

The Development of a Smart Magnetic Resonance Imaging and Chemical Exchange Saturation Transfer Contrast Agent for the Imaging of Sulfatase Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
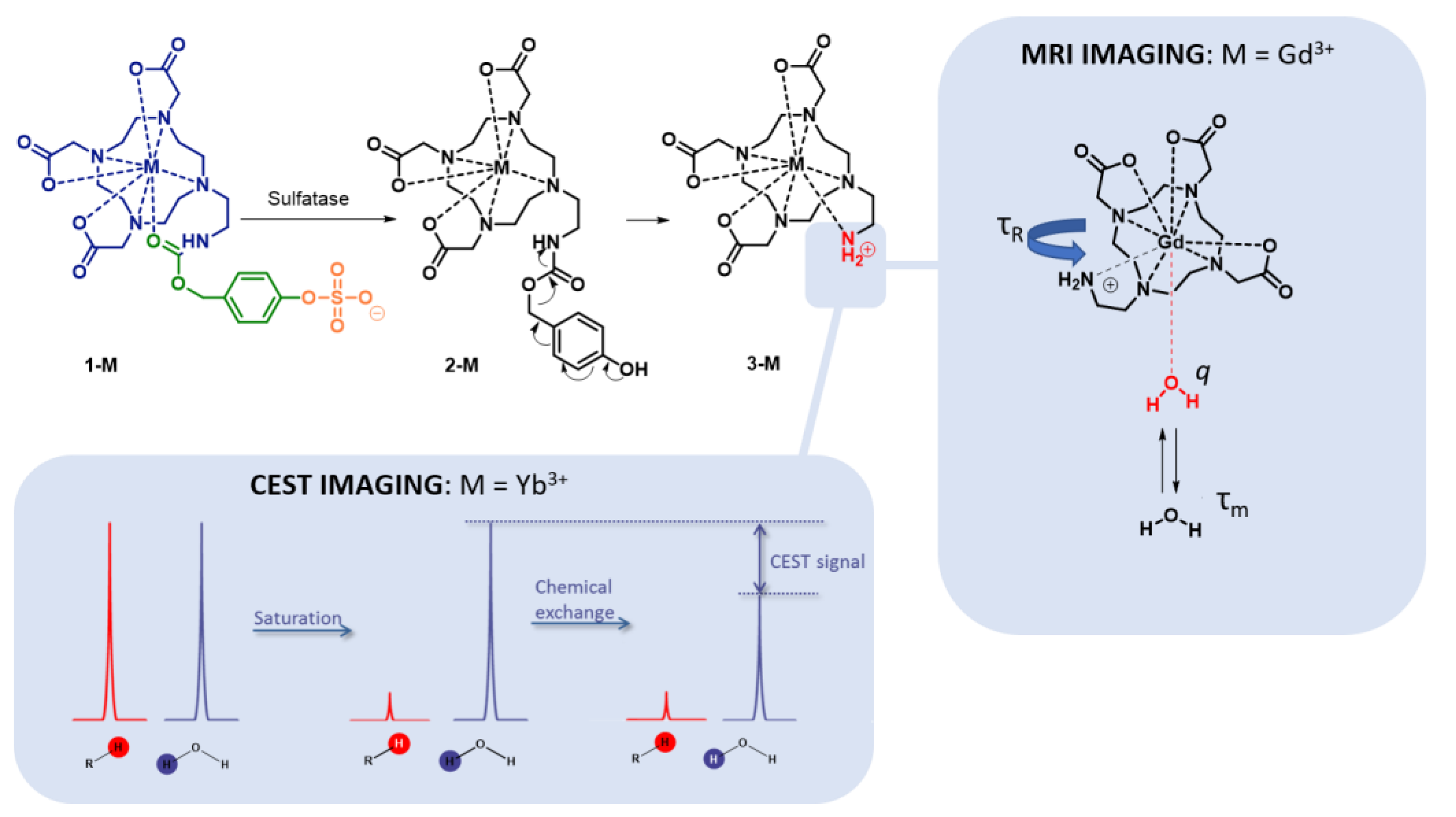
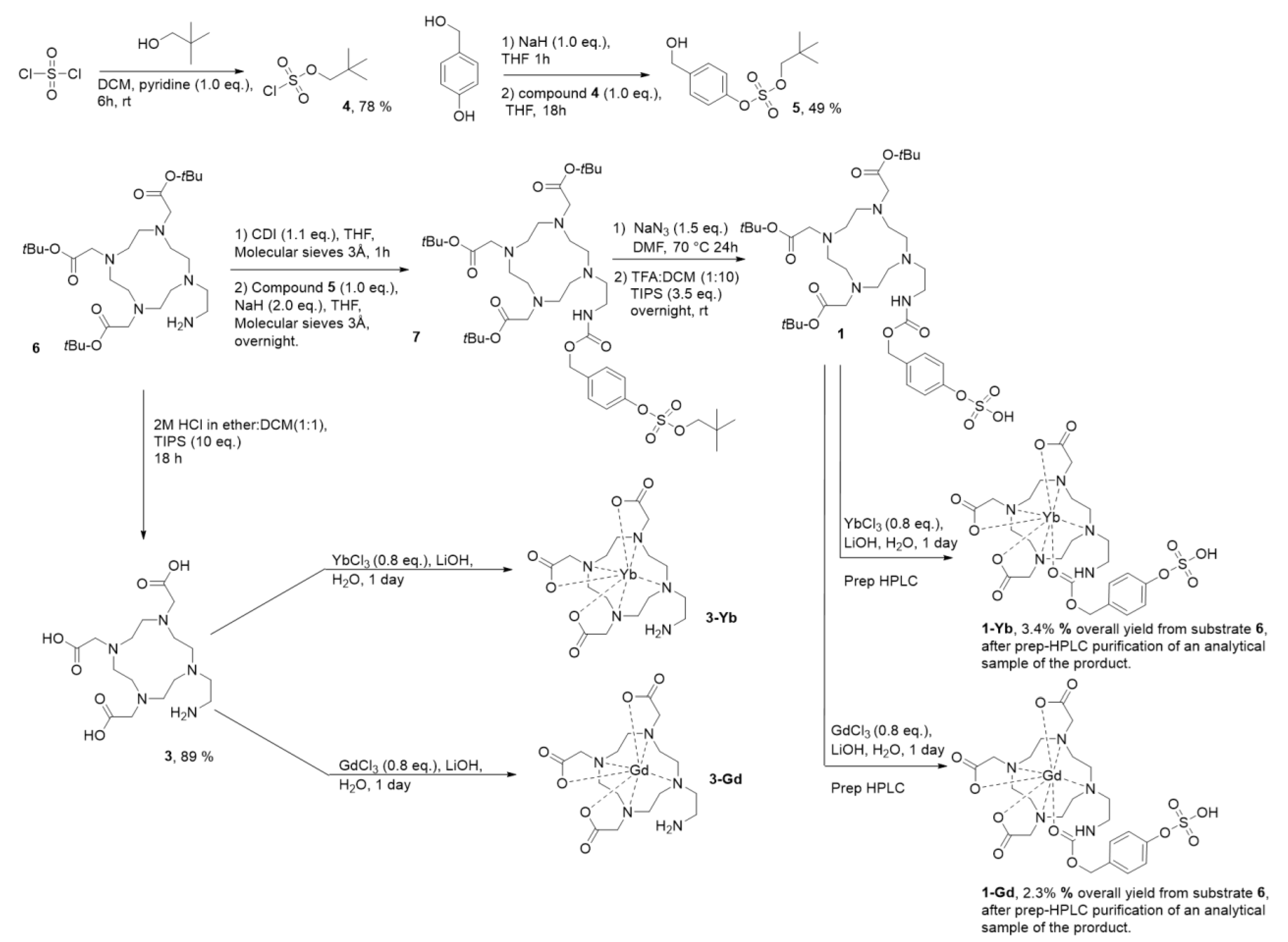
2.1. Design and Synthesis of Contrast Agents 1-Gd and 1-Yb
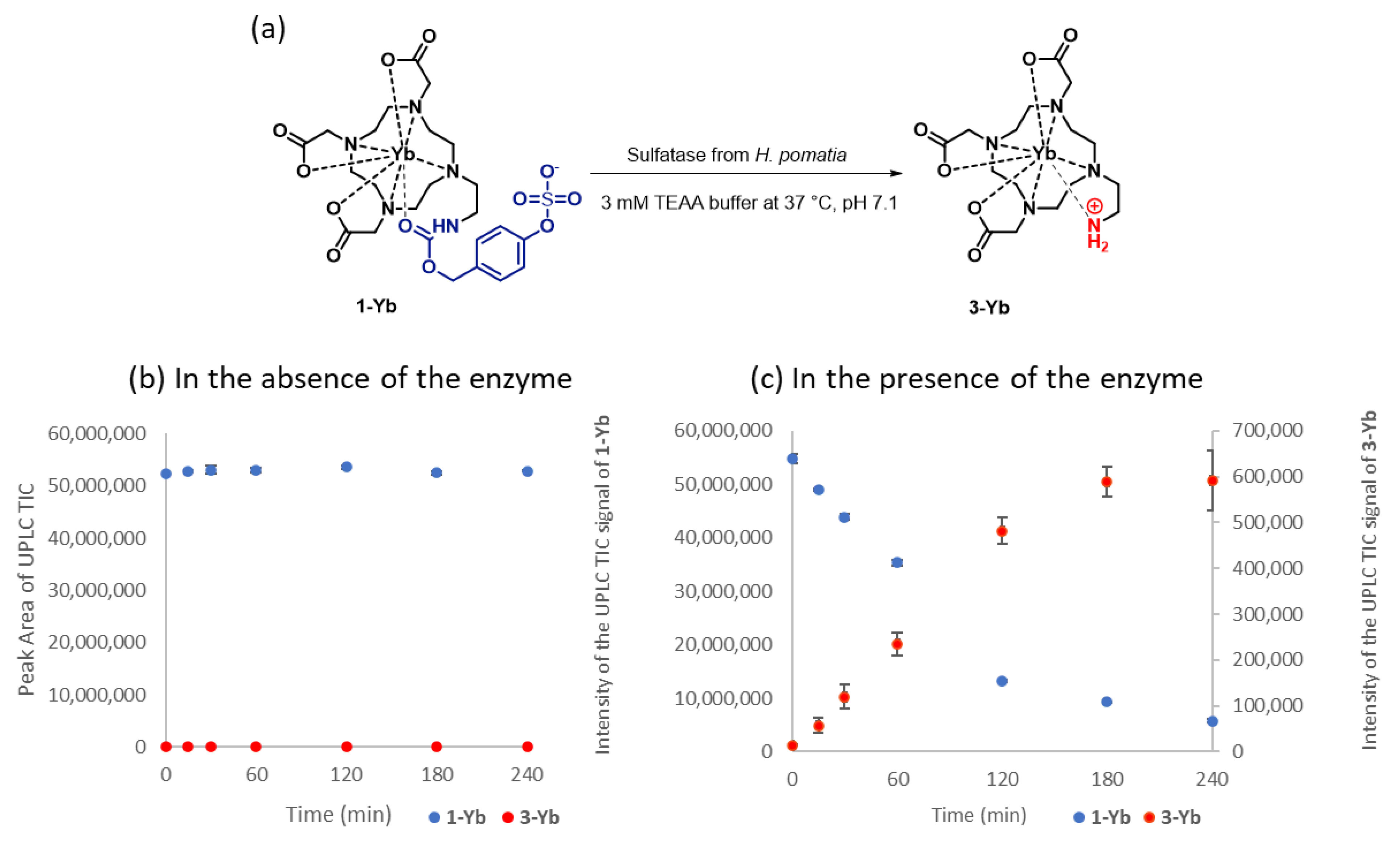
2.2. Enzymatic Hydrolysis
2.3. A Fast Field Cycling NMR Relaxometry Analysis of MRI Contrast Agent 1-Gd
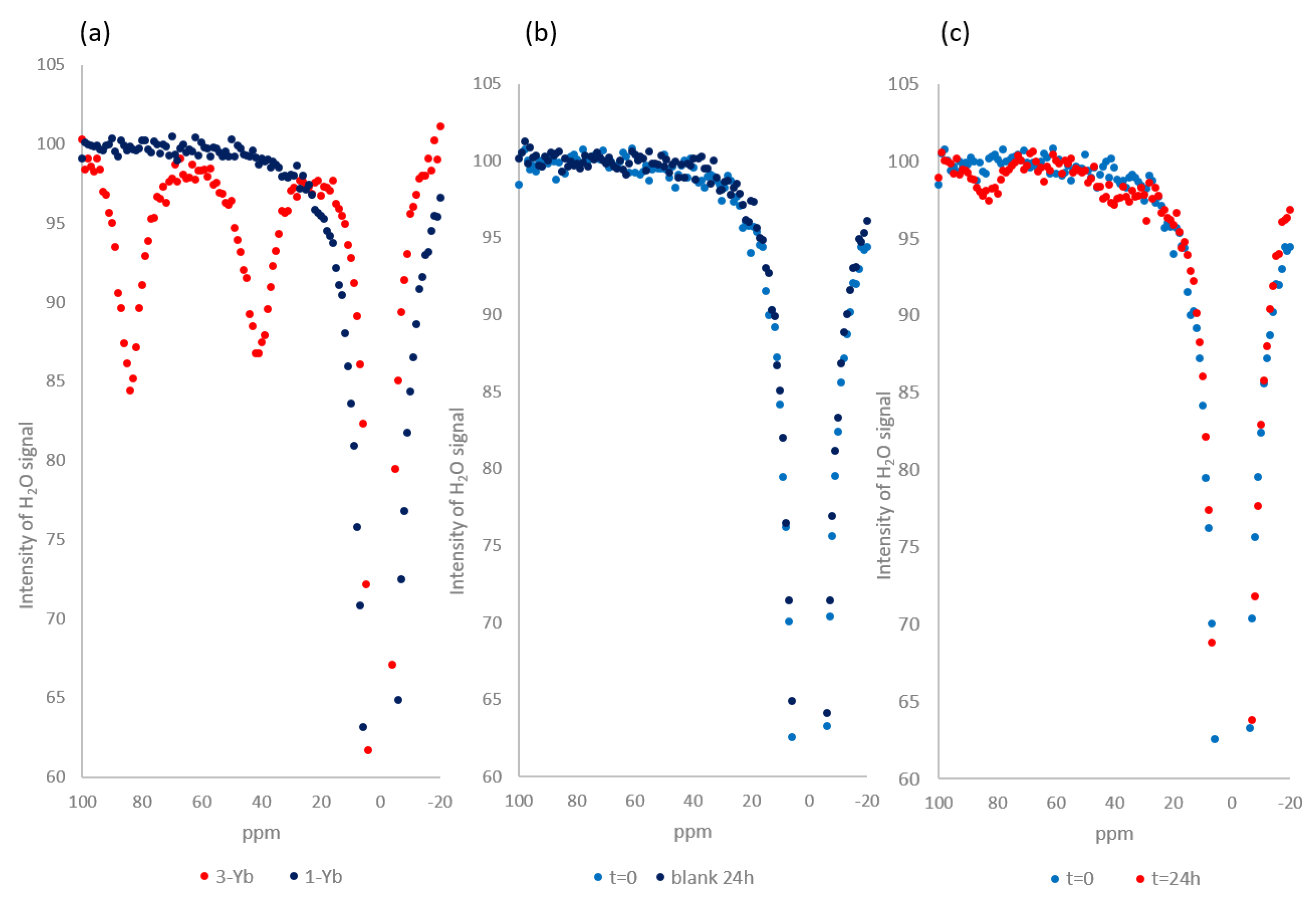
2.4. Z-Spectra Activity Analysis of 1-Yb for CEST Applications
3. Discussion
4. Materials and Methods
4.1. Synthetic Procedures and Spectroscopic Data
4.2. Enzymatic Hydrolysis
4.3. FFC Relaxometry
4.4. Z-spectra
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, M.L.; Gambhir, S.S. A molecular imaging primer: Modalities, imaging agents, and applications. Physiol. Rev. 2012, 92, 897–965. [Google Scholar] [CrossRef]
- Hengerer, J.G.A.; Grimm, J. Molecular magnetic resonance imaging. Biomed. Imaging Interv. J. 2006, 2, e8. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, Y.; Zhao, C.; Chen, Z.; Su, L.; Yang, H.; Song, J. Activated molecular probes for enzyme recognition and detection. Theranostics 2022, 12, 1459–1485. [Google Scholar] [CrossRef]
- Mishani, E.; Hagooly, A. Strategies for molecular imaging of epidermal growth factor receptor tyrosine kinase in cancer. J. Nucl. Med. 2009, 50, 1199–1202. [Google Scholar] [CrossRef]
- Reed, M.J.; Purohit, A.; Woo, L.W.L.; Newman, S.P.; Potter, B.V.L. Steroid sulfatase: Molecular biology, regulation, and inhibition. Endocr. Rev. 2005, 26, 171–202. [Google Scholar] [CrossRef]
- Lübke, T.; Damme, M. Lysosomal sulfatases: A growing family. Biochem. J. 2020, 477, 3963–3983. [Google Scholar] [CrossRef]
- Rižner, T.L.; Romano, A. Targeting the formation of estrogens for treatment of hormone dependent diseases–current status. Front. Pharmacol. 2023, 14, 1155558. [Google Scholar] [CrossRef]
- Anbar, H.S.; Isa, Z.; Elounais, J.J.; Jameel, M.A.; Zib, J.H.; Samer, A.M.; Jawad, A.F.; El-Gamal, M.I. Steroid sulfatase inhibitors: The current landscape. Expert Opin. Ther. Pat. 2021, 31, 453–472. [Google Scholar] [CrossRef]
- Rempel, B.P.; Price, E.W.; Phenix, C.P. Molecular Imaging of Hydrolytic Enzymes Using PET and SPECT. Mol. Imaging 2017, 16, 1536012117717852. [Google Scholar] [CrossRef]
- Brust, P.; Rodig, H.; Römer, J.; Kasch, H.; Bergmann, R.; Füchtner, F.; Zips, D.; Baumann, M.; Steinbach, J.; Johannsen, B. Distribution of 16α-[18F]fluoro-estradiol-3,17β-disulfamate in rats, tumour-bearing mice and piglets. Appl. Radiat. Isot. 2002, 57, 687–695. [Google Scholar] [CrossRef]
- Wang, M.; Mickens, J.; Gao, M.; Miller, K.D.; Sledge, G.W.; Hutchins, G.D.; Zheng, Q. Design and synthesis of carbon-11-labeled dual aromatase–steroid sulfatase inhibitors as new potential PET agents for imaging of aromatase and steroid sulfatase expression in breast cancer. Steroids 2009, 74, 896–905. [Google Scholar] [CrossRef]
- Xiang, M.; Jiang, Z.; Zhao, W.; Zhang, E.; Xia, L.; Kong, R.; Zhao, Y.; Kong, W.; Liu, X.; Qu, F.; et al. Activatable Near-Infrared Fluorescent and Photoacoustic Dual-Modal Probe for Highly Sensitive Imaging of Sulfatase In Vivo. ACS. Sens. 2023, 8, 2021–2029. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, M.; Zhang, W.; Liu, T.; Ren, X.; Gu, Y.; Ran, C.; Yang, J.; Wang, P. A sulfatase-activatable AIEgen nanoprobe for inhalation imaging-guided surgical excision of lung cancer. Chem. Eng. J. 2022, 428, 132514. [Google Scholar] [CrossRef]
- Ekanger, L.A.; Allen, M.J. Overcoming the concentration-dependence of responsive probes for magnetic resonance imaging. Metallomics 2015, 7, 405–421. [Google Scholar] [CrossRef]
- Alford, R.; Ogawa, M.; Choyke, P.L.; Kobayashi, H. Molecular probes for the in vivo imaging of cancer. Mol. Biosyst. 2009, 5, 1279–1291. [Google Scholar] [CrossRef]
- Hingorani, D.V.; Yoo, B.; Bernstein, A.S.; Pagel, M.D. Detecting enzyme activities with exogenous MRI contrast agents. Chem. A Eur. J. 2014, 20, 9840–9850. [Google Scholar] [CrossRef]
- Hingorani, D.V.; Bernstein, A.S.; Pagel, M.D. A review of responsive MRI contrast agents: 2005–2014. Contrast Media Mol. Imaging 2015, 10, 245–265. [Google Scholar] [CrossRef]
- Tóth, E.; Helm, L.; Merbach, A. Relaxivity of gadolinium (III) complexes: Theory and mechanism. In The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed.; Merbach, A., Helm, L., Tóth, E., Eds.; John Wiley & Sons: Chichester, UK, 2013; pp. 25–76. [Google Scholar]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI contrast agents: Current challenges and new frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef]
- Moats, R.A.; Fraser, S.E.; Meade, T.J. A ‘Smart’ Magnetic Resonance Imaging Agent That Reports on Specific Enzymatic Activity. Angen. Chem. Int. Ed. 1997, 36, 726–728. [Google Scholar] [CrossRef]
- Tang, J.H.; Li, H.; Yuan, C.; Parigi, G.; Luchinat, C.; Meade, J.H. Molecular Engineering of Self-Immolative Bioresponsive MR Probes. J. Am. Chem. Soc. 2023, 145, 10045–10050. [Google Scholar] [CrossRef]
- Nivorozhkin, A.L.; Kolodziej, A.F.; Caravan, P.; Greenfield, M.T.; Lauffer, R.B.; McMurry, T.J. Enzyme-Activated Gd3+ Magnetic Resonance Imaging Contrast Agents with a Prominent Receptor-Induced Magnetization Enhancement. Angew. Chem. 2001, 113, 2987–2990. [Google Scholar] [CrossRef]
- Milne, M.; Wu, Y.; Sherry, A.D. ParaCest Agents: Design, discovery and implementation. In Chemical Exchange Saturation Transfer Imaging: Advances and Applications, 1st ed.; McMahon, M.T., Gilad, A.A., Bulte, J.W.M., van Zijl, P.C.M., Eds.; Pan Standford Publishing: Singapore, 2017; pp. 219–252. [Google Scholar]
- Sinharay, S.; Fernández-Cuervo, G.; Acfalle, J.P.; Pagel, M.D. Detection of Sulfatase Enzyme Activity with a CatalyCEST MRI Contrast Agent. Chem. A Eur. J. 2016, 22, 6491–6495. [Google Scholar] [CrossRef]
- Chauvin, T.; Durand, P.; Bernier, M.; Meudal, H.; Doan, B.; Noury, F.; Badet, B.; Beloeil, J.; Tóth, E. Detection of enzymatic activity by PARACEST MRI: A general approach to target a large variety of enzymes. Angew. Chem. 2008, 47, 4370–4372. [Google Scholar] [CrossRef]
- Duimstra, J.A.; Femia, F.J.; Meade, T.J. A gadolinium chelate for detection of β-glucuronidase: A self-immolative approach. J. Am. Chem. Soc. 2005, 127, 12847–12855. [Google Scholar] [CrossRef]
- Giovenzana, G.B.; Negri, R.; Rolla, G.A.; Tei, L. Gd-Aminoethyl-DO3A Complexes: A Novel Class of pH-Sensitive MRI Contrast Agents. Eur. J. Inorg. Chem. 2012, 2012, 2035–2039. [Google Scholar] [CrossRef]
- Krchová, T.; Kotek, J.; Jirák, D.; Havlíčková, J.; Císařová, I.; Hermann, P. Lanthanide(iii) complexes of aminoethyl-DO3A as PARACEST contrast agents based on decoordination of the weakly bound amino group. Dalt. Trans. 2013, 42, 15735–15747. [Google Scholar] [CrossRef]
- Romanov-Michailidis, F.; Viton, F.; Fumeaux, R.; Lévèques, A.; Actis-Goretta, L.; Rein, M.; Williamson, G.; Barron, D. Epicatechin B-ring conjugates: First enantioselective synthesis and evidence for their occurrence in human biological fluids. Org. Lett. 2012, 14, 3902–3905. [Google Scholar] [CrossRef]
- Simpson, L.S.; Widlanski, T.S. A comprehensive approach to the synthesis of sulfate esters. J. Am. Chem. Soc. 2006, 128, 1605–1610. [Google Scholar] [CrossRef]
- De La Reberdière, A.; Lachaud, F.; Chuburu, F.; Cadiou, C.; Lemercier, G. Synthesis of a new family of protected 1,4,7,10-tetraazacyclododecane-1,4, 7-triacetic acid derivatives with thioctic acid pending arms. Tetrahedron Lett. 2012, 53, 6115–6118. [Google Scholar] [CrossRef]
- Muller, R.N.; Vander Elst, L.; Rinck, P.A.; Vallet, P.; Maton, F.; Fischer, H.; Roch, A.; Van Haverbeke, Y. The Importance of Nuclear Magnetic Relaxation Dispersion (NMRD) Profiles in MRI Contrast Media Development. Investig. Radiol. 1988, 23, S229–S231. [Google Scholar] [CrossRef]
- Aime, S.; Botta, M.; Esteban-Gómez, D.; Platas-Iglesias, C. Characterisation of magnetic resonance imaging (MRI) contrast agents using NMR relaxometry. Mol. Phys. 2019, 117, 898–909. [Google Scholar] [CrossRef]
- Reeßing, F.; Huijsse, S.E.M.; Dierckx, R.A.J.O.; Feringa, B.L.; Borra, R.J.H.; Szymański, W. A photocleavable contrast agent for light-responsive MRI. Pharmaceuticals 2020, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Liu, M.; Zhang, J.; Li, Y.; Jiang, L. Synthesis and characterization of DOTA-mono-adamantan-1-ylamide. Res. Chem. Intermed. 2015, 41, 5109–5119. [Google Scholar] [CrossRef]
- Barge, A.; Cravotto, G.; Gianolio, E.; Fedeli, F. How to determine free Gd and free ligand in solution of Gd chelates. A technical note. Contrast Media Mol. Imaging 2006, 1, 184–188. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welleman, I.M.; Reeβing, F.; Boersma, H.H.; Dierckx, R.A.J.O.; Feringa, B.L.; Szymanski, W. The Development of a Smart Magnetic Resonance Imaging and Chemical Exchange Saturation Transfer Contrast Agent for the Imaging of Sulfatase Activity. Pharmaceuticals 2023, 16, 1439. https://doi.org/10.3390/ph16101439
Welleman IM, Reeβing F, Boersma HH, Dierckx RAJO, Feringa BL, Szymanski W. The Development of a Smart Magnetic Resonance Imaging and Chemical Exchange Saturation Transfer Contrast Agent for the Imaging of Sulfatase Activity. Pharmaceuticals. 2023; 16(10):1439. https://doi.org/10.3390/ph16101439
Chicago/Turabian StyleWelleman, Ilse M., Friederike Reeβing, Hendrikus H. Boersma, Rudi A. J. O. Dierckx, Ben L. Feringa, and Wiktor Szymanski. 2023. "The Development of a Smart Magnetic Resonance Imaging and Chemical Exchange Saturation Transfer Contrast Agent for the Imaging of Sulfatase Activity" Pharmaceuticals 16, no. 10: 1439. https://doi.org/10.3390/ph16101439
APA StyleWelleman, I. M., Reeβing, F., Boersma, H. H., Dierckx, R. A. J. O., Feringa, B. L., & Szymanski, W. (2023). The Development of a Smart Magnetic Resonance Imaging and Chemical Exchange Saturation Transfer Contrast Agent for the Imaging of Sulfatase Activity. Pharmaceuticals, 16(10), 1439. https://doi.org/10.3390/ph16101439