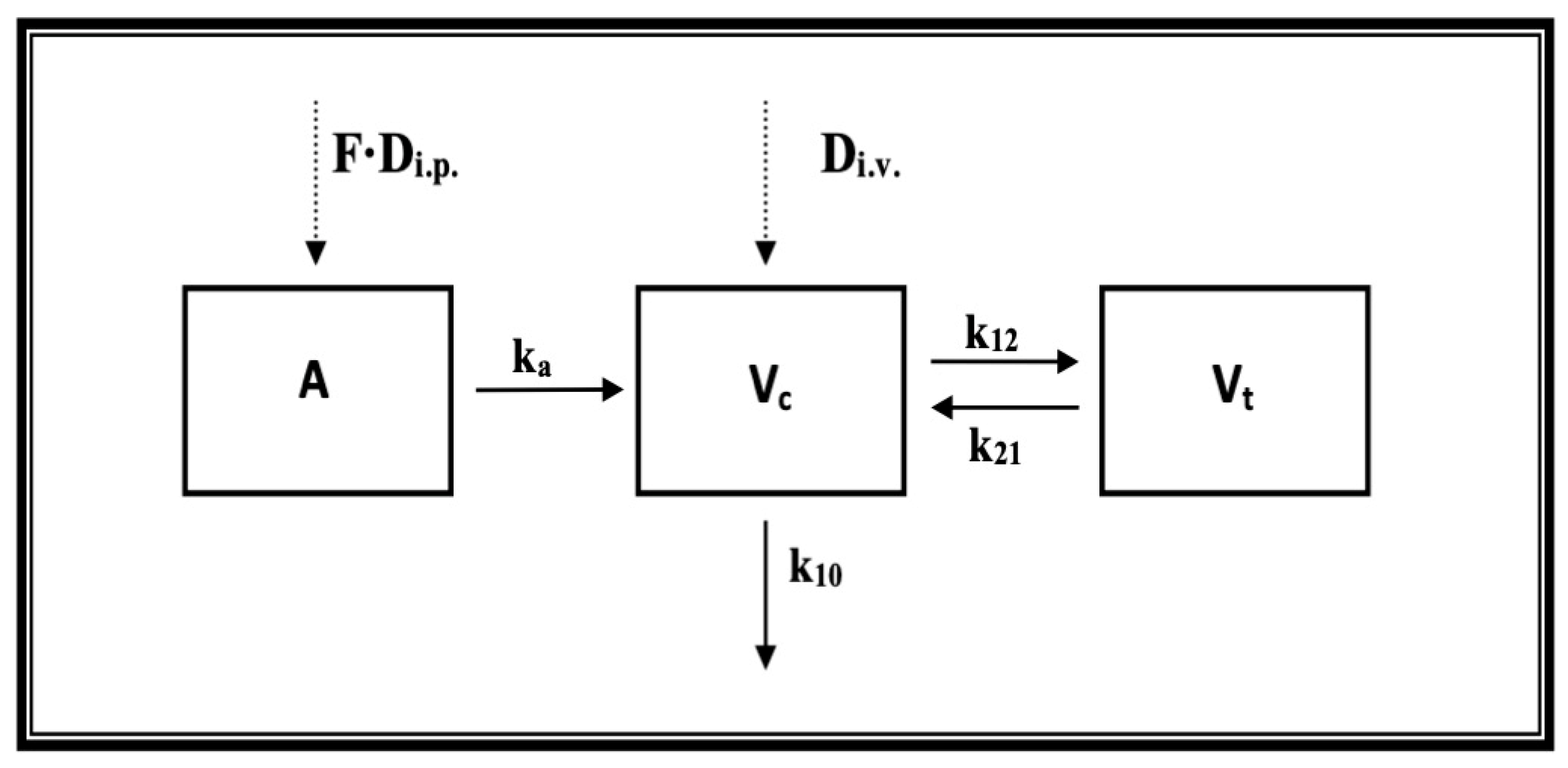
4.4.4. Pharmacokinetic Analysis
4.5. In Vivo Pharmacological Studies
4.5.1. Hot Plate Test
4.5.2. Formalin Test
4.5.3. Capsaicin Test
4.5.4. Carrageenan-Induced Inflammatory Pain and Edema
4.5.5. Oxaliplatin-Induced Neuropathy
4.5.6. Histamine-, Chloroquine- and SLIGR-L-Induced Pruritus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fornasari, D. Pharmacotherapy for Neuropathic Pain: A Review. Pain Ther. 2017, 6, 25–33. [Google Scholar] [CrossRef]
- Liu, T.; Ji, R.-R. New insights into the mechanisms of itch: Are pain and itch controlled by distinct mechanisms? Pflug. Arch-Eur. J. Physiol. 2013, 465, 1671–1685. [Google Scholar] [CrossRef] [PubMed]
- Damiani, G.; Kridin, K.; Pacifico, A.; Malagoli, P.; Pigatto, P.D.M.; Finelli, R.; Taccone, F.S.; Peluso, L.; Conic, R.R.Z.; Bragazzi, N.L.; et al. Antihistamines-refractory chronic pruritus in psoriatic patients undergoing biologics: Aprepitant vs antihistamine double dosage, a real-world data. J. Dermatol. Treat. 2022, 33, 1554–1557. [Google Scholar] [CrossRef]
- Dong, X.; Dong, X. Peripheral and Central Mechanisms of Itch. Neuron 2018, 98, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M. Modulation of Pain and Itch by Spinal Glia. Neurosci. Bull. 2018, 34, 178–185. [Google Scholar] [CrossRef]
- Han, L.; Ma, C.; Liu, Q.; Weng, H.-J.; Cui, Y.; Tang, Z.; Kim, Y.; Nie, H.; Qu, L.; Patel, K.N.; et al. A subpopulation of nociceptors specifically linked to itch. Nat. Neurosci. 2013, 16, 174–182. [Google Scholar] [CrossRef]
- Okuse, K. Pain signalling pathways: From cytokines to ion channels. Int. J. Biochem. Cell Biol. 2007, 39, 490–496. [Google Scholar] [CrossRef]
- Schaible, H.G. Peripheral and central mechanisms of pain generation. Analgesia 2007, 177, 3–28. [Google Scholar]
- Stein, C.; Clark, J.D.; Oh, U.; Vasko, M.R.; Wilcox, G.L.; Overland, A.C.; Vanderah, T.W.; Spencer, R.H. Peripheral mechanisms of pain and analgesia. Brain Res. Rev. 2009, 60, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.Z.; Bautista, D.M. Getting in Touch with Mechanical Pain Mechanisms. Trends Neurosci. 2020, 43, 311–325. [Google Scholar] [CrossRef]
- Besson, J.M. The neurobiology of pain. Lancet. 1999, 353, 1610–1615. [Google Scholar] [CrossRef]
- Linley, J.E.; Rose, K.; Ooi, L.; Gamper, N. Understanding inflammatory pain: Ion channels contributing to acute and chronicnociception. Pflug. Arch-Eur. J. Physiol. 2010, 459, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Boyce-Rustay, J.M.; Jarvis, M.F. Neuropathic Pain: Models and Mechanisms. Curr. Pharm. Des. 2009, 15, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Ru, F.; Sun, H.; Jurcakova, D.; Herbstsomer, R.A.; Meixong, J.; Dong, X.; Undem, B.J. Mechanisms of pruritogen-induced activation of itch nerves in isolated mouse skin. J. Physiol. 2017, 595, 3651–3666. [Google Scholar] [CrossRef] [PubMed]
- Meixiong, J.; Vasavda, C.; Snyder, S.H.; Dong, X. MRGPRX4 is a G protein-coupled receptor activated by bile acids that may contribute to cholestatic pruritus. Proc. Natl. Acad. Sci. USA 2019, 116, 10525–10530. [Google Scholar] [CrossRef]
- Wu, G.Y.; Tang, Z.X. Mrgprs: An Essential Role in Itch. Biomed. J. 2018, 2, 9. [Google Scholar]
- Chen, X.-J.; Sun, Y.-G. Central circuit mechanisms of itch. Nat. Commun. 2020, 11, 3052. [Google Scholar] [CrossRef] [PubMed]
- Lay, M.; Dong, X. Neural Mechanisms of Itch. Annu. Rev. Neurosci. 2020, 43, 187–205. [Google Scholar] [CrossRef]
- Koch, S.C.; Acton, D.; Goulding, M. Spinal Circuits for Touch, Pain, and Itch. Annu. Rev. Physiol. 2018, 80, 189–217. [Google Scholar] [CrossRef]
- Ji, R.-R.; Donnelly, C.R.; Nedergaard, M. Astrocytes in chronic pain and itch. Nat. Rev. Neurosci. 2019, 20, 677–685. [Google Scholar] [CrossRef]
- Nelson, T.S.; Taylor, B.K. Targeting spinal neuropeptide Y1 receptor-expressing interneurons to alleviate chronic pain and itch. Prog. Neurobiol. 2020, 196, 101894. [Google Scholar] [CrossRef]
- Anzelc, M.; Burkhart, C.G. Pain and Pruritus: A study of their similarities and differences. Int. J. Dermatol. 2020, 59, 159–164. [Google Scholar] [CrossRef]
- Łażewska, D.; Kaleta, M.; Schwed, J.S.; Karcz, T.; Mogilski, S.; Latacz, G.; Olejarz, A.; Siwek, A.; Kubacka, M.; Lubelska, A.; et al. Biphenyloxy-alkyl-piperidine and azepane derivatives as histamine H3 receptor ligands. Bioorg. Med. Chem. 2017, 25, 5341–5354. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Zaręba, P.; Godyń, J.; Doroz-Płonka, A.; Frank, A.; Reiner-Link, D.; Bajda, M.; Stary, D.; Mogilski, S.; Olejarz-Maciej, A.; et al. Biphenylalkoxyamine Derivatives-Histamine H3 Receptor Ligands with Butyrylcholinesterase Inhibitory Activity. Molecules 2021, 26, 3580. [Google Scholar] [CrossRef] [PubMed]
- Riddy, D.M.; Cook, A.E.; Shackleford, D.M.; Pierce, T.L.; Mocaer, E.; la Cour, C.M.; Sors, A.; Charman, W.N.; Summers, R.J.; Sexton, P.M.; et al. Drug-receptor kinetics and sigma-1 receptor affinity differentiate clinically evaluated histamine H3 receptor antagonists. Neuropharmacology 2019, 144, 244–255. [Google Scholar] [CrossRef]
- Muley, M.M.; Krustev, E.; McDougall, J.J. Preclinical Assessment of Inflammatory Pain. CNS Neurosci. Ther. 2015, 22, 88–101. [Google Scholar] [CrossRef]
- Entrena, J.M.; Sánchez-Fernández, C.; Nieto, F.R.; González-Cano, R.; Yeste, S.; Cobos, E.J.; Baeyens, J.M. Sigma-1 Receptor Agonism Promotes Mechanical Allodynia After Priming the Nociceptive System with Capsaicin. Sci. Rep. 2016, 6, 37835. [Google Scholar] [CrossRef] [PubMed]
- Alachkar, A.; Łażewska, D.; Kieć-Kononowicz, K.; Sadek, B. The Histamine H3 Receptor Antagonist E159 Reverses Memory Deficits Induced by Dizocilpine in Passive Avoidance and Novel Object Recognition Paradigm in Rats. Front. Pharmacol. 2017, 8, 709. [Google Scholar] [CrossRef]
- Haas, H.L.; Panula, P. Histamine receptors. Neuropharmacology 2016, 106, 1–2. [Google Scholar] [CrossRef]
- Panula, P.; Sundvik, M.; Karlstedt, K. Developmental roles of brain histamine. Trends Neurosci. 2014, 37, 159–168. [Google Scholar] [CrossRef]
- Tiligada, E.; Ennis, M. Histamine pharmacology: From Sir Henry Dale to the 21st century. Br. J. Pharmacol. 2020, 177, 469–489. [Google Scholar] [CrossRef]
- Obara, I.; Telezhkin, V.; Alrashdi, I.; Chazot, P.L. Histamine, histamine receptors, and neuropathic pain relief. Br. J. Pharmacol. 2019, 174, S17–S21. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Gooshchi, N.; Tamaddonfard, E.; Farshid, A.A. Effects of microinjection of histamine into the anterior cingulate cortex on pain-related behaviors induced by formalin in rats. Pharmacol. Rep. 2015, 67, 593–599. [Google Scholar] [CrossRef] [PubMed]
- MacGlashan, D. Histamine: A mediator of inflammation. J. Allergy Clin. Immunol. 2003, 112, S53–S59. [Google Scholar] [CrossRef] [PubMed]
- Köchling, H.K.; Schaper, K.; Wilzopolski, J.; Gutzmer, R.; Werfel, T.; Bäumer, W.; Kietzmann, M.; Rossbach, K. Combined treatment with H1 and H4 receptor antagonists reduces inflammation in a mouse model of atopic dermatitis. J. Dermatol. Sci. 2017, 87, 130–137. [Google Scholar] [CrossRef]
- Rossbach, K.; Nassenstein, C.; Gschwandtner, M.; Schnell, D.; Sander, K.; Seifert, R.; Stark, H.; Kietzmann, M.; Bäumer, W. Histamine H1, H3 and H4 receptors are involved in pruritus. Neuroscience 2011, 190, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Simons, F.E.R. Advances in H 1-Antihistamines. N. Engl. J. Med. 2004, 351, 2203–2217. [Google Scholar] [CrossRef]
- Kajihara, Y.; Murakami, M.; Imagawa, T.; Otsuguro, K.; Ito, S.; Ohta, T. Histamine potentiates acid-induced responses mediating transient receptor potential V1 in mouse primary sensory neurons. Neuroscience 2010, 166, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Mogilski, S.; Kubacka, M.; Redzicka, A.; Kazek, G.; Dudek, M.; Malinka, W.; Filipek, B. Antinociceptive, anti-inflammatory and smooth muscle relaxant activities of the pyrrolo [3,4-d]pyridazinone derivatives: Possible mechanisms of action. Pharmacol. Biochem. Behav. 2015, 133, 99–110. [Google Scholar] [CrossRef]
- Chaumette, T.; Chapuy, E.; Berrocoso, E.; Llorca-Torralba, M.; Bravo, L.; Mico, J.A.; Chalus, M.; Eschalier, A.; Ardid, D.; Marchand, F.; et al. Effects of S 38093, an antagonist/inverse agonist of histamine H3 receptors, in models of neuropathic pain in rats. Eur. J. Pain 2017, 22, 127–141. [Google Scholar] [CrossRef]
- Wei, H.; Jin, C.-Y.; Viisanen, H.; You, H.-J.; Pertovaara, A. Histamine in the locus coeruleus promotes descending noradrenergic inhibition of neuropathic hypersensitivity. Pharmacol. Res. 2014, 90, 58–66. [Google Scholar] [CrossRef]
- Hough, L.B.; Rice, F.L. H3 Receptors and Pain Modulation: Peripheral, Spinal, and Brain Interactions. J. Pharmacol. Exp. Ther. 2010, 336, 30–37. [Google Scholar] [CrossRef]
- Blandina, P. Histamine neurons in the tuberomamillary nucleus: A whole center or distinct subpopulations? Front. Syst. Neurosci. 2012, 6, 33. [Google Scholar] [CrossRef]
- Munari, L.; Provensi, G.; Passani, M.B.; Blandina, P. Selective brain region activation by histamine H3 receptor antagonist/inverse agonist ABT-239 enhances acetylcholine and histamine release and increases c-Fos expression. Neuropharmacology 2013, 70, 131–140. [Google Scholar] [CrossRef]
- Cannon, K.E.; Hough, L.B. Inhibition of chemical and low-intensity mechanical nociception by activation of histamine H3 receptors. J. Pain 2005, 6, 193–200. [Google Scholar] [CrossRef]
- Hasanein, P. Effects of histamine H3 receptors on chemical hyperalgesia in diabetic rats. Neuropharmacology 2011, 60, 886–891. [Google Scholar] [CrossRef]
- Lu, C.-W.; Lin, T.-Y.; Chang, C.-Y.; Huang, S.-K.; Wang, S.-J. Ciproxifan, a histamine H3 receptor antagonist and inverse agonist, presynaptically inhibits glutamate release in rat hippocampus. Toxicol. Appl. Pharmacol. 2017, 319, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Medhurst, A.D.; Briggs, M.A.; Bruton, G.; Calver, A.R.; Chessell, I.; Crook, B.; Davis, J.B.; Davis, R.P.; Foley, A.G.; Heslop, T.; et al. Structurally novel histamine H3 receptor antagonists GSK207040 and GSK334429 improve scopolamine-induced memory impairment and capsaicin-induced secondary allodynia in rats. Biochem. Pharmacol. 2007, 73, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Łażewska, D.; Latacz, G.; Olejarz, A.; Makuch, W.; Stark, H.; Kieć-Kononowicz, K.; Mika, J. Antinociceptive effects of novel histamine H3 and H4 receptor antagonists and their influence on morphine analgesia of neuropathic pain in the mouse. Br. J. Pharmacol. 2018, 175, 2897–2910. [Google Scholar] [CrossRef] [PubMed]
- Medhurst, A.D.; Atkins, A.R.; Beresford, I.J.; Brackenborough, K.; Briggs, M.A.; Calver, A.R.; Cilia, J.; Cluderay, J.E.; Crook, B.; Davis, J.B.; et al. GSK189254, a Novel H3 Receptor Antagonist That Binds to Histamine H3 Receptors in Alzheimer’s Disease Brain and Improves Cognitive Performance in Preclinical Models. J. Pharmacol. Exp. Ther. 2007, 321, 1032–1045. [Google Scholar] [CrossRef]
- Milano, J.; Oliveira, S.M.; Rossato, M.F.; Sauzem, P.D.; Machado, P.; Beck, P.; Zanatta, N.; Martins, M.A.P.; Mello, C.F.; Rubin, M.A.; et al. Antinociceptive effect of novel trihalomethyl-substituted pyrazoline methyl esters in formalin and hot-plate tests in mice. Eur. J. Pharmacol. 2008, 581, 86–96. [Google Scholar] [CrossRef]
- Salinas-Abarca, A.B.; Avila-Rojas, S.H.; Barragán-Iglesias, P.; Pineda-Farias, J.B.; Granados-Soto, V. Formalin injection produces long-lasting hypersensitivity with characteristics of neuropathic pain. Eur. J. Pharmacol. 2017, 797, 83–93. [Google Scholar] [CrossRef]
- Joseph, E.K.; Levine, J.D. Comparison of Oxaliplatin- and Cisplatin-Induced Painful Peripheral Neuropathy in the Rat. J. Pain 2009, 10, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Planas, E.; Poveda, R.; Sánchez, S.; Pol, O.; Puig, M.M. Anti-exudative effects of opioid receptor agonists in a rat model of carrageenan-induced acute inflammation of the paw. Eur. J. Pharmacol. 2005, 511, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Galici, R.; Boggs, J.D.; Aluisio, L.; Fraser, I.C.; Bonaventure, P.; Lord, B.; Lovenberg, T.W. JNJ-10181457, a selective non-imidazole histamine H3 receptor antagonist, normalizes acetylcholine neurotransmission and has efficacy in translational rat models of cognition. Neuropharmacology 2009, 56, 1131–1137. [Google Scholar] [CrossRef]
- Gawel, K.; Gibula-Bruzda, E.; Dziedzic, M.; Jenda-Wojtanowska, M.; Marszalek-Grabska, M.; Silberring, J.; Kotlinska, J.H. Cholinergic activation affects the acute and chronic antinociceptive effects of morphine. Physiol. Behav. 2017, 169, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.A.; Sugimoto, Y.; Kayasuga, R.; Kamei, C. Involvement of histamine H3 receptors in scratching behaviour in mast cell-deficient mice. Br. J. Dermatol. 2003, 149, 17–22. [Google Scholar] [CrossRef]
- McGaraughty, S.; Chu, K.L.; Cowart, M.D.; Brioni, J.D. Antagonism of Supraspinal Histamine H3 Receptors Modulates Spinal Neuronal Activity in Neuropathic Rats. J. Pharmacol. Exp. Ther. 2012, 343, 13–20. [Google Scholar] [CrossRef]
- Sahn, J.J.; Mejia, G.L.; Ray, P.R.; Martin, S.F.; Price, T.J. Sigma 2 Receptor/Tmem97 Agonists Produce Long Lasting Antineuropathic Pain Effects in Mice. ACS Chem. Neurosci. 2017, 8, 1801–1811. [Google Scholar] [CrossRef]
- Bravo-Caparrós, I.; Perazzoli, G.; Yeste, S.; Cikes, D.; Baeyens, J.M.; Cobos, E.J.; Nieto, F.R. Sigma-1 Receptor Inhibition Reduces Neuropathic Pain Induced by Partial Sciatic Nerve Transection in Mice by Opioid-Dependent and -Independent Mechanisms. Front. Pharmacol. 2019, 10, 613. [Google Scholar] [CrossRef]
- Ruiz-Cantero, M.C.; González-Cano, R.; Tejada, M.Á.; Santos-Caballero, M.; Perazzoli, G.; Nieto, F.R.; Cobos, E.J. Sigma-1 receptor: A drug target for the modulation of neuroimmune and neuroglial interactions during chronic pain. Pharmacol. Res. 2021, 163, 105339. [Google Scholar] [CrossRef] [PubMed]
- Montilla-García, Á.; Tejada, M.Á.; Ruiz-Cantero, M.C.; Bravo-Caparrós, I.; Yeste, S.; Zamanillo, D.; Cobos, E.J. Modulation by Sigma-1 Receptor of Morphine Analgesia and Tolerance: Nociceptive Pain, Tactile Allodynia and Grip Strength Deficits During Joint Inflammation. Front. Pharmacol. 2019, 10, 136. [Google Scholar] [CrossRef]
- Cortés-Montero, E.; Sánchez-Blázquez, P.; Onetti, Y.; Merlos, M.; Garzón, J. Ligands Exert Biased Activity to Regulate Sigma 1 Receptor Interactions with Cationic TRPA1, TRPV1, and TRPM8 Channels. Front. Pharmacol. 2019, 10, 634. [Google Scholar] [CrossRef]
- Gris, G.; Merlos, M.; Vela, J.M.; Zamanillo, D.; Portillo-Salido, E. S1RA, a selective sigma-1 receptor antagonist, inhibits inflammatory pain in the carrageenan and complete Freund’s adjuvant models in mice. Behav. Pharmacol. 2014, 25, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Torres, A.; Fernández-Pastor, B.; Carceller, A.; Vela, J.M.; Merlos, M.; Zamanillo, D. Supraspinal and Peripheral, but Not Intrathecal, σ1R Blockade by S1RA Enhances Morphine Antinociception. Front. Pharmacol. 2019, 10, 422. [Google Scholar] [CrossRef]
- Motawe, Z.Y.; Abdelmaboud, S.S.; Cuevas, J.; Breslin, J.W. PRE-084 as a tool to uncover potential therapeutic applications for selective sigma-1 receptor activation. Int. J. Biochem. Cell Biol. 2020, 126, 105803. [Google Scholar] [CrossRef]
- Ortíz-Rentería, M.; Juárez-Contreras, R.; González-Ramírez, R.; Islas, L.D.; Sierra-Ramírez, F.; Llorente, I.; Simon, S.A.; Hiriart, M.; Rosenbaum, T.; Morales-Lázaro, S.L. TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain. Proc. Natl. Acad. Sci. USA 2018, 115, E1657–E1666. [Google Scholar] [CrossRef] [PubMed]
- Bannon, A.W.; Malmberg, A.B. Models of nociception: Hot-plate, tail-flick, and formalin tests in rodents. Curr. Protoc. Neurosci. 2007, 41, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.; Mogilski, S.; Abram, M.; Rapacz, A.; Latacz, G.; Szulczyk, B.; Walczak, M.; Kuś, K.; Matyjaszczyk, K.; Kamiński, R.M. KA-104, a new multitargeted anticonvulsant with potent antinociceptive activity in preclinical models. Epilepsia 2020, 61, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Mogilski, S.; Kubacka, M.; Łażewska, D.; Więcek, M.; Głuch-Lutwin, M.; Tyszka-Czochara, M.; Bukowska-Strakova, K.; Filipek, B.; Kieć-Kononowicz, K. Aryl-1,3,5-triazine ligands of histamine H4 receptor attenuate inflammatory and nociceptive response to carrageen, zymosan and lipopolysaccharide. Inflamm. Res. 2017, 66, 79–95. [Google Scholar]
- Sałat, K.; Kołaczkowski, M.; Furgała, A.; Rojek, A.; Śniecikowska, J.; Varney, M.A.; Newman-Tancredi, A. Antinociceptive, antiallodynic and antihyperalgesic effects of the 5-HT1A receptor selective agonist, NLX-112 in mouse models of pain. Neuropharmacology 2017, 125, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; McQueen, D.S.; Rees, J.L. Involvement of histamine H 4and H 1receptors in scratching induced by histamine receptor agonists in BalbC mice. Br. J. Pharmacol. 2004, 142, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.-M.; Cheng, R.-X.; Wang, S.; Huang, Y.; Gao, Y.-J.; Zhou, Y.; Liu, T.-T.; Wang, X.-L.; Chen, L.-H.; Liu, T. Antioxidants Attenuate Acute and Chronic Itch: Peripheral and Central Mechanisms of Oxidative Stress in Pruritus. Neurosci. Bull. 2016, 33, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Coavoy-Sánchez, S.A.; Rodrigues, L.; Teixeira, S.A.; Soares, A.G.; Torregrossa, R.; Wood, M.E.; Whiteman, M.; Costa, S.K.P.; Muscará, M.N. Hydrogen sulfide donors alleviate itch secondary to the activation of type-2 protease activated receptors (PAR-2) in mice. Pharmacol. Res. 2016, 113, 686–694. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Histamine H3 Receptor a | Human Sigma Receptors | Human Opioid Receptors | |||
|---|---|---|---|---|---|
| Sigma 1 b | Sigma 2 c | Kappa d | Delta e | μ f | |
| Ki = 33.9 ± 8.1 nM g | IC50 = 2.4 nM (Ki = 1.2 nM) | (101.7%) h | (52.1%) h | (−5.3%) h | (11.2%) h |
| Parameter (Unit) | Estimate | CV (%) |
|---|---|---|
| Vc (L/kg) | 6.39 | 19.30 |
| k10 (h−1) | 1.00 | 23.61 |
| k01 (h−1) | 2.32 | 28.14 |
| k12 (h−1) | 0.33 | 28.66 |
| k21 (h−1) | 0.19 | 72.20 |
| F | 0.90 | 11.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mogilski, S.; Kubacka, M.; Świerczek, A.; Wyska, E.; Szczepańska, K.; Sapa, J.; Kieć-Kononowicz, K.; Łażewska, D. Efficacy of the Multi-Target Compound E153 in Relieving Pain and Pruritus of Different Origins. Pharmaceuticals 2023, 16, 1481. https://doi.org/10.3390/ph16101481
Mogilski S, Kubacka M, Świerczek A, Wyska E, Szczepańska K, Sapa J, Kieć-Kononowicz K, Łażewska D. Efficacy of the Multi-Target Compound E153 in Relieving Pain and Pruritus of Different Origins. Pharmaceuticals. 2023; 16(10):1481. https://doi.org/10.3390/ph16101481
Chicago/Turabian StyleMogilski, Szczepan, Monika Kubacka, Artur Świerczek, Elżbieta Wyska, Katarzyna Szczepańska, Jacek Sapa, Katarzyna Kieć-Kononowicz, and Dorota Łażewska. 2023. "Efficacy of the Multi-Target Compound E153 in Relieving Pain and Pruritus of Different Origins" Pharmaceuticals 16, no. 10: 1481. https://doi.org/10.3390/ph16101481
APA StyleMogilski, S., Kubacka, M., Świerczek, A., Wyska, E., Szczepańska, K., Sapa, J., Kieć-Kononowicz, K., & Łażewska, D. (2023). Efficacy of the Multi-Target Compound E153 in Relieving Pain and Pruritus of Different Origins. Pharmaceuticals, 16(10), 1481. https://doi.org/10.3390/ph16101481