
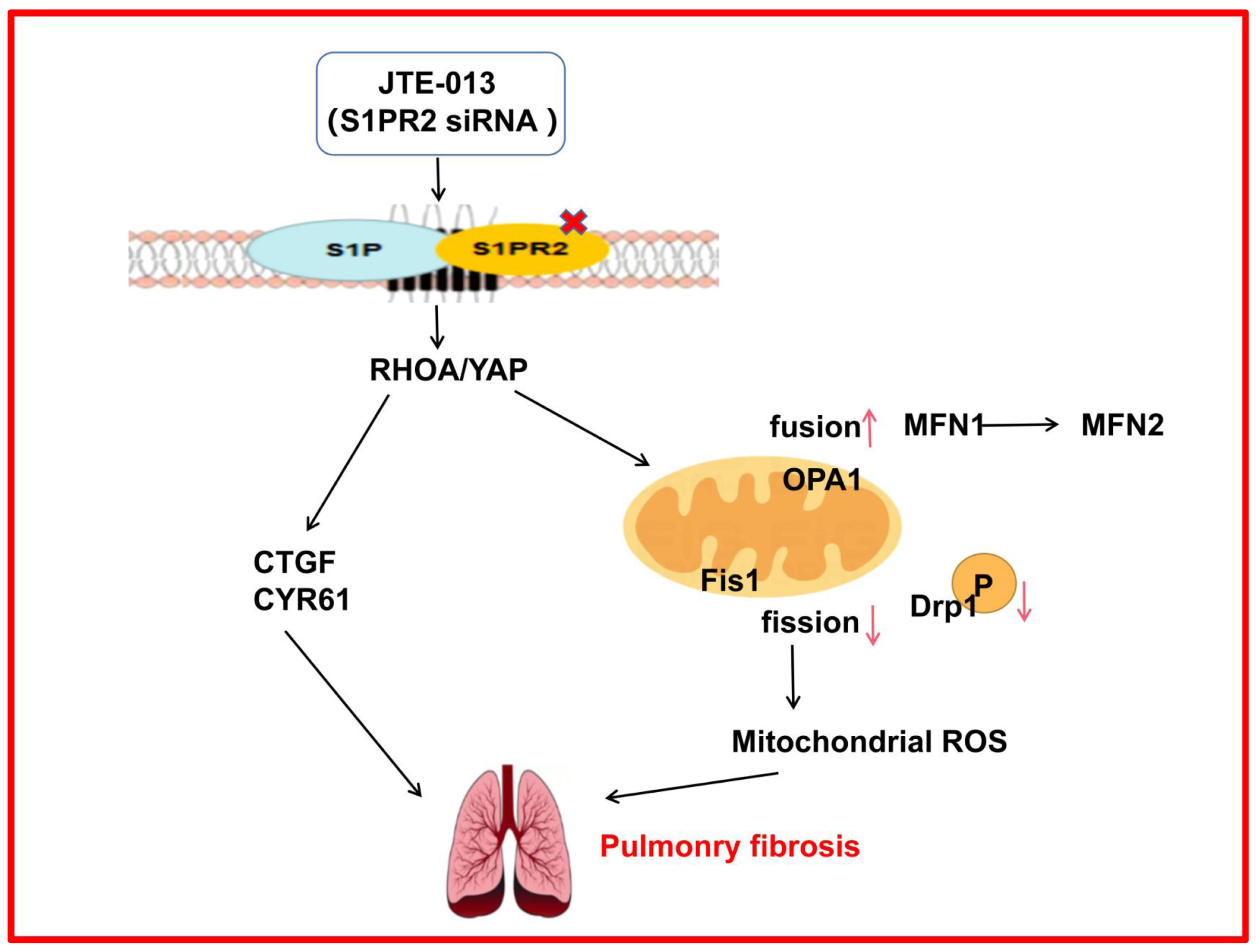
JTE-013 Alleviates Pulmonary Fibrosis by Affecting the RhoA/YAP Pathway and Mitochondrial Fusion/Fission

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
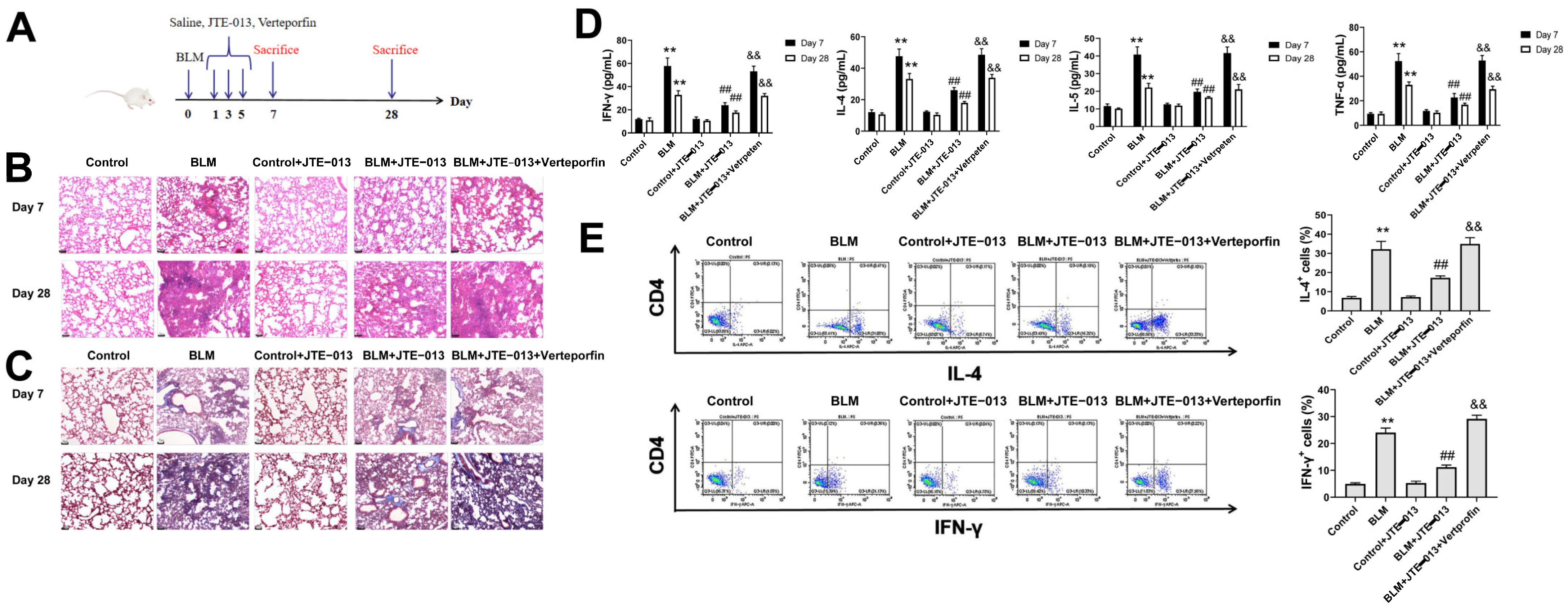
2.1. JTE-013 Alleviates BLM-Induced Pathophysiological Changes in Mice with Pulmonary Fibrosis
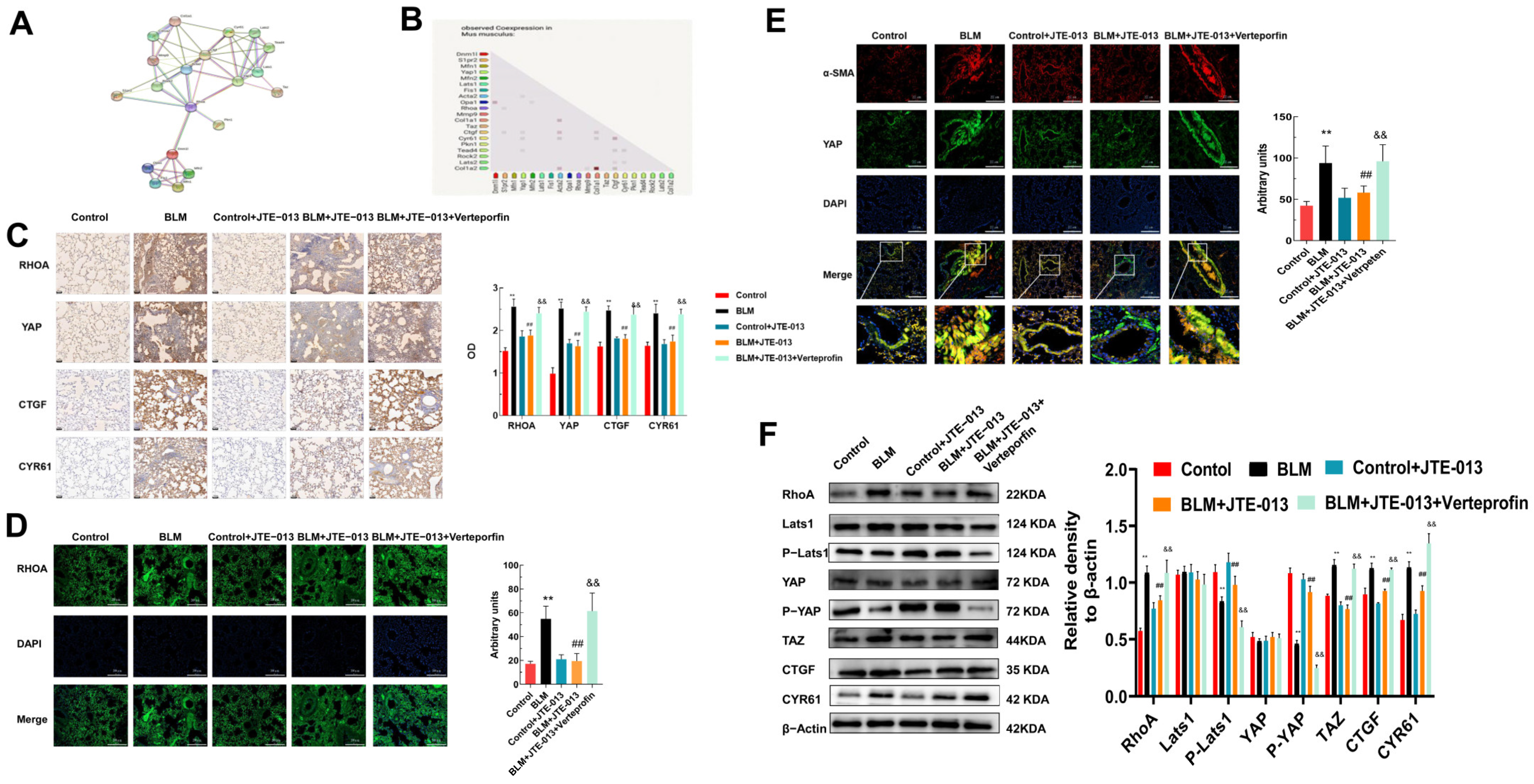
2.2. JTE-013 Inhibits the Expression of RHOA/YAP Pathway Proteins in Lung Tissues of Mice
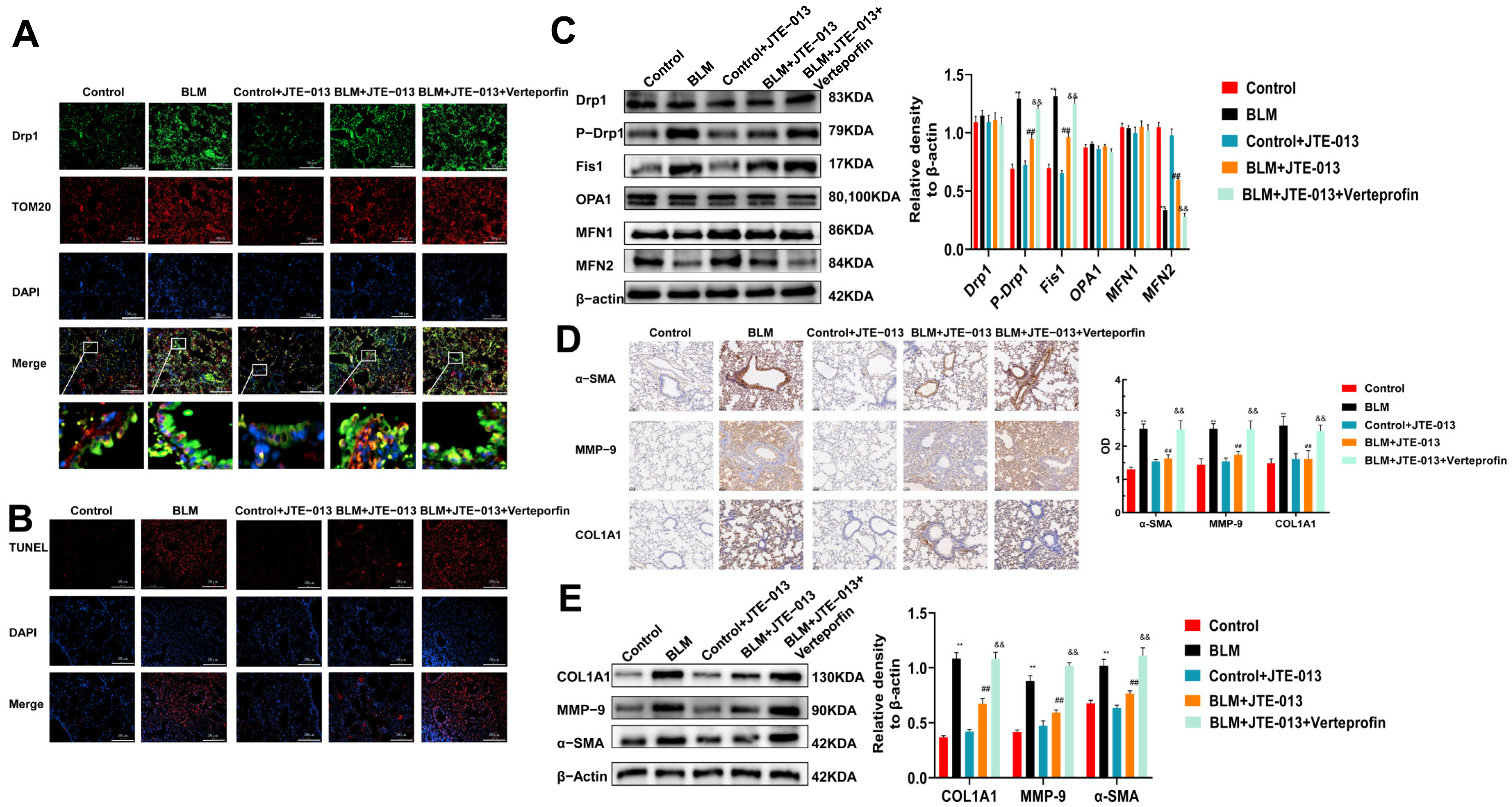
2.3. JTE-013 Restores the Expression of Mitochondrial Fusion/Fission and Fibrosis-Marker Proteins in Lung Tissues of Mice
2.4. JTE-013 Inhibits the Expression of RHOA/YAP Pathway and Fibrosis Marker Proteins in Alveolar Epithelial MLE-12 Cells
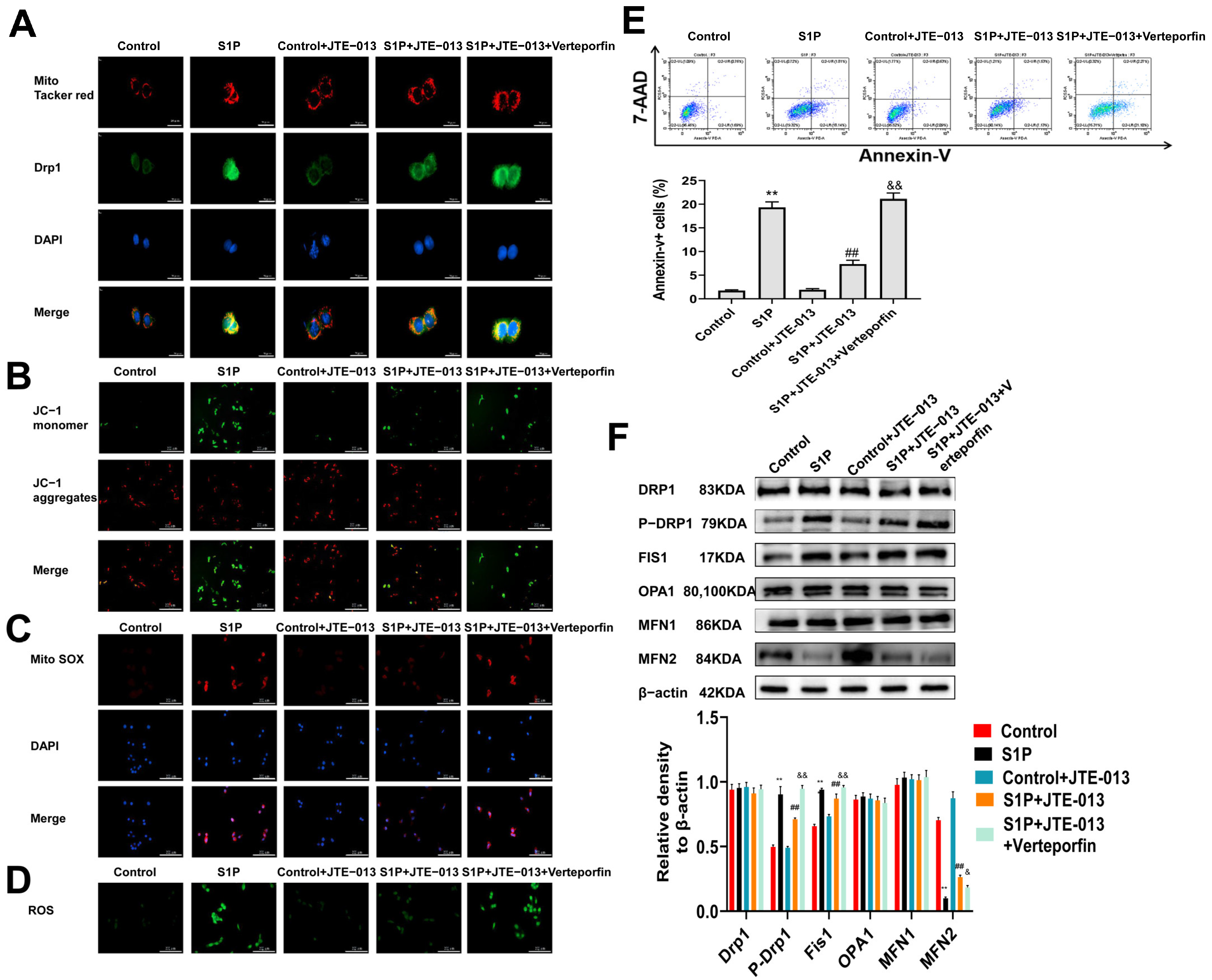
2.5. Regulation of S1PR2 Knockdown on Mitochondrial Fusion/Fission and Mitochondrial Membrane Potential in MLE-12 Cells
2.6. Regulation of JTE-013 on Mitochondrial Fusion/Fission and Mitochondrial Membrane Potential in MLE-12 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Animal Treatment and Grouping
4.4. Sample Collection
4.5. Histological Analysis
4.6. Bioinformatics Analysis
4.7. Immunohistochemistry
4.8. TUNEL Assay
4.9. Cell Culture and Treatment
4.10. Western Blot
4.11. MTT Assay
4.12. Immunofluorescence Staining
4.13. Mitochondrial Fission Analysis and Mitochondrial Membrane Potential Assay
4.14. Total ROS and Mitochondrial ROS Detection
4.15. Flow Cytometry Analysis
4.16. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ley, B.; Collard, H.R.; King, T.E.J. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Mathai, S.K.; Schwartz, D.A. Translational research in pulmonary fibrosis. Transl. Res. J. Lab. Clin. Med. 2019, 209, 1–13. [Google Scholar] [CrossRef]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef]
- King, A.; Houlihan, D.D.; Kavanagh, D.; Haldar, D.; Luu, N.; Owen, A.; Suresh, S.; Than, N.N.; Reynolds, G.; Penny, J.; et al. Sphingosine-1-Phosphate Prevents Egress of Hematopoietic Stem Cells From Liver to Reduce Fibrosis. Gastroenterology 2017, 153, 233.e216–248.e216. [Google Scholar] [CrossRef]
- Yang, L.; Yue, S.; Yang, L.; Liu, X.; Han, Z.; Zhang, Y.; Li, L. Sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis is involved in liver fibrosis-associated angiogenesis. J. Hepatol. 2013, 59, 114–123. [Google Scholar] [CrossRef]
- Im, H.; Park, J.H.; Im, S.; Han, J.; Kim, K.; Lee, Y.H. Regulatory roles of G-protein coupled receptors in adipose tissue metabolism and their therapeutic potential. Arch. Pharmacal Res. 2021, 44, 133–145. [Google Scholar] [CrossRef]
- Uhlig, S.; Gulbins, E. Sphingolipids in the lungs. Am. J. Respir. Crit. Care Med. 2008, 178, 1100–1114. [Google Scholar] [CrossRef]
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1312–1318. [Google Scholar] [CrossRef]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef]
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Macrophage Sphingosine 1-Phosphate Receptor 2 Blockade Attenuates Liver Inflammation and Fibrogenesis Triggered by NLRP3 Inflammasome. Front. Immunol. 2020, 11, 1149. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, S.; You, H.; Liu, X.; Lin, M.; Yang, L.; Li, L. Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J. Hepatol. 2011, 54, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Terashita, T.; Kobayashi, K.; Nagano, T.; Kawa, Y.; Tamura, D.; Nakata, K.; Yamamoto, M.; Tachihara, M.; Kamiryo, H.; Nishimura, Y. Administration of JTE013 abrogates experimental asthma by regulating proinflammatory cytokine production from bronchial epithelial cells. Respir. Res. 2016, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Im, D.S. Blockage of sphingosine-1-phosphate receptor 2 attenuates allergic asthma in mice. Br. J. Pharmacol. 2019, 176, 938–949. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Gaspar, P.; Tapon, N. Sensing the local environment: Actin architecture and Hippo signalling. Curr. Opin. Cell Biol. 2014, 31, 74–83. [Google Scholar] [CrossRef]
- Speight, P.; Kofler, M.; Szászi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFβ-regulated Smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef]
- Loria, R.; Laquintana, V.; Scalera, S.; Fraioli, R.; Caprara, V.; Falcone, I.; Bazzichetto, C.; Di Martile, M.; Rosanò, L.; Del Bufalo, D.; et al. SEMA6A/RhoA/YAP axis mediates tumor-stroma interactions and prevents response to dual BRAF/MEK inhibition in BRAF-mutant melanoma. J. Exp. Clin. Cancer Res. CR 2022, 41, 148. [Google Scholar] [CrossRef]
- Zhang, W.; Han, H. Targeting matrix stiffness-induced activation of retinal pigment epithelial cells through the RhoA/YAP pathway ameliorates proliferative vitreoretinopathy. Exp. Eye Res. 2021, 209, 108677. [Google Scholar] [CrossRef]
- Shin, Y.; Jung, W.; Kim, M.Y.; Shin, D.; Kim, G.H.; Kim, C.H.; Park, S.H.; Cho, E.H.; Choi, D.W.; Han, C.J.; et al. NPFFR2 Contributes to the Malignancy of Hepatocellular Carcinoma Development by Activating RhoA/YAP Signaling. Cancers 2022, 14, 5850. [Google Scholar] [CrossRef]
- Li, S.; Li, C.; Zhang, Y.; He, X.; Chen, X.; Zeng, X.; Liu, F.; Chen, Y.; Chen, J. Targeting Mechanics-Induced Fibroblast Activation through CD44-RhoA-YAP Pathway Ameliorates Crystalline Silica-Induced Silicosis. Theranostics 2019, 9, 4993–5008. [Google Scholar] [CrossRef] [PubMed]
- Emelyanov, V.V. Mitochondrial connection to the origin of the eukaryotic cell. Eur. J. Biochem. 2003, 270, 1599–1618. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Larson Casey, J.L.; Andrabi, S.A.; Lee, J.H.; Meza-Perez, S.; Randall, T.D.; Carter, A.B. Mitochondrial calcium uniporter regulates PGC-1α expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019, 26, 101307. [Google Scholar] [CrossRef]
- Chung, K.P.; Hsu, C.L.; Fan, L.C.; Huang, Z.; Bhatia, D.; Chen, Y.J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Guo, T.; Jiang, C.S.; Yang, S.Z.; Zhu, Y.; He, C.; Carter, A.B.; Antony, V.B.; Peng, H.; Zhou, Y. Mitochondrial fission and bioenergetics mediate human lung fibroblast durotaxis. JCI Insight 2023, 8, e157348. [Google Scholar] [CrossRef]
- Mao, C.; Zhang, J.; Lin, S.; Jing, L.; Xiang, J.; Wang, M.; Wang, B.; Xu, P.; Liu, W.; Song, X.; et al. MiRNA-30a inhibits AECs-II apoptosis by blocking mitochondrial fission dependent on Drp-1. J. Cell. Mol. Med. 2014, 18, 2404–2416. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, P.; Wang, Y.; Wang, M.; Li, H.; Lin, S.; Mao, C.; Wang, B.; Song, X.; Lv, C. Astaxanthin prevents pulmonary fibrosis by promoting myofibroblast apoptosis dependent on Drp1-mediated mitochondrial fission. J. Cell. Mol. Med. 2015, 19, 2215–2231. [Google Scholar] [CrossRef]
- Bhatia, D.; Capili, A.; Nakahira, K.; Muthukumar, T.; Torres, L.K.; Choi, A.M.K.; Choi, M.E. Conditional deletion of myeloid-specific mitofusin 2 but not mitofusin 1 promotes kidney fibrosis. Kidney Int. 2022, 101, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 18. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef]
- Inoue, R.; Yasuma, T.; Fridman D’Alessandro, V.; Toda, M.; Ito, T.; Tomaru, A.; D’Alessandro-Gabazza, C.N.; Tsuruga, T.; Okano, T.; Takeshita, A.; et al. Amelioration of Pulmonary Fibrosis by Matrix Metalloproteinase-2 Overexpression. Int. J. Mol. Sci. 2023, 24, 6695. [Google Scholar] [CrossRef]
- Baratella, E.; Ruaro, B.; Giudici, F.; Wade, B.; Santagiuliana, M.; Salton, F.; Confalonieri, P.; Simbolo, M.; Scarpa, A.; Tollot, S.; et al. Evaluation of Correlations between Genetic Variants and High-Resolution Computed Tomography Patterns in Idiopathic Pulmonary Fibrosis. Diagnostics 2021, 11, 762. [Google Scholar] [CrossRef]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Suzuki, A.; Maher, T.M. Interstitial lung diseases. Lancet 2022, 400, 769–786. [Google Scholar] [CrossRef]
- Oskeritzian, C.A.; Hait, N.C.; Wedman, P.; Chumanevich, A.; Kolawole, E.M.; Price, M.M.; Falanga, Y.T.; Harikumar, K.B.; Ryan, J.J.; Milstien, S.; et al. The sphingosine-1-phosphate/sphingosine-1-phosphate receptor 2 axis regulates early airway T-cell infiltration in murine mast cell-dependent acute allergic responses. J. Allergy Clin. Immunol. 2015, 135, 1008.e1001–1018.e1001. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, Z.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Yang, L.; Li, L. NLRP3 inflammasome priming and activation in cholestatic liver injury via the sphingosine 1-phosphate/S1P receptor 2/Gα((12/13))/MAPK signaling pathway. J. Mol. Med. 2021, 99, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tang, X.; Liang, Z.; Chen, M.; Sun, L. Taurocholic Acid Promotes Hepatic Stellate Cell Activation via S1PR2/p38 MAPK/YAP Signaling under Cholestatic Conditions. Clin. Mol. Hepatol. 2023, 29, 465. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Suzuki, K.; Uechi, M.; Kurihara, E.; Goto, K.; Sakai, H.; Misawa, M. Downregulation of sphingosine-1-phosphate receptors in bronchial smooth muscle of mouse experimental asthma. Pharmacol. Res. 2010, 62, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cui, K.; Feng, H.; Li, R.; Lin, H.; Chen, Y.; Zhang, Y.; Chen, Z.; Yuan, H.; Li, M.; et al. JTE-013 supplementation improves erectile dysfunction in rats with streptozotocin-induced type|diabetes through the inhibition of the rho-kinase pathway, fibrosis, and apoptosis. Andrology 2020, 8, 497–508. [Google Scholar] [CrossRef]
- Seyedsadr, M.S.; Weinmann, O.; Amorim, A.; Ineichen, B.V.; Egger, M.; Mirnajafi-Zadeh, J.; Becher, B.; Javan, M.; Schwab, M.E. Inactivation of sphingosine-1-phosphate receptor 2 (S1PR2) decreases demyelination and enhances remyelination in animal models of multiple sclerosis. Neurobiol. Dis. 2019, 124, 189–201. [Google Scholar] [CrossRef]
- Chen, Q.; Rehman, J.; Chan, M.; Fu, P.; Dudek, S.M.; Natarajan, V.; Malik, A.B.; Liu, Y. Angiocrine Sphingosine-1-Phosphate Activation of S1PR2-YAP Signaling Axis in Alveolar Type II Cells Is Essential for Lung Repair. Cell Rep. 2020, 31, 107828. [Google Scholar] [CrossRef]
- Warren, R.; Lyu, H.; Klinkhammer, K.; De Langhe, S.P.J. Hippo signaling impairs alveolar epithelial regeneration in pulmonary fibrosis. eLife 2023, 12, e85092. [Google Scholar] [CrossRef]
- Park, S.J.; Im, D.S. Blockage of sphingosine-1-phosphate receptor 2 attenuates 2,4-dinitrochlorobenzene-induced atopic dermatitis in mice. Acta Pharmacol. Sin. 2020, 41, 1487–1496. [Google Scholar] [CrossRef]
- Sun, X.; Sun, P.; Zhen, D.; Xu, X.; Yang, L.; Fu, D.; Wei, C.; Niu, X.; Tian, J.; Li, H. Melatonin alleviates doxorubicin-induced mitochondrial oxidative damage and ferroptosis in cardiomyocytes by regulating YAP expression. Toxicol. Appl. Pharmacol. 2022, 437, 115902. [Google Scholar] [CrossRef]
- Yan, H.; Qiu, C.; Sun, W.; Gu, M.; Xiao, F.; Zou, J.; Zhang, L. Yap regulates gastric cancer survival and migration via SIRT1/Mfn2/mitophagy. Oncol. Rep. 2018, 39, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Zhang, Y.; Liu, L.; Zhou, K.; Xia, S.; Peng, M.; Yan, H.; Tang, X.; Chen, Z.; Zhang, D.; et al. Yap1 modulates cardiomyocyte hypertrophy via impaired mitochondrial biogenesis in response to chronic mechanical stress overload. Theranostics 2022, 12, 7009–7031. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xiang, H.; Chen, R.; Yang, J.; Yang, X.; Zhou, J.; Liu, H.; Zhao, S.; Xiao, J.; Chen, P.; et al. S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol. 2019, 20, 135. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, J.; Xiang, H.; Chen, W.; Zhong, H.; Yang, G.; Fang, T.; Deng, H.; Yuan, H.; Chen, A.F.; et al. Role of sphingosine-1-phosphate receptor 1 and sphingosine-1-phosphate receptor 2 in hyperglycemia-induced endothelial cell dysfunction. Int. J. Mol. Med. 2015, 35, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Hirao, H.; Kojima, H.; Dery, K.J.; Nakamura, K.; Kadono, K.; Zhai, Y.; Farmer, D.G.; Kaldas, F.M.; Kupiec-Weglinski, J.W. Neutrophil CEACAM1 determines susceptibility to NETosis by regulating the S1PR2/S1PR3 axis in liver transplantation. J. Clin. Investig. 2023, 133, e162940. [Google Scholar] [CrossRef]
- Liu, H.; Li, L.; Chen, Z.; Song, Y.; Liu, W.; Gao, G.; Li, L.; Jiang, J.; Xu, C.; Yan, G.; et al. S1PR2 Inhibition Attenuates Allergic Asthma Possibly by Regulating Autophagy. Front. Pharmacol. 2020, 11, 598007. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Song, Y.; Wang, X.; Li, X.; Liu, C.; Tian, C.; Wang, C.; Li, L.; Yan, G.; Cui, H. JTE-013 Alleviates Pulmonary Fibrosis by Affecting the RhoA/YAP Pathway and Mitochondrial Fusion/Fission. Pharmaceuticals 2023, 16, 1444. https://doi.org/10.3390/ph16101444
Zhou J, Song Y, Wang X, Li X, Liu C, Tian C, Wang C, Li L, Yan G, Cui H. JTE-013 Alleviates Pulmonary Fibrosis by Affecting the RhoA/YAP Pathway and Mitochondrial Fusion/Fission. Pharmaceuticals. 2023; 16(10):1444. https://doi.org/10.3390/ph16101444
Chicago/Turabian StyleZhou, Jiaxu, Yilan Song, Xingmei Wang, Xinrui Li, Chang Liu, Chenchen Tian, Chongyang Wang, Liangchang Li, Guanghai Yan, and Hong Cui. 2023. "JTE-013 Alleviates Pulmonary Fibrosis by Affecting the RhoA/YAP Pathway and Mitochondrial Fusion/Fission" Pharmaceuticals 16, no. 10: 1444. https://doi.org/10.3390/ph16101444
APA StyleZhou, J., Song, Y., Wang, X., Li, X., Liu, C., Tian, C., Wang, C., Li, L., Yan, G., & Cui, H. (2023). JTE-013 Alleviates Pulmonary Fibrosis by Affecting the RhoA/YAP Pathway and Mitochondrial Fusion/Fission. Pharmaceuticals, 16(10), 1444. https://doi.org/10.3390/ph16101444