
Montelukast and Acute Coronary Syndrome: The Endowed Drug

 ,
,  ,
,  , and
, and 
Abstract
{kind=link}
{kind=link}
{kind=link}
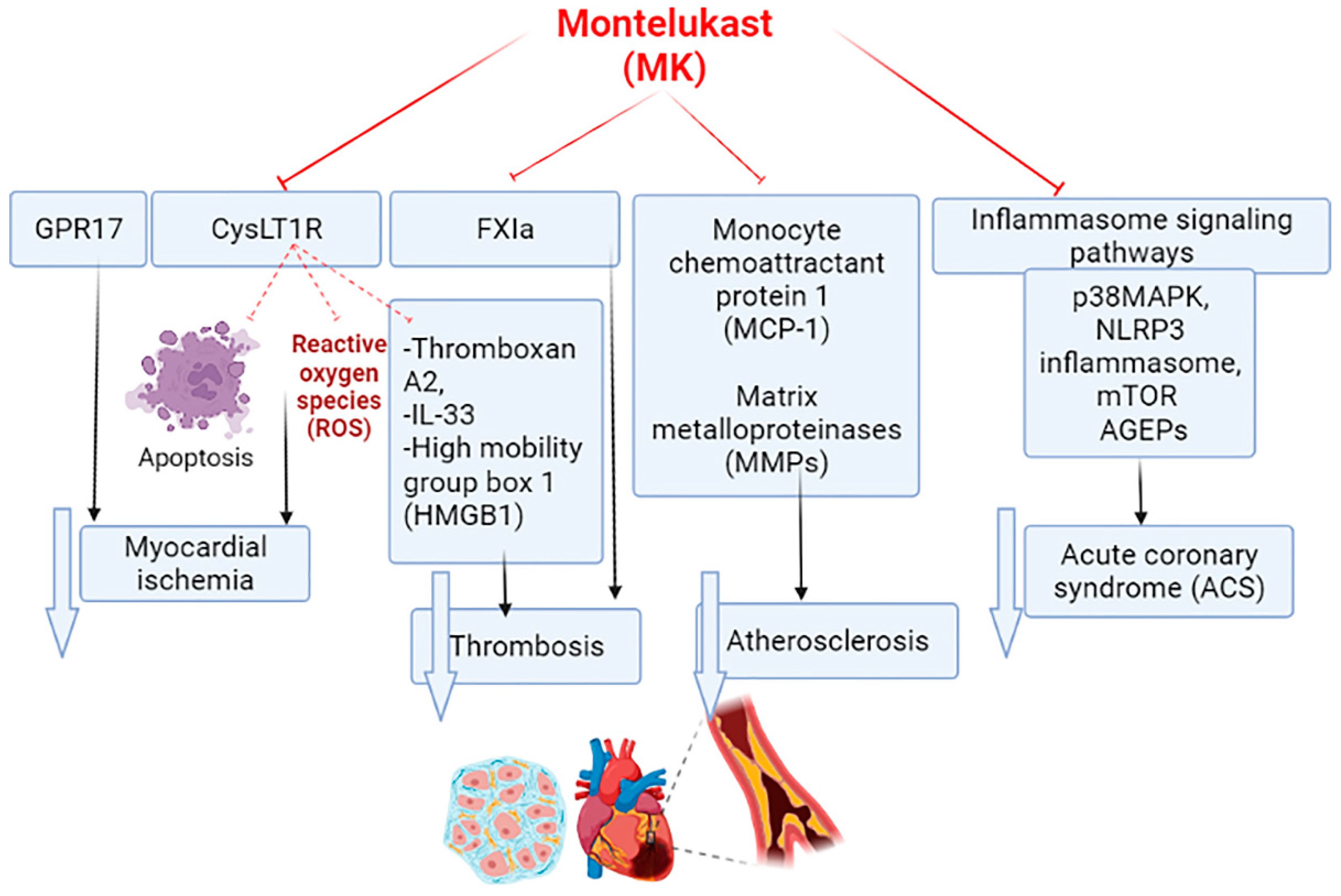{kind=link}
1. Introduction
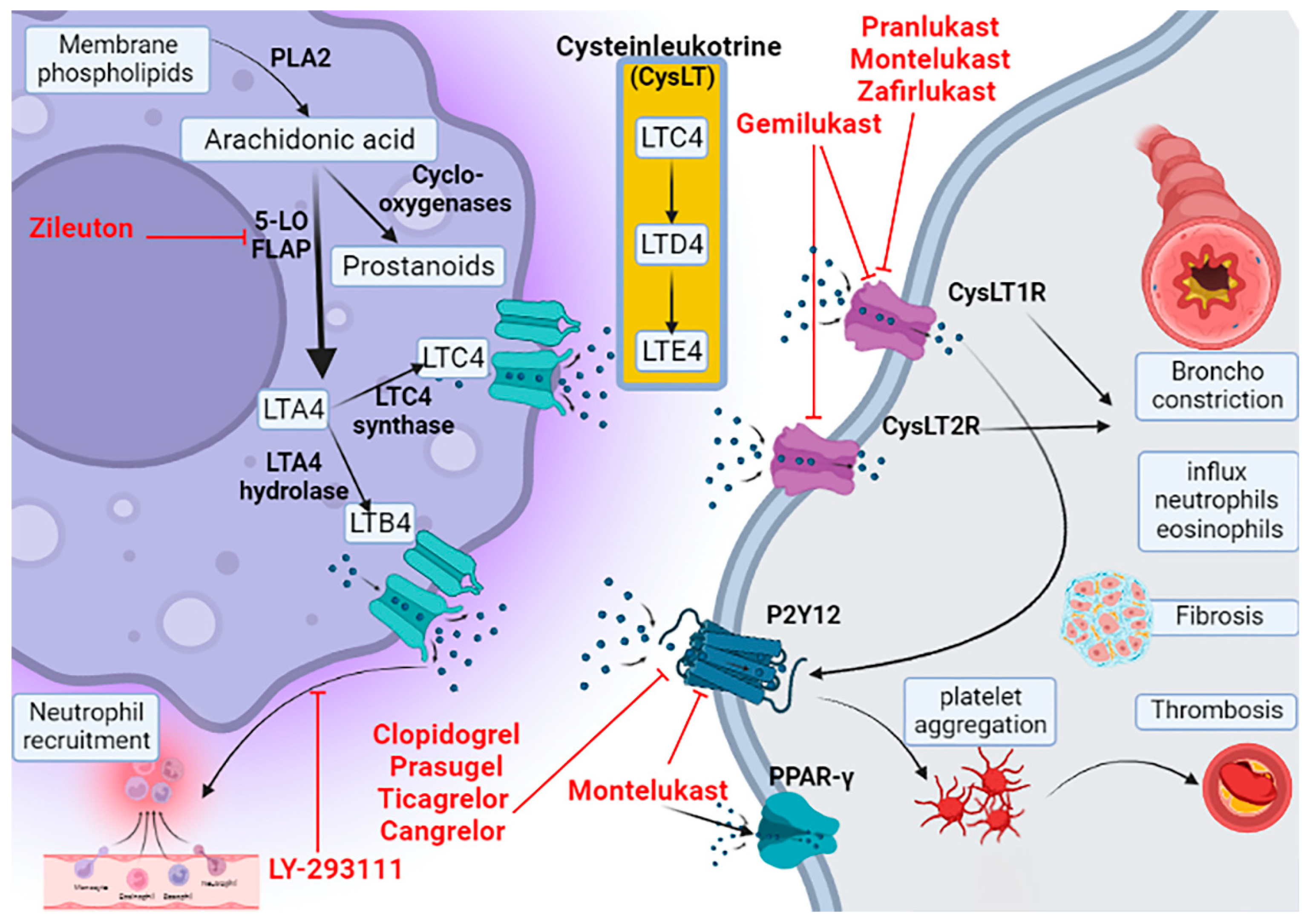
2. Leukotriene Pathway
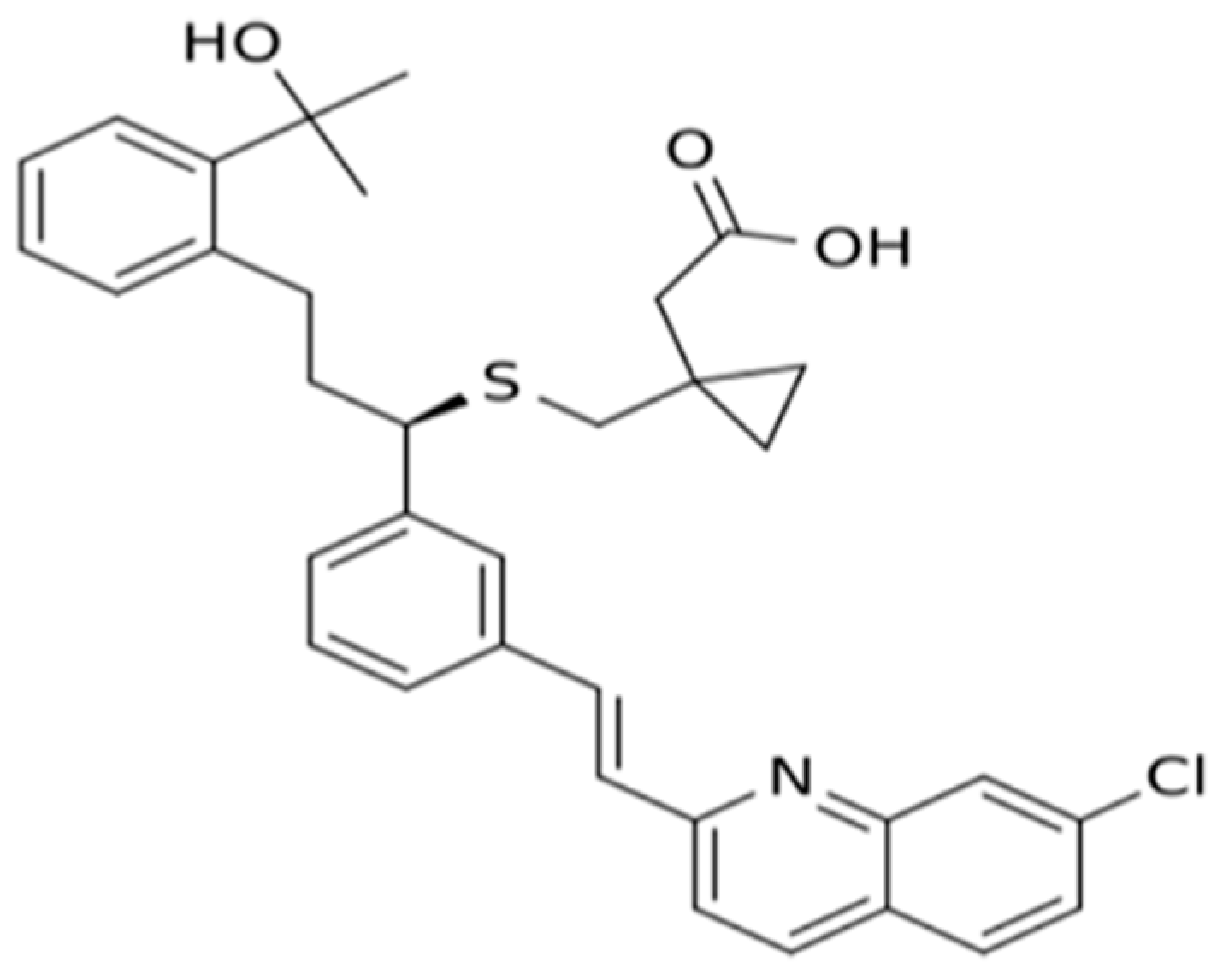
3. Pharmacology of Montelukast
4. Montelukast and Cardiovascular Complications
5. Montelukast and Acute Coronary Syndrome
6. Montelukast and Myocardial Infarction
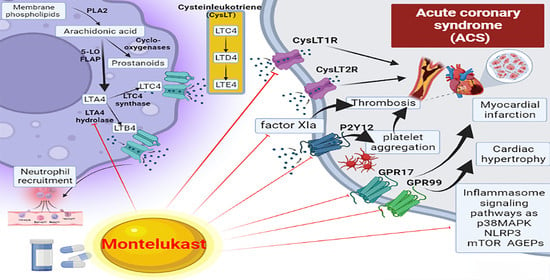
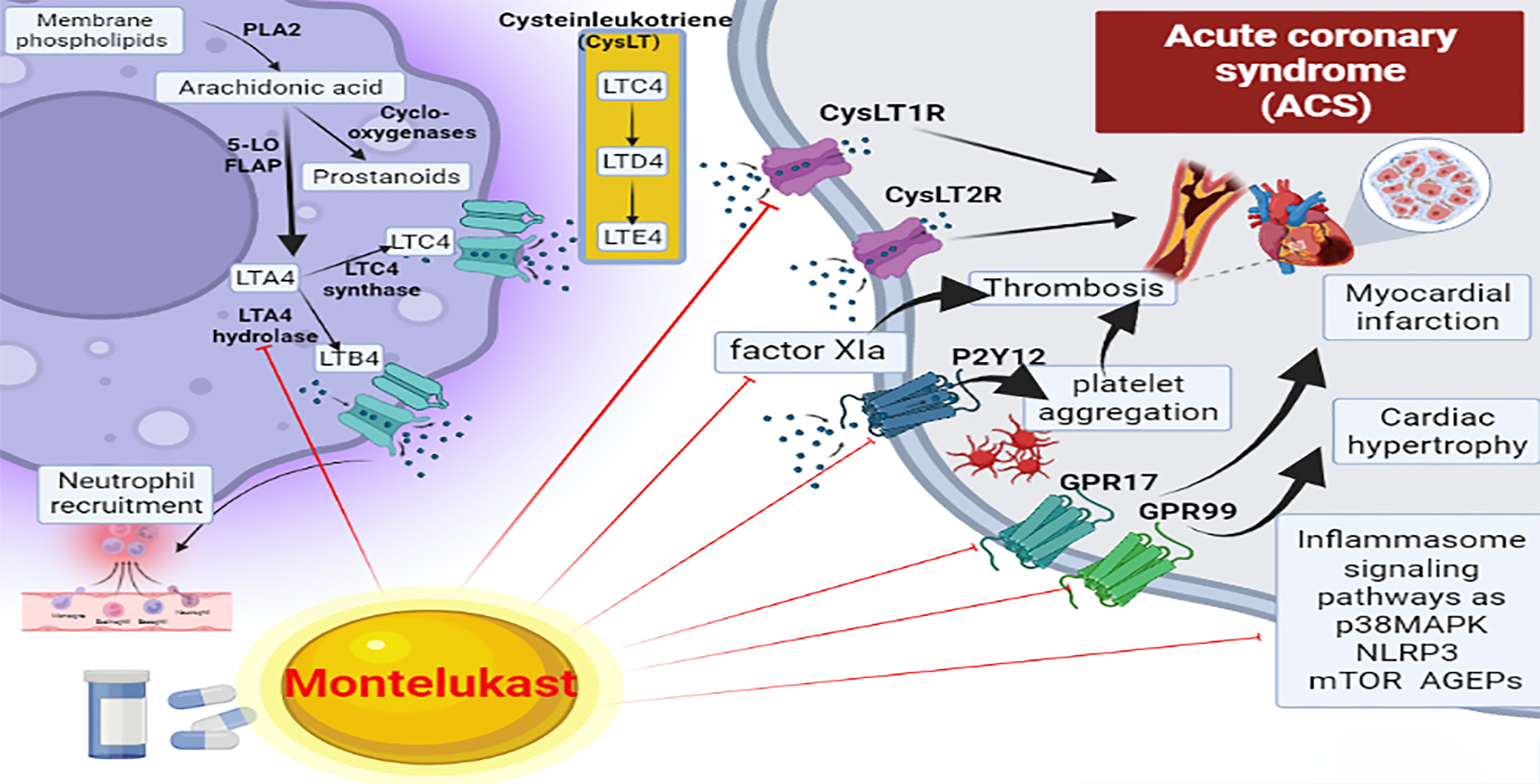
7. Effects of Montelukast on the Inflammatory Signaling Pathway in Acute Coronary Syndrome
8. Montelukast and Thrombosis in Acute Coronary Syndrome
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Al-Buhadilly, A.K. Rosuvastatin improves vaspin serum levels in obese patients with acute coronary syndrome. Diseases 2018, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Samy, O.M. Statin therapy improves serum Annexin A1 levels in patients with acute coronary syndrome: A case-controlled study. Int. J. Crit. Illn. Inj. Sci. 2021, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Awad, M.S.; Alrifai, S.B. Assessment of serum prolactin levels in acute myocardial infarction: The role of pharmacotherapy. Indian J. Endocrinol. Metab. 2016, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Welson, N.N.; Batiha, G.E.-S. Trimetazidine and COVID-19-induced acute cardiac injury: A missed key. Int. J. Clin. Pharm. 2022, 44, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Sami, O.M.; Al-kuraishy, H.M.; Al-Gareeb, A.I. Differential effects of statins on annexin A1 serum level in patients with acute coronary syndrome: A pleiotropic update. Ann. Trop. Med. Public Health 2021, 24, 394–402. [Google Scholar] [CrossRef]
- Mason, P.J.; Shah, B.; Tamis-Holland, J.E.; Bittl, J.A.; Cohen, M.G.; Safirstein, J.; Drachman, D.E.; Valle, J.A.; Rhodes, D.; Gilchrist, I.C. An update on radial artery access and best practices for transradial coronary angiography and intervention in acute coronary syndrome: A scientific statement from the American Heart Association. Circ. Cardiovasc. Interv. 2018, 11, e000035. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Hoxha, M.; Rovati, G.E.; Cavanillas, A.B. The leukotriene receptor antagonist montelukast and its possible role in the cardiovascular field. Eur. J. Clin. Pharmacol. 2017, 73, 799–809. [Google Scholar] [CrossRef]
- Kellaway, C.; Trethewie, E. The liberation of a slow-reacting smooth muscle-stimulating substance in anaphylaxis. Q. J. Exp. Physiol. Cogn. Med. Sci. Transl. Integr. 1940, 30, 121–145. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Almulaiky, Y.Q.; Cruz-Martins, N.; Batiha, G.E.-S. Role of leukotriene pathway and montelukast in pulmonary and extrapulmonary manifestations of Covid-19: The enigmatic entity. Eur. J. Pharmacol. 2021, 904, 174196. [Google Scholar] [CrossRef]
- Bankova, L.G.; Dwyer, D.F.; Yoshimoto, E.; Ualiyeva, S.; McGinty, J.W.; Raff, H.; von Moltke, J.; Kanaoka, Y.; Frank Austen, K.; Barrett, N.A. The cysteinyl leukotriene 3 receptor regulates expansion of IL-25–producing airway brush cells leading to type 2 inflammation. Sci. Immunol. 2018, 3, eaat9453. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Chen, Y.; Cai, Q. The role of the LTB4-BLT1 axis in health and disease. Pharmacol. Res. 2020, 158, 104857. [Google Scholar] [CrossRef] [PubMed]
- Alorabi, M.; Cavalu, S.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Mostafa-Hedeab, G.; Negm, W.A.; Youssef, A.; El-Kadem, A.H.; Saad, H.M.; Batiha, G.E.-S. Pentoxifylline and berberine mitigate diclofenac-induced acute nephrotoxicity in male rats via modulation of inflammation and oxidative stress. Biomed. Pharmacother. 2022, 152, 113225. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Fukutomi, Y.; Mitsui, C.; Kajiwara, K.; Watai, K.; Kamide, Y.; Nakamura, Y.; Hamada, Y.; Tomita, Y.; Sekiya, K. Omalizumab for aspirin hypersensitivity and leukotriene overproduction in aspirin-exacerbated respiratory disease. A randomized controlled trial. Am. J. Respir. Crit. Care Med. 2020, 201, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Batiha, G.E.-S.; Al-Gareeb, A.I.; Saad, H.M.; Al-Kuraishy, H.M. COVID-19 and corticosteroids: A narrative review. Inflammopharmacology 2022, 30, 1189–1205. [Google Scholar] [CrossRef]
- Möller, I.; Murali, R.; Müller, H.; Wiesner, T.; Jackett, L.A.; Scholz, S.L.; Cosgarea, I.; van de Nes, J.A.; Sucker, A.; Hillen, U. Activating cysteinyl leukotriene receptor 2 (CYSLTR2) mutations in blue nevi. Mod. Pathol. 2017, 30, 350–356. [Google Scholar] [CrossRef]
- Tantisira, K.G.; Drazen, J.M. Genetics and pharmacogenetics of the leukotriene pathway. J. Allergy Clin. Immunol. 2009, 124, 422–427. [Google Scholar] [CrossRef]
- Park, J.S.; Chang, H.S.; Park, C.-S.; Lee, J.-H.; Lee, Y.M.; Choi, J.H.; Park, H.-S.; Kim, L.H.; Park, B.L.; Choi, Y.H. Association analysis of cysteinyl-leukotriene receptor 2 (CYSLTR2) polymorphisms with aspirin intolerance in asthmatics. Pharmacogenet. Genom. 2005, 15, 483–492. [Google Scholar] [CrossRef][Green Version]
- Mostafa-Hedeab, G.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Jeandet, P.; Saad, H.M.; Batiha, G.E.-S. A raising dawn of pentoxifylline in management of inflammatory disorders in Covid-19. Inflammopharmacology 2022, 30, 799–809. [Google Scholar] [CrossRef]
- Itadani, S.; Yashiro, K.; Aratani, Y.; Sekiguchi, T.; Kinoshita, A.; Moriguchi, H.; Ohta, N.; Takahashi, S.; Ishida, A.; Tajima, Y. Discovery of gemilukast (ONO-6950), a dual CysLT1 and CysLT2 antagonist as a therapeutic agent for asthma. J. Med. Chem. 2015, 58, 6093–6113. [Google Scholar] [CrossRef]
- Calderón-Zamora, L.; Canizalez-Román, A.; León-Sicairos, N.; Aguilera-Mendez, A.; Huang, F.; Hong, E.; Villafaña, S. Changes in expression of orphan receptors GPR99 and GPR107 during the development and establishment of hypertension in spontaneously hypertensive rats. J. Recept. Signal Transduct. 2021, 41, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Raffaele, S.; Abbracchio, M.P.; Fumagalli, M. Regulation and signaling of the GPR17 receptor in oligodendroglial cells. Glia 2020, 68, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Steg, P.G.; Bhatt, D.L.; Capodanno, D.; Angiolillo, D.J. Cangrelor: Clinical data, contemporary use, and future perspectives. J. Am. Heart Assoc. 2021, 10, e022125. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.; Bachelot-Loza, C.; Nesseler, N.; Gaussem, P.; Gouin-Thibault, I. P2Y12 inhibition beyond thrombosis: Effects on inflammation. Int. J. Mol. Sci. 2020, 21, 1391. [Google Scholar] [CrossRef]
- Kang, J.H.; Lim, H.; Lee, D.S.; Yim, M. Montelukast inhibits RANKL-induced osteoclast formation and bone loss via CysLTR1 and P2Y12. Mol. Med. Rep. 2018, 18, 2387–2398. [Google Scholar] [CrossRef]
- Göbel, T.; Diehl, O.; Heering, J.; Merk, D.; Angioni, C.; Wittmann, S.K.; Buscato, E.l.; Kottke, R.; Weizel, L.; Schader, T. Zafirlukast is a dual modulator of human soluble epoxide hydrolase and peroxisome proliferator-activated receptor γ. Front. Pharmacol. 2019, 10, 263. [Google Scholar] [CrossRef]
- Fuse809 (talk)—Own work, Public Domain. Available online: https://commons.wikimedia.org/w/index.php?curid=31613539 (accessed on 18 August 2022).
- Sansing-Foster, V.; Haug, N.; Mosholder, A.; Cocoros, N.M.; Bradley, M.; Ma, Y.; Pennap, D.; Dee, E.C.; Toh, S.; Pestine, E. Risk of psychiatric adverse events among montelukast users. J. Allergy Clin. Immunol. Pract. 2021, 9, 385–393.e312. [Google Scholar] [CrossRef]
- Glockler-Lauf, S.D.; Finkelstein, Y.; Zhu, J.; Feldman, L.Y.; To, T. Montelukast and neuropsychiatric events in children with asthma: A nested case–control study. J. Pediatr. 2019, 209, 176–182.e174. [Google Scholar] [CrossRef]
- Fox, C.W.; Khaw, C.L.; Gerke, A.K.; Lund, B.C. Montelukast and neuropsychiatric events–a sequence symmetry analysis. J. Asthma 2021, 1–7. [Google Scholar] [CrossRef]
- Hirvensalo, P.; Tornio, A.; Neuvonen, M.; Tapaninen, T.; Paile-Hyvärinen, M.; Kärjä, V.; Männistö, V.T.; Pihlajamäki, J.; Backman, J.T.; Niemi, M. Comprehensive pharmacogenomic study reveals an important role of UGT1A3 in montelukast pharmacokinetics. Clin. Pharmacol. Ther. 2018, 104, 158–168. [Google Scholar] [CrossRef]
- Colazzo, F.; Gelosa, P.; Tremoli, E.; Sironi, L.; Castiglioni, L. Role of the cysteinyl leukotrienes in the pathogenesis and progression of cardiovascular diseases. Mediat. Inflamm. 2017, 2017, 2432958. [Google Scholar] [CrossRef] [PubMed]
- Folco, G.; Rossoni, G.; Buccellati, C.; Berti, F.; Maclouf, J.; Sala, A. Leukotrienes in cardiovascular diseases. Am. J. Respir. Crit. Care Med. 2000, 161, S112–S116. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Amin, E.F.; Ibrahim, S.A.; Abdelzaher, W.Y.; Abdelrahman, A.M. Montelukast and irbesartan ameliorate metabolic and hepatic disorders in fructose-induced metabolic syndrome in rats. Eur. J. Pharmacol. 2014, 724, 204–210. [Google Scholar] [CrossRef]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef]
- Şener, G.; Şehirli, Ö.; Velioğlu-Öğünç, A.; Çetinel, Ş.; Gedik, N.; Caner, M.; Sakarcan, A.; Yeğen, B.Ç. Montelukast protects against renal ischemia/reperfusion injury in rats. Pharmacol. Res. 2006, 54, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Zhou, G.; Cheng, S.; Liu, D.; Xu, J.; Xu, G.; Liu, X. Anti-atherogenic effects of montelukast associated with reduced MCP-1 expression in a rabbit carotid balloon injury model. Atherosclerosis 2009, 205, 74–79. [Google Scholar] [CrossRef]
- Liu, D.; Ge, S.; Zhou, G.; Xu, G.; Zhang, R.; Zhu, W.; Liu, Z.; Cheng, S.; Liu, X. Montelukast inhibits matrix metalloproteinases expression in atherosclerotic rabbits. Cardiovasc. Drugs Ther. 2009, 23, 431–437. [Google Scholar] [CrossRef]
- Wang, Y.-X.J.; Ulu, A.; Zhang, L.-N.; Hammock, B. Soluble epoxide hydrolase in atherosclerosis. Curr. Atheroscler. Rep. 2010, 12, 174–183. [Google Scholar] [CrossRef]
- Cipollone, F.; Mezzetti, A.; Fazia, M.L.; Cuccurullo, C.; Iezzi, A.; Ucchino, S.; Spigonardo, F.; Bucci, M.; Cuccurullo, F.; Prescott, S.M. Association between 5-lipoxygenase expression and plaque instability in humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1665–1670. [Google Scholar] [CrossRef]
- Allayee, H.; Hartiala, J.; Lee, W.; Mehrabian, M.; Irvin, C.G.; Conti, D.V.; Lima, J.J. The effect of montelukast and low-dose theophylline on cardiovascular disease risk factors in asthmatics. Chest 2007, 132, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc. Drugs Ther. 2009, 23, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Manolescu, A.; Thorleifsson, G.; Gretarsdottir, S.; Jonsdottir, H.; Thorsteinsdottir, U.; Samani, N.J.; Gudmundsson, G.; Grant, S.F.; Thorgeirsson, G. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat. Genet. 2004, 36, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Mehrabian, M.; Allayee, H.; Wong, J.; Shih, W.; Wang, X.-P.; Shaposhnik, Z.; Funk, C.D.; Lusis, A.J. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ. Res. 2002, 91, 120–126. [Google Scholar] [CrossRef]
- Zhou, G.; Ge, S.; Liu, D.; Xu, G.; Zhang, R.; Yin, Q.; Zhu, W.; Chen, J.; Liu, X. Atorvastatin reduces plaque vulnerability in an atherosclerotic rabbit model by altering the 5-lipoxygenase pathway. Cardiology 2010, 115, 221–228. [Google Scholar] [CrossRef]
- Bevan, S.; Lorenz, M.W.; Sitzer, M.; Markus, H.S. Genetic variation in the leukotriene pathway and carotid intima-media thickness: A 2-stage replication study. Stroke 2009, 40, 696–701. [Google Scholar] [CrossRef]
- Dwyer, J.H.; Allayee, H.; Dwyer, K.M.; Fan, J.; Wu, H.; Mar, R.; Lusis, A.J.; Mehrabian, M. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N. Engl. J. Med. 2004, 350, 29–37. [Google Scholar] [CrossRef]
- Nobili, E.; Salvado, M.D.; Folkersen, L.; Castiglioni, L.; Kastrup, J.; Wetterholm, A.; Tremoli, E.; Hansson, G.K.; Sironi, L.; Haeggström, J.Z. Cysteinyl leukotriene signaling aggravates myocardial hypoxia in experimental atherosclerotic heart disease. PLoS ONE 2012, 7, e41786. [Google Scholar] [CrossRef]
- Duah, E.; Adapala, R.K.; Al-Azzam, N.; Kondeti, V.; Gombedza, F.; Thodeti, C.K.; Paruchuri, S. Cysteinyl leukotrienes regulate endothelial cell inflammatory and proliferative signals through CysLT2 and CysLT1 receptors. Sci. Rep. 2013, 3, 3274. [Google Scholar] [CrossRef]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef]
- Papayianni, A.; Serhan, C.N.; Brady, H.R. Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells. J. Immunol. 1996, 156, 2264–2272. [Google Scholar] [PubMed]
- Uzonyi, B.; Lötzer, K.; Jahn, S.; Kramer, C.; Hildner, M.; Bretschneider, E.; Radke, D.; Beer, M.; Vollandt, R.; Evans, J.F. Cysteinyl leukotriene 2 receptor and protease-activated receptor 1 activate strongly correlated early genes in human endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6326–6331. [Google Scholar] [CrossRef] [PubMed]
- Omede, A. Role of Alpha-Ketoglutarate Receptor G-Protein Coupled Receptor 99 (GPR99) in Cardiac Hypertrophy; The University of Manchester: Manchester, UK, 2015. [Google Scholar]
- Nguyen, A.V.; Bagood, M.D.; Wang, M.; Caryotakis, S.E.; Smith, G.; Yee, S.; Shen, H.; Isseroff, R.R.; Soulika, A.M. Montelukast, an Antagonist of Cysteinyl Leukotriene Signaling, Impairs Burn Wound Healing. Plast. Reconstr. Surg. 2022, 150, 92e–104e. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Liu, Z.; Shi, T.; Wang, Q.; Li, D.; Wu, Y.; Han, J.; Guo, S.; Tang, B.; et al. DanQi Pill protects against heart failure through the arachidonic acid metabolism pathway by attenuating different cyclooxygenases and leukotrienes B4. BMC Complement. Altern. Med. 2014, 14, 67. [Google Scholar] [CrossRef]
- Zhao, X.; Du, J.-Q.; Xu, D.-Y.; Zhao, S.-P. Effects of soluble epoxide hydrolase inhibitor on the expression of fatty acid synthase in peripheral blood mononuclear cell in patients with acute coronary syndrome. Lipids Heal. Dis. 2013, 12, 3. [Google Scholar] [CrossRef]
- Oni-Orisan, A.; Cresci, S.; Jones, P.G.; Theken, K.N.; Spertus, J.A.; Lee, C.R. Association between the EPHX2 p. Lys55Arg polymorphism and prognosis following an acute coronary syndrome. Prostaglandins Other Lipid Mediat. 2018, 138, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Motoki, A.; Merkel, M.J.; Packwood, W.H.; Cao, Z.; Liu, L.; Iliff, J.; Alkayed, N.J.; Van Winkle, D.M. Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am. J. Physiol.-Heart Circ. Physiol. 2008, 295, H2128–H2134. [Google Scholar] [CrossRef]
- Libby, P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001, 104, 365–372. [Google Scholar] [CrossRef]
- Ali, S.A.-J.; Al-Shalah, H.; Al-Salihi, O. Assessment of circulating PPAR-γ Level as a risk and diagnostic biomarker in Acute Coronary Syndrome. J. Univ. Babylon Pure Appl. Sci. 2018, 26, 210–218. [Google Scholar] [CrossRef]
- Arat, A.; Yılmaz, Ü.; Yılmaz, N.; Fazlıoğulları, O.; Çelik, F.; Başaran, C.; Zeybek, Ü. Effects of Leptin, Resistin, and PPAR-Gama Gene Variants on Obese Patients with Acute Coronary Syndrome in the Turkish Population. JAREM. J. Acad. Res. Med. 2020, 10, 166. [Google Scholar] [CrossRef]
- Fujimori, K.; Uno, S.; Kuroda, K.; Matsumoto, C.; Maehara, T. Leukotriene C4 synthase is a novel PPARγ target gene, and leukotriene C4 and D4 activate adipogenesis through cysteinyl LT1 receptors in adipocytes. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119203. [Google Scholar] [CrossRef] [PubMed]
- Iovannisci, D.M.; Lammer, E.J.; Steiner, L.; Cheng, S.; Mahoney, L.T.; Davis, P.H.; Lauer, R.M.; Burns, T.L. Association between a leukotriene C4 synthase gene promoter polymorphism and coronary artery calcium in young women: The Muscatine Study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D.; Feinmark, S.J.; Cannon, P.J. Leukotriene biosynthesis by canine and human coronary arteries. J. Pharmacol. Exp. Ther. 1987, 241, 763–770. [Google Scholar] [PubMed]
- Vigorito, C.; Giordano, A.; Cirillo, R.; Genovese, A.; Rengo, F.; Marone, G. Metabolic and hemodynamic effects of peptide leukotriene C4 and D4 in man. Int. J. Clin. Lab. Res. 1997, 27, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Eaton, A.; Nagy, E.; Pacault, M.; Fauconnier, J.; Bäck, M. Cysteinyl leukotriene signaling through perinuclear CysLT1 receptors on vascular smooth muscle cells transduces nuclear calcium signaling and alterations of gene expression. J. Mol. Med. 2012, 90, 1223–1231. [Google Scholar] [CrossRef]
- Loötzer, K.; Spanbroek, R.; Hildner, M.; Urbach, A.; Heller, R.; Bretschneider, E.; Galczenski, H.; Evans, J.F.; Habenicht, A.J. Differential Leukotriene Receptor Expression and Calcium Responses in Endothelial Cells and Macrophages Indicate 5-Lipoxygenase–Dependent Circuits of Inflammation and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, e32–e36. [Google Scholar]
- Peters-Golden, M.; Henderson, W.R., Jr. Leukotrienes. N. Engl. J. Med. 2007, 357, 1841–1854. [Google Scholar] [CrossRef]
- Allen, S.P.; Sampson, A.P.; Piper, P.J.; Chester, A.H.; Ohri, S.K.; Yacoub, M.H. Enhanced excretion of urinary leukotriene E4 in coronary artery disease and after coronary artery bypass surgery. Coron. Artery Dis. 1993, 4, 899–904. [Google Scholar] [CrossRef]
- Ni, N.C.; Ballantyne, L.L.; Mewburn, J.D.; Funk, C.D. Multiple-site activation of the cysteinyl leukotriene receptor 2 is required for exacerbation of ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 321–330. [Google Scholar] [CrossRef]
- Gross, G.J.; Falck, J.R.; Gross, E.R.; Isbell, M.; Moore, J.; Nithipatikom, K. Cytochrome P450 and arachidonic acid metabolites: Role in myocardial ischemia/reperfusion injury revisited. Cardiovasc. Res. 2005, 68, 18–25. [Google Scholar] [CrossRef]
- Scott, W.; Pawlowski, N.; Andreach, M.; Cohn, Z. Resting macrophages produce distinct metabolites from exogenous arachidonic acid. J. Exp. Med. 1982, 155, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Becher, U.M.; Ghanem, A.; Tiyerili, V.; Fürst, D.O.; Nickenig, G.; Mueller, C.F. Inhibition of leukotriene C4 action reduces oxidative stress and apoptosis in cardiomyocytes and impedes remodeling after myocardial injury. J. Mol. Cell. Cardiol. 2011, 50, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tian, R.; Neubauer, S.; Gaudron, P.; Hu, K.; Ertl, G. Effects of LTD4 and its specific antagonist L-660,711 in isolated rat hearts with chronic myocardial infarction. Am. J. Physiol.-Heart Circ. Physiol. 1994, 266, H2068–H2073. [Google Scholar] [CrossRef]
- Ni, N.C.; Yan, D.; Ballantyne, L.L.; Barajas-Espinosa, A.; Amand, T.S.; Pratt, D.A.; Funk, C.D. A selective cysteinyl leukotriene receptor 2 antagonist blocks myocardial ischemia/reperfusion injury and vascular permeability in mice. J. Pharmacol. Exp. Ther. 2011, 339, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hall, S.R.; Moos, M.P.; Cao, R.Y.; Ishii, S.; Ogunyankin, K.O.; Melo, L.G.; Funk, C.D. Endothelial cysteinyl leukotriene 2 receptor expression mediates myocardial ischemia-reperfusion injury. Am. J. Pathol. 2008, 172, 592–602. [Google Scholar] [CrossRef]
- Qi, A.-D.; Harden, T.K.; Nicholas, R.A. Is GPR17 a P2Y/leukotriene receptor? examination of uracil nucleotides, nucleotide sugars, and cysteinyl leukotrienes as agonists of GPR17. J. Pharmacol. Exp. Ther. 2013, 347, 38–46. [Google Scholar] [CrossRef]
- Carry, M.; Korley, V.; Willerson, J.T.; Weigelt, L.; Ford-Hutchinson, A.W.; Tagari, P. Increased urinary leukotriene excretion in patients with cardiac ischemia. In vivo evidence for 5-lipoxygenase activation. Circulation 1992, 85, 230–236. [Google Scholar] [CrossRef]
- Prescott, E.; Pernow, J.; Saraste, A.; Åkerblom, A.; Angerås, O.; Erlinge, D.; Grove, E.L.; Hedman, M.; Jensen, L.O.; Svedlund, S. Design and rationale of FLAVOUR: A phase IIa efficacy study of the 5-lipoxygenase activating protein antagonist AZD5718 in patients with recent myocardial infarction. Contemp. Clin. Trials Commun. 2020, 19, 100629. [Google Scholar] [CrossRef]
- Lee, C.C.; Appleyard, R.F.; Byrne, J.G.; Cohn, L.H. Leukotrienes D4 and E4 produced in myocardium impair coronary flow and ventricular function after two hours of global ischaemia in rat heart. Cardiovasc. Res. 1993, 27, 770–773. [Google Scholar] [CrossRef]
- Rossoni, G.; Sala, A.; Berti, F.; Testa, T.; Buccellati, C.; Molta, C.; Muller-Peddinghaus, R.; Maclouf, J.; Folco, G. Myocardial protection by the leukotriene synthesis inhibitor BAY X1005: Importance of transcellular biosynthesis of cysteinyl-leukotrienes. J. Pharmacol. Exp. Ther. 1996, 276, 335–341. [Google Scholar]
- Shekher, A.; Singh, M. Role of eicosanoid inhibition of ischemia reperfusion injury: Intact and isolated rat heart studies. Methods Find. Exp. Clin. Pharmacol. 1997, 19, 223–229. [Google Scholar] [PubMed]
- Hahn, R.A.; MacDonald, B.R.; Simpson, P.J.; Potts, B.D.; Parli, C.J. Antagonism of leukotriene B4 receptors does not limit canine myocardial infarct size. J. Pharmacol. Exp. Ther. 1990, 253, 58–66. [Google Scholar] [PubMed]
- Helgadottir, A.; Manolescu, A.; Helgason, A.; Thorleifsson, G.; Thorsteinsdottir, U.; Gudbjartsson, D.F.; Gretarsdottir, S.; Magnusson, K.P.; Gudmundsson, G.; Hicks, A. A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat. Genet. 2006, 38, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Nakaya, M.; Ohara, H.; Nishihara, H.; Watari, K.; Nagasaka, A.; Nakaya, T.; Sugiura, Y.; Okuno, T.; Koga, T. Leukotriene B4 receptor 1 exacerbates inflammation following myocardial infarction. FASEB J. 2020, 34, 8749–8763. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, M.; Lewis-Mikhael, A.-M.; Bueno-Cavanillas, A. Potential role of leukotriene receptor antagonists in reducing cardiovascular and cerbrovascular risk: A systematic review of human clinical trials and in vivo animal studies. Biomed. Pharmacother. 2018, 106, 956–965. [Google Scholar] [CrossRef]
- Khodir, A.; Ghoneim, H.; Rahim, M.; Suddek, G. Montelukast attenuates lipopolysaccharide-induced cardiac injury in rats. Hum. Exp. Toxicol. 2016, 35, 388–397. [Google Scholar] [CrossRef]
- Stodółkiewicz, E.; Sokołowska, B.; Rzeszutko, M.; Tomala, M.; Chrustowicz, A.; Żmudka, K.; Sanak, M.; Szczeklik, W. Leukotriene biosynthesis in coronary artery disease: Results of the Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) study. Pol. Arch. Med. Wewnętrznej Pol. Arch. Intern. Med. 2018, 128, 43–51. [Google Scholar] [CrossRef]
- Vroegindewey, M.M.; Buljubasic, N.; Oemrawsingh, R.M.; Kardys, I.; Asselbergs, F.W.; van der Harst, P.; Umans, V.A.; Kietselaer, B.; Lenderink, T.; Liem, A. MAPK-cascade stimulating biomarkers in relation to recurrent coronary events following an acute coronary syndrome. In The Prognostic Value of Coronary Imaging and Biomarkers in Ischemic Heart Disease; Gildeprint: Enschede, The Netherlands, 2019; p. 123. [Google Scholar]
- Wang, L.; Li, D.; Yang, K.; Hu, Y.; Zeng, Q. Toll-like receptor-4 and mitogen-activated protein kinase signal system are involved in activation of dendritic cells in patients with acute coronary syndrome. Immunology 2008, 125, 122–130. [Google Scholar] [CrossRef]
- Indolfi, C.; Gasparri, C.; Vicinanza, C.; De Serio, D.; Boncompagni, D.; Mongiardo, A.; Spaccarotella, C.; Agosti, V.; Torella, D.; Curcio, A. Mitogen-activated protein kinases activation in T lymphocytes of patients with acute coronary syndromes. Basic Res. Cardiol. 2011, 106, 667–679. [Google Scholar] [CrossRef]
- Wang, S.; Su, X.; Xu, L.; Chang, C.; Yao, Y.; Komal, S.; Cha, X.; Zang, M.; Ouyang, X.; Zhang, L. Glycogen synthase kinase-3β inhibition alleviates activation of the NLRP3 inflammasome in myocardial infarction. J. Mol. Cell. Cardiol. 2020, 149, 82–94. [Google Scholar] [CrossRef]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 inflammasome in acute myocardial infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhou, Z.; Liu, W.; Chang, Q.; Sun, G.; Dai, Y. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int. J. Biochem. Cell Biol. 2017, 84, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Suhara, T.; Baba, Y.; Shimada, B.K.; Higa, J.K.; Matsui, T. The mTOR signaling pathway in myocardial dysfunction in type 2 diabetes mellitus. Curr. Diab. Rep. 2017, 17, 38. [Google Scholar] [CrossRef]
- Blackburn, N.J.R.; Vulesevic, B.; McNeill, B.; Cimenci, C.E.; Ahmadi, A.; Gonzalez-Gomez, M.; Ostojic, A.; Zhong, Z.; Brownlee, M.; Beisswenger, P.J.; et al. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res. Cardiol. 2017, 112, 57. [Google Scholar] [CrossRef] [PubMed]
- Mansour, R.M.; Ahmed, M.A.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Shi, X.; Huang, H.; Zhu, Y.; Wu, Y. Montelukast attenuates neuropathic pain through inhibiting p38 mitogen-activated protein kinase and nuclear factor-kappa B in a rat model of chronic constriction injury. Anesth. Analg. 2014, 118, 1090–1096. [Google Scholar] [CrossRef]
- Fei, Z.; Zhang, L.; Wang, L.; Jiang, H.; Peng, A. Montelukast ameliorated pemetrexed-induced cytotoxicity in hepatocytes by mitigating endoplasmic reticulum (ER) stress and nucleotide oligomerization domain-like receptor protein 3 (NLRP3) activation. Bioengineered 2022, 13, 7894–7903. [Google Scholar] [CrossRef]
- El-Kashef, D.H.; Zaghloul, R.A. Ameliorative effect of montelukast against carbon tetrachloride-induced hepatotoxicity: Targeting NLRP3 inflammasome pathway. Life Sci. 2022, 304, 120707. [Google Scholar] [CrossRef]
- Tong, J.; Yu, Q.; Xu, W.; Yu, W.; Wu, C.; Wu, Y.; Yan, H. Montelukast enhances cytocidal effects of carfilzomib in multiple myeloma by inhibiting mTOR pathway. Cancer Biol. Ther. 2019, 20, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Wu, S.; Wen, C.; Li, Y.; Liu, N.; Huang, J.; Li, L.; Fu, M.; Liu, J. Zafirlukast attenuates advanced glycation end-products (AGEs)-induced degradation of articular extracellular matrix (ECM). Int. Immunopharmacol. 2019, 68, 68–73. [Google Scholar] [CrossRef]
- Abbate, R.; Cioni, G.; Ricci, I.; Miranda, M.; Gori, A.M. Thrombosis and acute coronary syndrome. Thromb. Res. 2012, 129, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Inflammation, thrombosis and acute coronary syndromes. Diabetes Vasc. Dis. Res. 2005, 2, 113–121. [Google Scholar] [CrossRef]
- Duman, H.; Çinier, G.; Bakırcı, E.M.; Duman, H.; Şimşek, Z.; Hamur, H.; Değirmenci, H.; Emlek, N. Relationship between C-reactive protein to albumin ratio and thrombus burden in patients with acute coronary syndrome. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618824418. [Google Scholar] [CrossRef] [PubMed]
- Gorog, D.A.; Price, S.; Sibbing, D.; Baumbach, A.; Capodanno, D.; Gigante, B.; Halvorsen, S.; Huber, K.; Lettino, M.; Leonardi, S.; et al. Antithrombotic therapy in patients with acute coronary syndrome complicated by cardiogenic shock or out-of-hospital cardiac arrest: A joint position paper from the European Society of Cardiology (ESC) Working Group on Thrombosis, in association with the Acute Cardiovascular Care Association (ACCA) and European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Babalghith, A.O.; Al-kuraishy, H.M.; Al-Gareeb, A.I.; De Waard, M.; Sabatier, J.-M.; Saad, H.M.; Batiha, G.E.-S. The Potential Role of Growth Differentiation Factor 15 in COVID-19: A Corollary Subjective Effect or Not? Diagnostics 2022, 12, 2051. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, Y.; Qi, Y.; Song, M.; Yao, H.; Liao, C.; Lin, H.; Huang, M.; Zhuo, D.; Jiang, L. Antithrombotic Effects of Montelukast by Targeting Coagulation Factor XIa. Preprint. 2021. [Google Scholar]
- Houard, X.; Ollivier, V.; Louedec, L.; Michel, J.B.; Back, M. Differential inflammatory activity across human abdominal aortic aneurysms reveals neutrophilderived leukotriene B4 as a major chemotactic factor released from the intraluminal thrombus. FASEB J. 2009, 23, 1376–1383. [Google Scholar] [CrossRef]
- Datta, Y.H.; Romano, M.; Jacobson, B.C.; Golan, D.E.; Serhan, C.N.; Ewenstein, B.M. Peptido-leukotrienes are potent agonists of von Willebrand factor secretion and P-selectin surface expression in human umbilical vein endothelial cells. Circulation 1995, 92, 3304–3311. [Google Scholar] [CrossRef]
- Chan, S.J.; Ng, M.P.; Zhao, H.; Ng, G.J.; De Foo, C.; Wong, P.T.-H.; Seet, R. Early and sustained increases in leukotriene B4 levels are associated with poor clinical outcome in ischemic stroke patients. Neurotherapeutics 2020, 17, 282–293. [Google Scholar] [CrossRef]
- Nichols, W.W.; Mehta, J.L.; Thompson, L.; Donnelly, W.H. Synergistic effects of LTC4 and TxA2 on coronary flow and myocardial function. Am. J. Physiol.-Heart Circ. Physiol. 1988, 255, H153–H159. [Google Scholar] [CrossRef]
- Al-kuraishy, H.M.; Batiha, G.E.-S.; Faidah, H.; Al-Gareeb, A.I.; Saad, H.M.; Simal-Gandara, J. Pirfenidone and post-Covid-19 pulmonary fibrosis: Invoked again for realistic goals. Inflammopharmacology 2022, 1–10. [Google Scholar] [CrossRef]
- Tang, X.; Fuchs, D.; Tan, S.; Trauelsen, M.; Schwartz, T.W.; Wheelock, C.E.; Li, N.; Haeggström, J.Z. Activation of metabolite receptor GPR91 promotes platelet aggregation and transcellular biosynthesis of leukotriene C4. J. Thromb. Haemost. 2020, 18, 976–984. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alomair, B.M.; Al-kuraishy, H.M.; Al-Gareeb, A.I.; Al-Hamash, S.M.; De Waard, M.; Sabatier, J.-M.; Saad, H.M.; El-Saber Batiha, G. Montelukast and Acute Coronary Syndrome: The Endowed Drug. Pharmaceuticals 2022, 15, 1147. https://doi.org/10.3390/ph15091147
Alomair BM, Al-kuraishy HM, Al-Gareeb AI, Al-Hamash SM, De Waard M, Sabatier J-M, Saad HM, El-Saber Batiha G. Montelukast and Acute Coronary Syndrome: The Endowed Drug. Pharmaceuticals. 2022; 15(9):1147. https://doi.org/10.3390/ph15091147
Chicago/Turabian StyleAlomair, Basil Mohammed, Hayder M. Al-kuraishy, Ali I. Al-Gareeb, Sadiq M. Al-Hamash, Michel De Waard, Jean-Marc Sabatier, Hebatallah M. Saad, and Gaber El-Saber Batiha. 2022. "Montelukast and Acute Coronary Syndrome: The Endowed Drug" Pharmaceuticals 15, no. 9: 1147. https://doi.org/10.3390/ph15091147
APA StyleAlomair, B. M., Al-kuraishy, H. M., Al-Gareeb, A. I., Al-Hamash, S. M., De Waard, M., Sabatier, J.-M., Saad, H. M., & El-Saber Batiha, G. (2022). Montelukast and Acute Coronary Syndrome: The Endowed Drug. Pharmaceuticals, 15(9), 1147. https://doi.org/10.3390/ph15091147