
Palladium(II) Complexes of Substituted Salicylaldehydes: Synthesis, Characterization and Investigation of Their Biological Profile

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
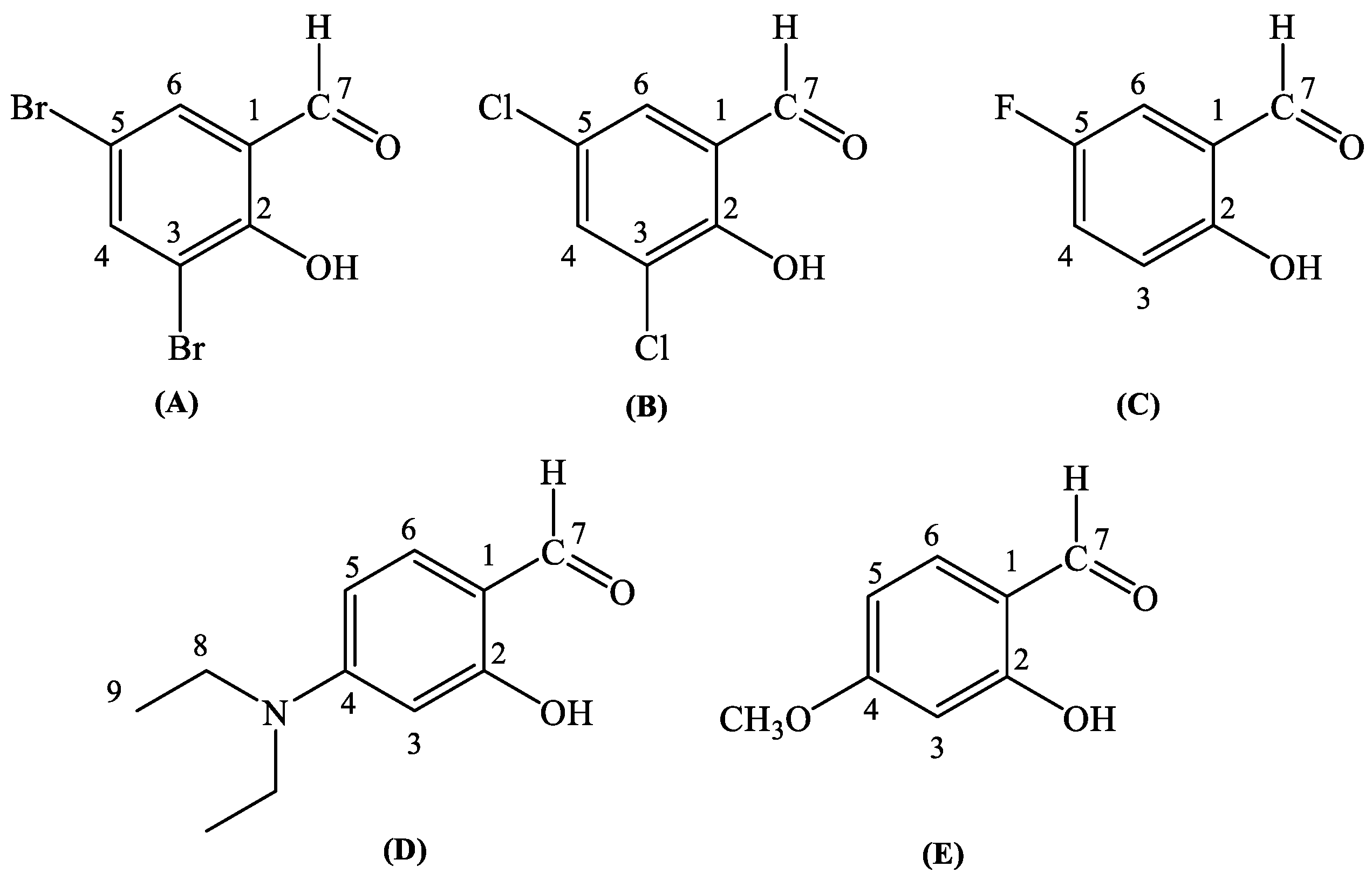
2.1. Synthesis and Characterization
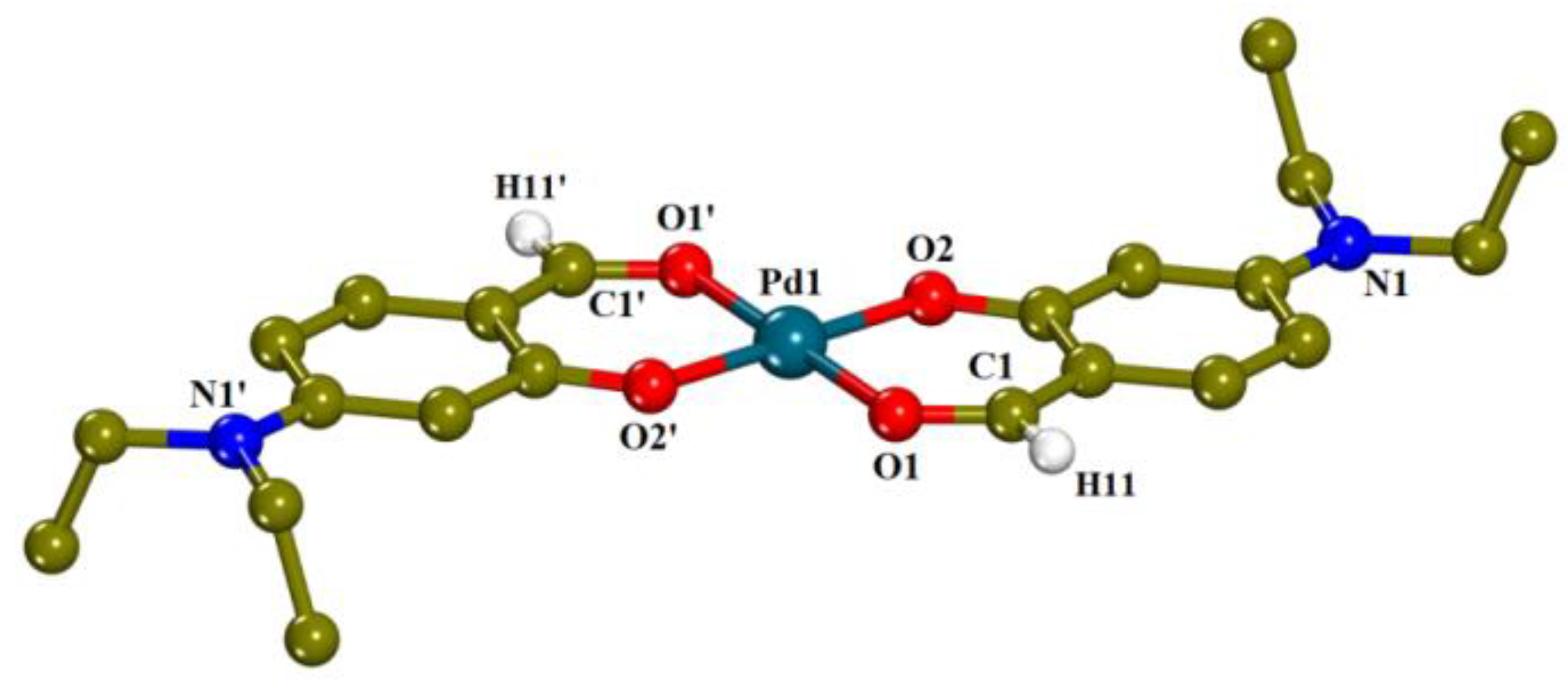
2.2. Structure of the Complexes
2.2.1. Description of the Crystal Structure of [Pd(4-Et2N-salo)2]·CH3CN, (1)·CH3CN
2.2.2. Proposed Structures for Complexes 2–5
2.3. Antioxidant Activity
2.4. Antimicrobial Activity
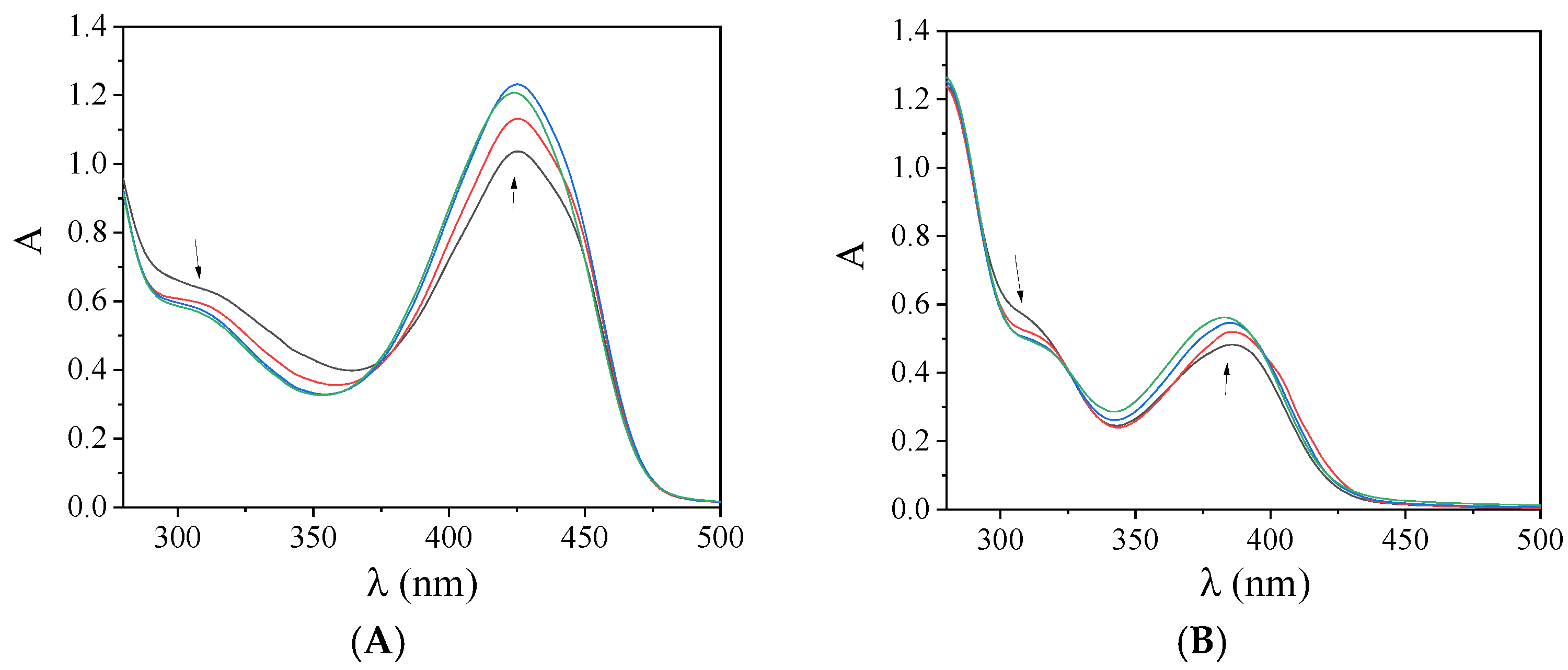
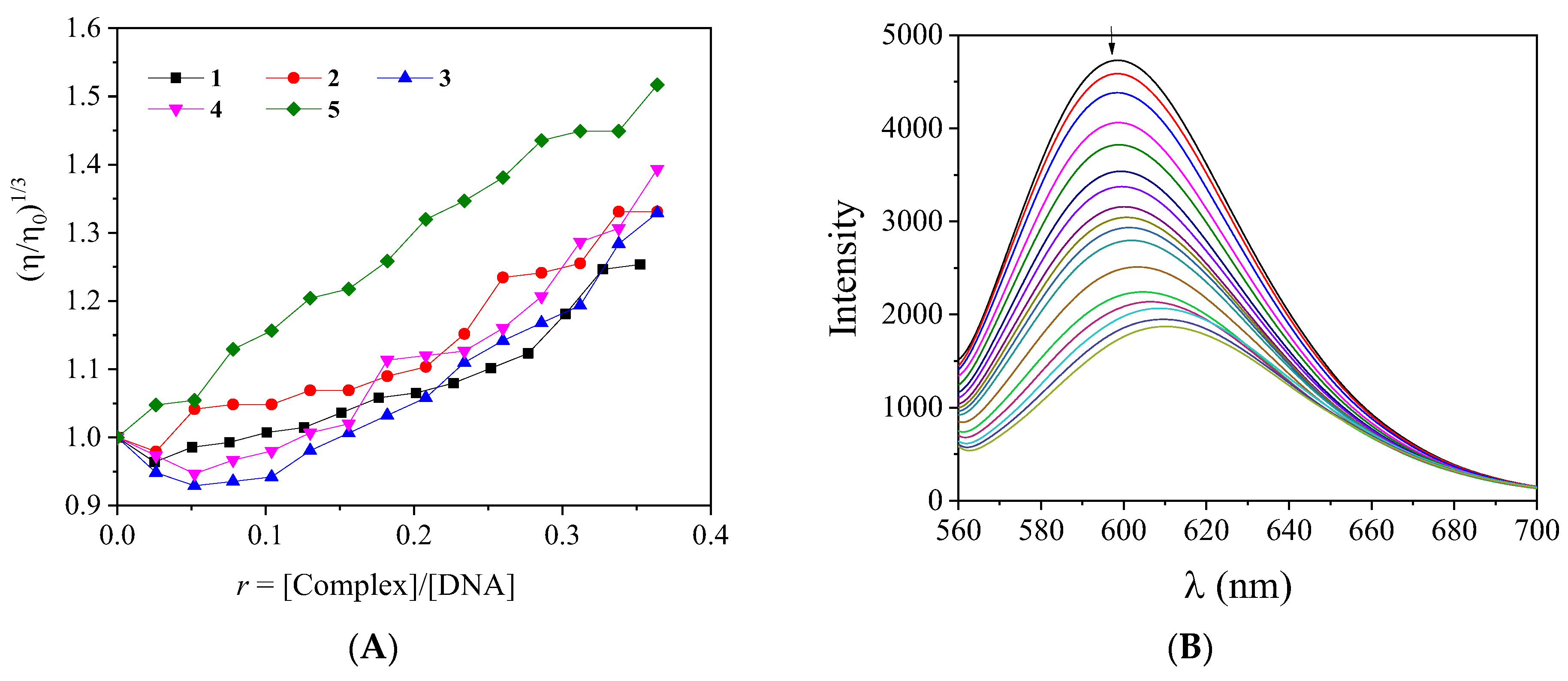
2.5. Interaction with CT DNA
2.6. Interaction with Serum Albumins
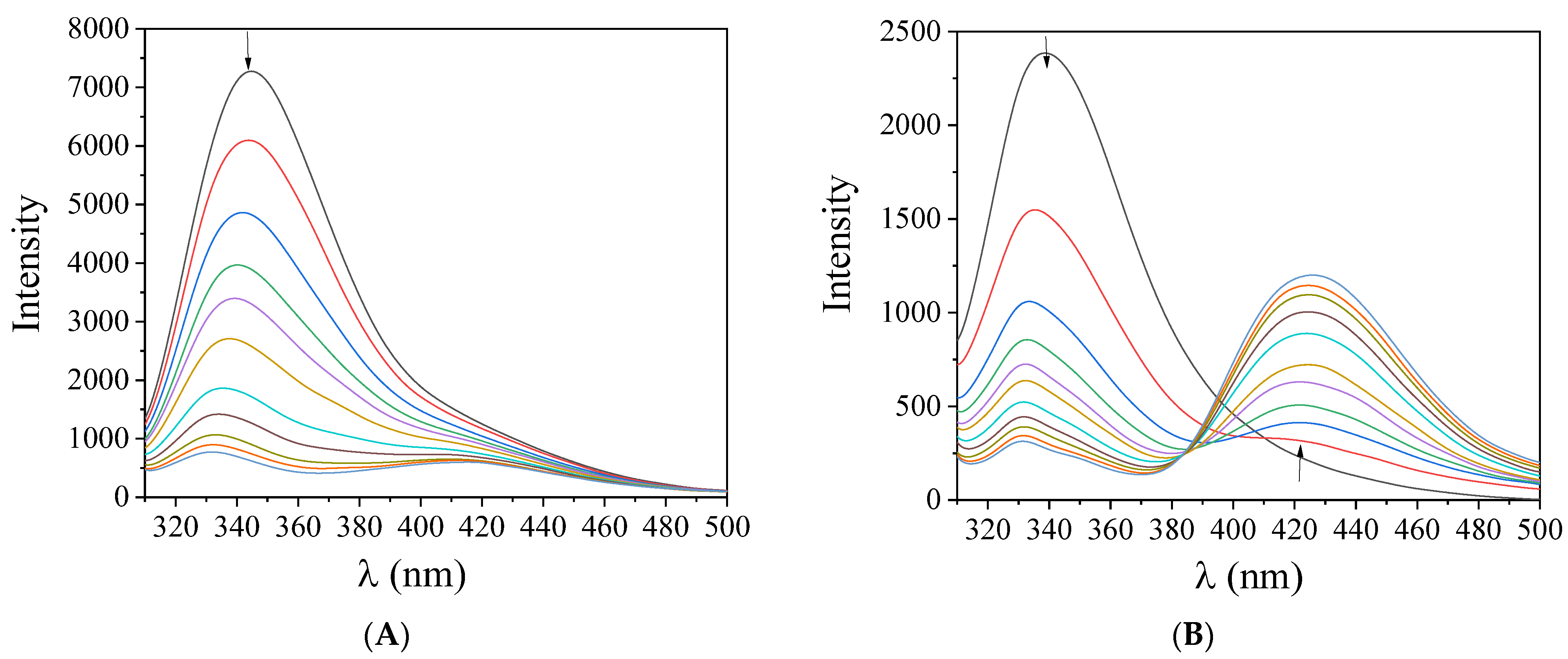
2.6.1. Affinity of the Compounds for BSA and HSA
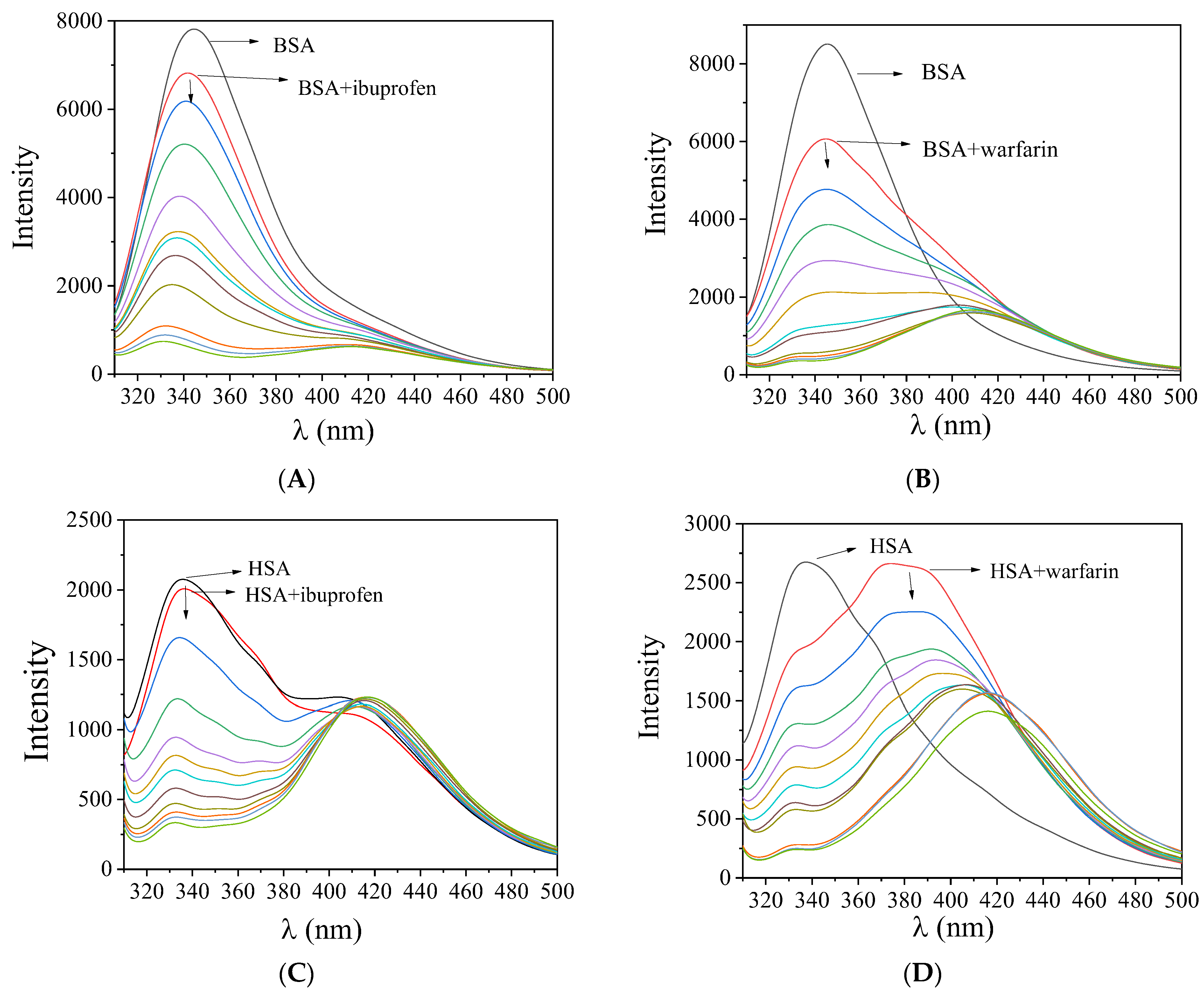
2.6.2. Location of the Albumin-Binding Site
2.7. In Silico Molecular Docking Studies
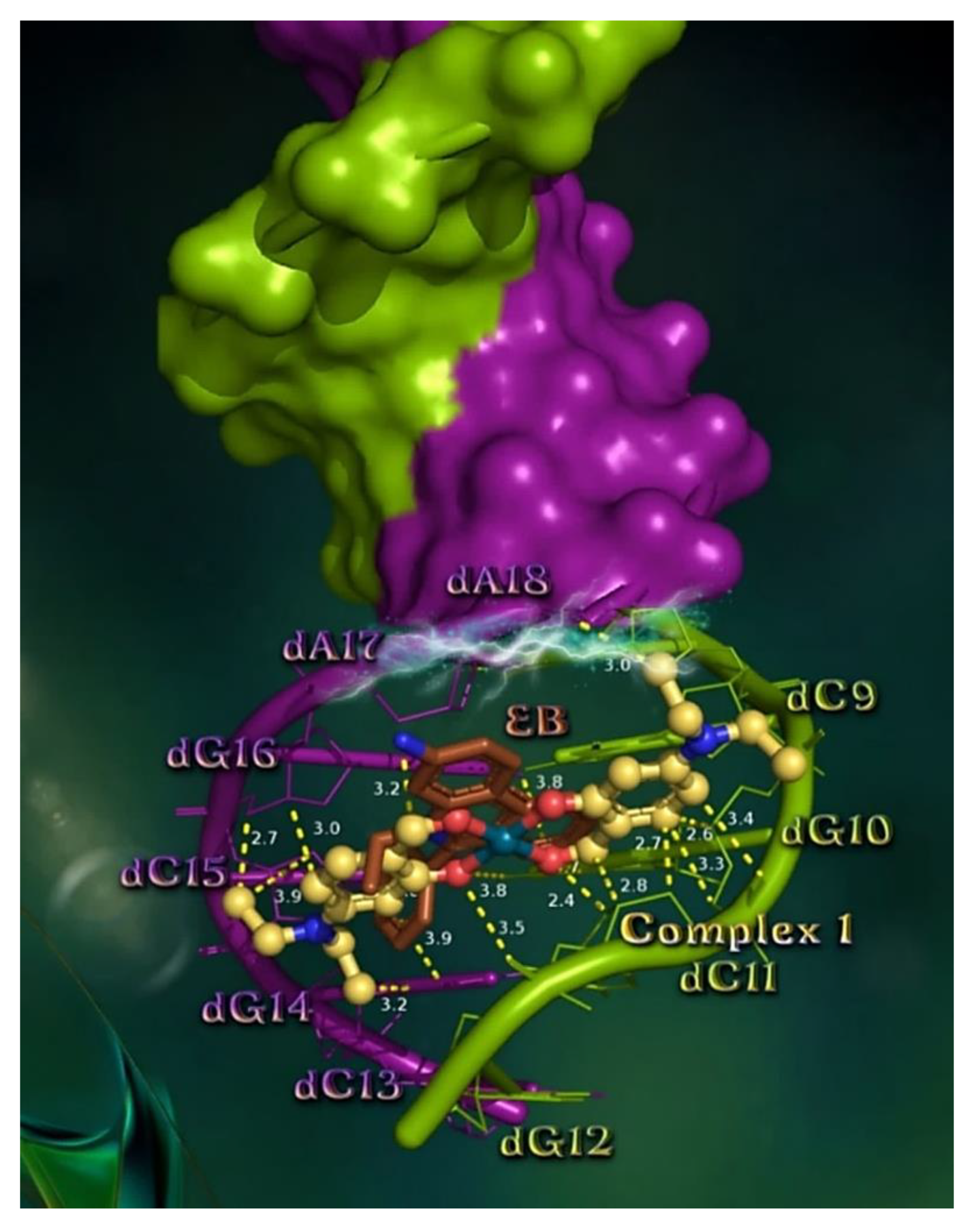
2.7.1. Docking Calculations on CT DNA
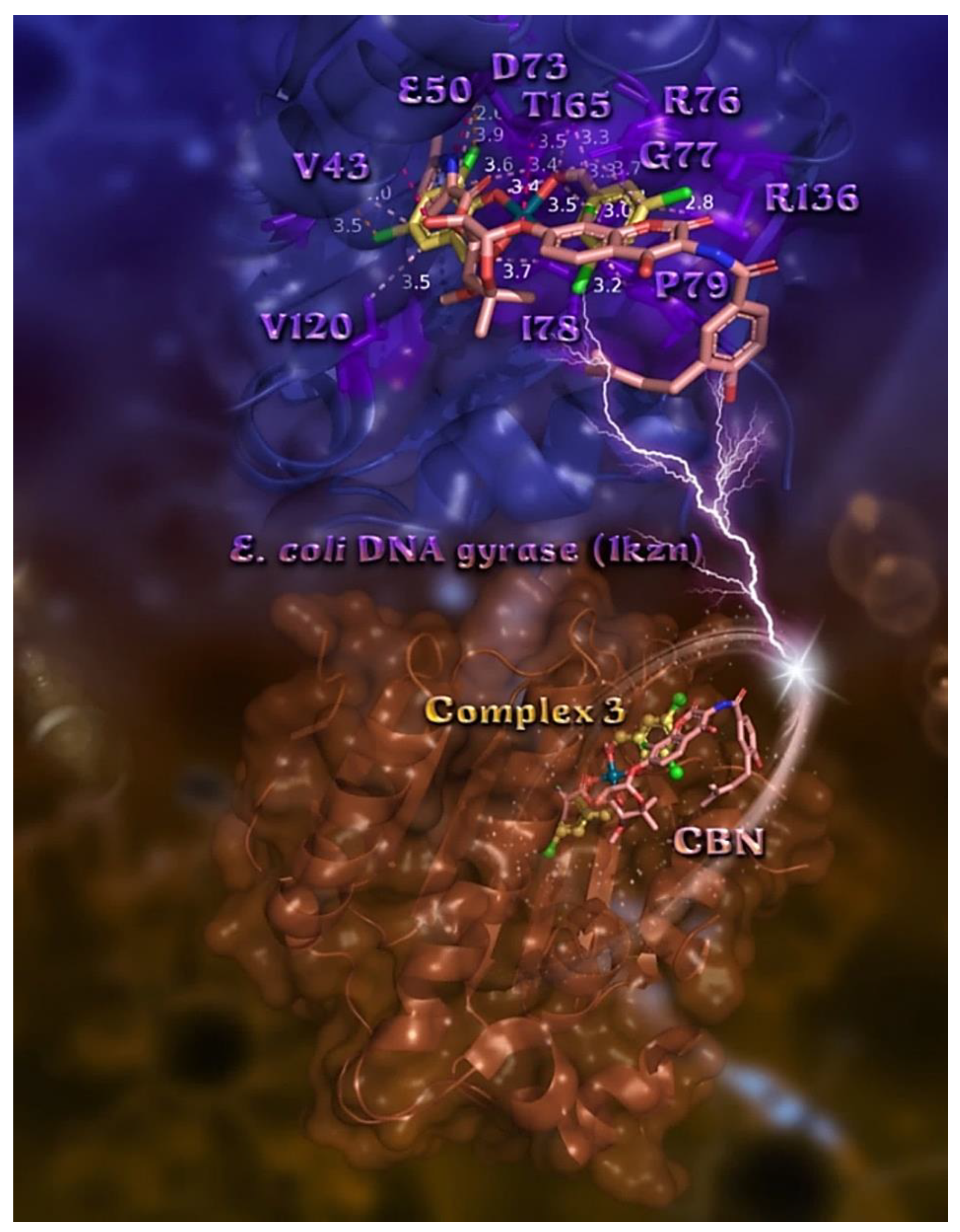
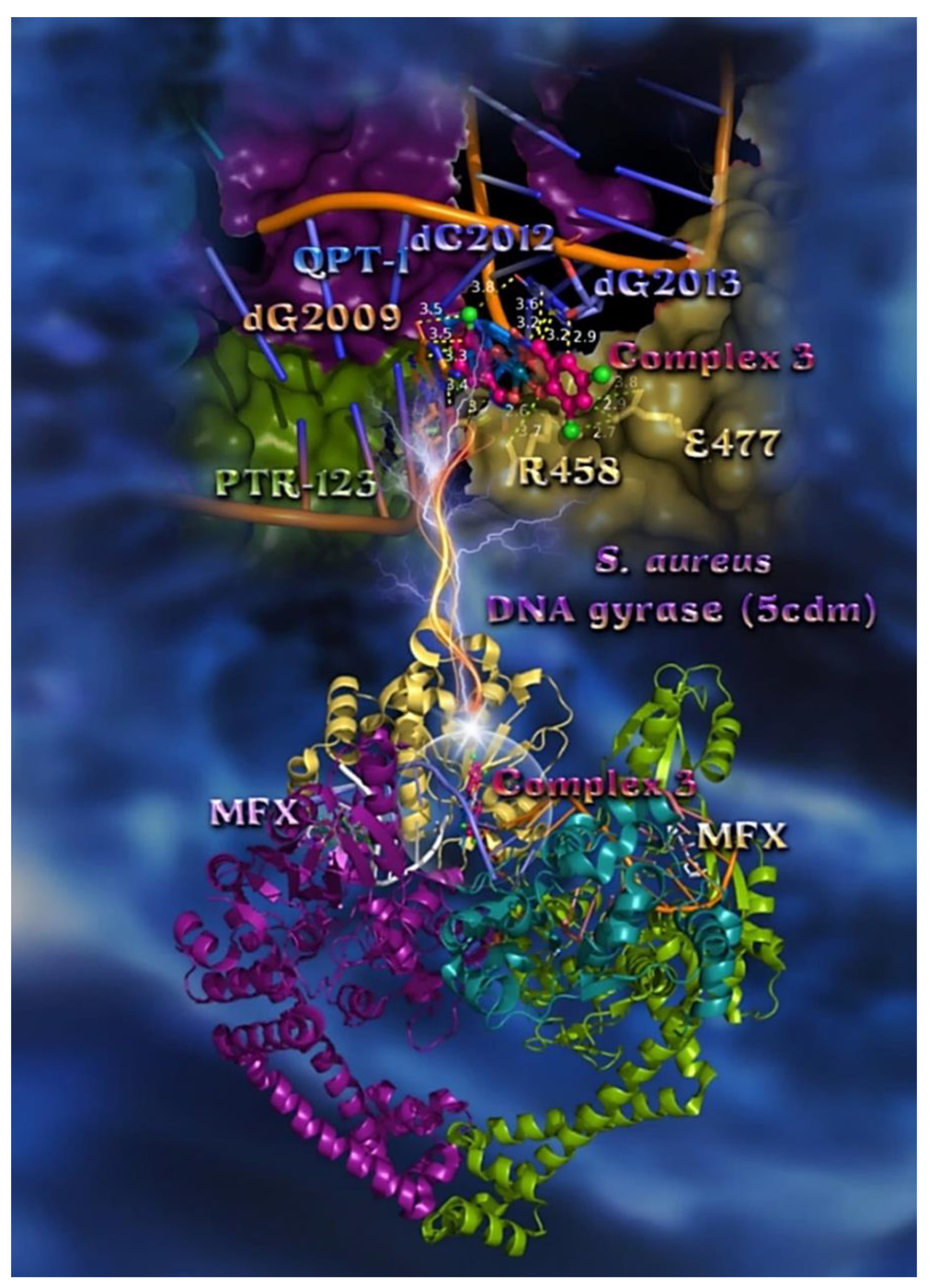
2.7.2. Docking Calculations on E. coli and S. aureus DNA-Gyrase
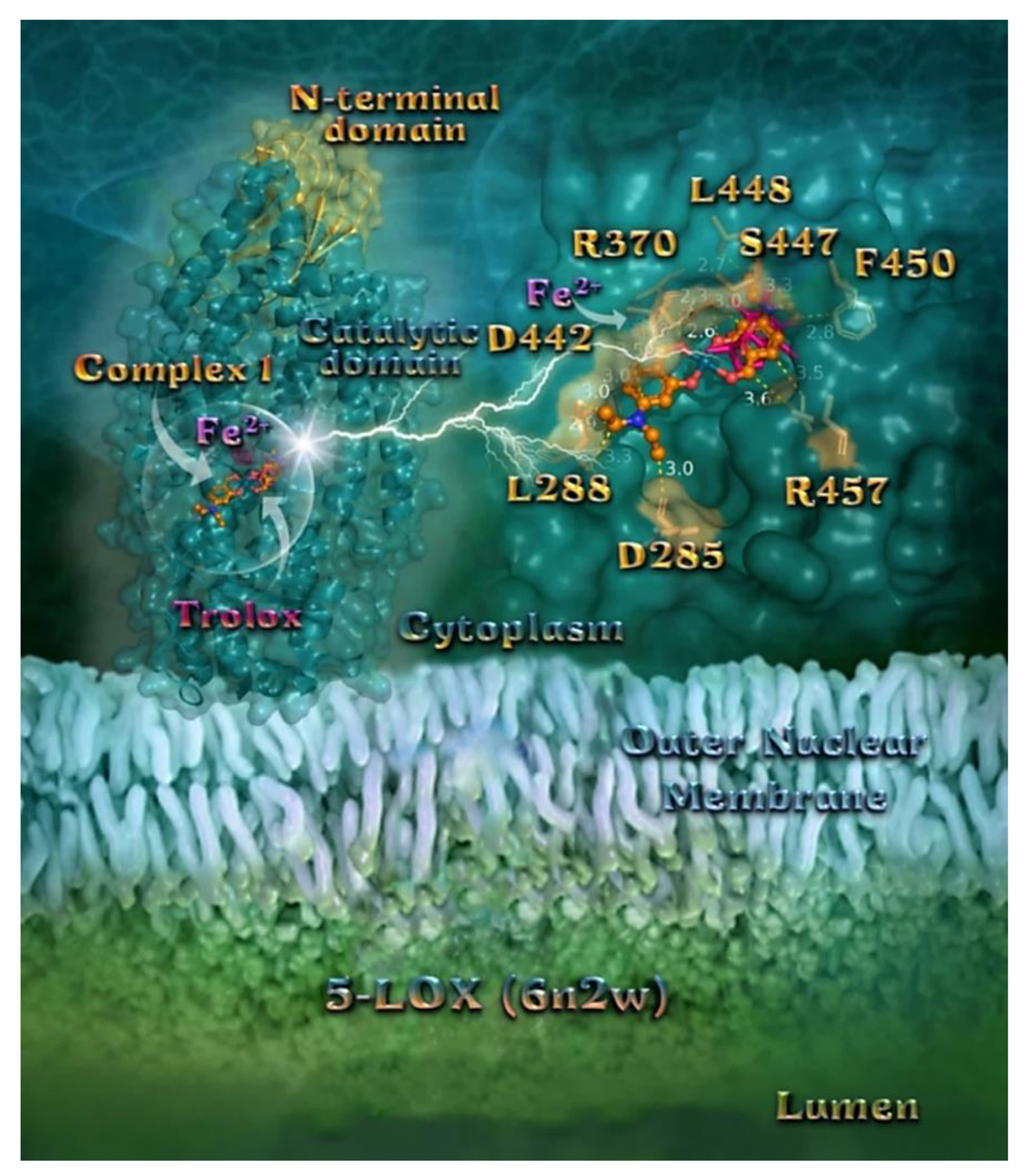
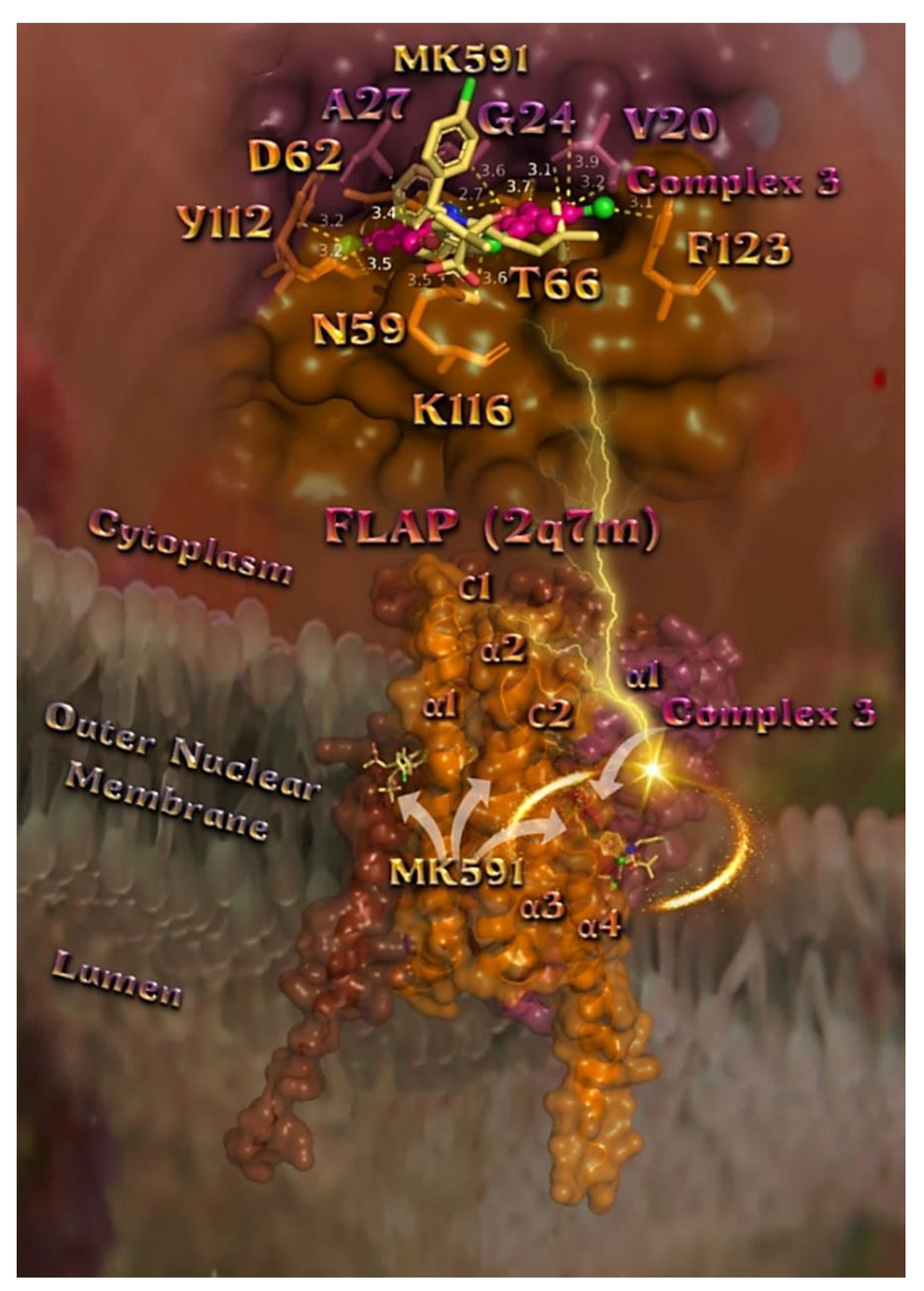
2.7.3. Docking Calculations on 5-LOX and FLAP Target Proteins
Docking Calculations on 5-LOX
Docking Calculations on FLAP
2.8. Chemical Reactivity
2.9. Molecular Pharmacokinetic Properties, Drug-Likeness, Target Proteins and Toxicity Predictions
2.9.1. Molecular Properties Prediction/Drug-Likeness
2.9.2. PASS Biological Activity Prediction Profile
Acute Rat Toxicity Prediction by GUSAR (on the Basis of PASS Prediction)
SMP: Prediction of Substrate/Metabolite Specificity (Pa > Pi)
ROSC-Pred: Rodent Organ-Specific Carcinogenicity Prediction
Quantitative Prediction of Anti-Target Interaction Profiles for Chemical Compounds by GUSAR Software
Environmental Ecotoxicity Predicted by GUSAR
CLC-Pred: In Silico Prediction of Cytotoxicity for Tumor and Non-Tumor Cell Lines
SOMP: Prediction of Sites of Metabolism
Activity Spectra Prediction
DIGEP-Pred: Prediction of Drug-Induced Changes of Gene Expression Profile
3. Materials and Methods
3.1. Materials, Instrumentation and Physical Measurements
3.2. Synthesis of the Complexes
3.3. X-ray Crystal Structure Determination
3.4. Study of the Biological Profile of the Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fanelli, M.; Formica, M.; Fusi, V.; Giorgi, L.; Micheloni, M.; Paoli, P. New trends in platinum and palladium complexes as antineoplastic agents. Coord. Chem. Rev. 2016, 310, 41–79. [Google Scholar] [CrossRef]
- Garoufis, A.; Hadjikakou, S.K.; Hadjiliadis, N. Palladium coordination compounds as anti-viral, anti-fungal, anti-microbial and anti-tumor agents. Coord. Chem. Rev. 2009, 253, 1384–1397. [Google Scholar] [CrossRef]
- Ayyannan, G.; Mohanraj, M.; Gopiraman, M.; Uthayamalar, R.; Raja, G.; Bhuvanesh, N.; Nandhakumar, R.; Jayabalakrishnan, C. New Palladium(II) complexes with ONO chelated hydrazone ligand: Synthesis, characterization, DNA/BSA interaction, antioxidant and cytotoxicity. Inorg. Chim. Acta 2020, 512, 119868. [Google Scholar] [CrossRef]
- Wei, X.; Yang, Y.; Ge, J.; Lin, X.; Liu, D.; Wang, S.; Zhang, J.; Zhou, G.; Li, S. Synthesis, characterization, DNA/BSA interactions and in vitro cytotoxicity study of palladium(II) complexes of hispolon derivatives. J. Inorg. Biochem. 2020, 202, 110857. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, M.; Saeidifar, M.; Mansouri-Torshizi, H. Synthesis, characterization, DNA binding, cytotoxicity, and molecular docking approaches of Pd(II) complex with N,O- donor ligands as a novel potent anticancer agent. J. Mol. Struct. 2020, 1215, 128212. [Google Scholar] [CrossRef]
- Fiori-Duarte, A.T.; Bergamini, F.R.G.; de Paiva, R.E.F.; Manzano, C.M.; Lustri, W.R.; Corbi, P.P. A new palladium(II) complex with ibuprofen: Spectroscopic characterization, DFT studies, antibacterial activities and interaction with biomolecules. J. Mol. Struct. 2019, 1186, 144–154. [Google Scholar] [CrossRef]
- Dheman, N.; Mahoney, N.; Cox, E.M.; Farley, J.J.; Amini, T.; Lanthier, M.L. An Analysis of Antibacterial Drug Development Trends in the United States, 1980–2019. Clin. Infect. Dis. 2021, 73, 4444–4450. [Google Scholar] [CrossRef]
- Dev, R.K.; Bhattarai, A.; Chaudhary, N.K.; Mishra, P. Synthesis, spectroscopic characterization and antibacterial assessment of zirconium(II) and palladium(II) complexes of TcSal mixed ligand (tetracycline (Tc) as primary ligand and salicylaldehyde as secondary ligand. Asian J. Chem. 2020, 32, 1473–1481. [Google Scholar] [CrossRef]
- Abu-Dief, A.M.; Abdel-Rahman, L.H.; Abdel-Mawgoud, A.A.H. A robust in vitro Anticancer, Antioxidant and Antimicrobial Agents Based on New Metal-Azomethine Chelates Incorporating Ag(I), Pd (II) and VO (II) Cations: Probing the Aspects of DNA Interaction. Appl. Organomet. Chem. 2020, 34, e5373. [Google Scholar] [CrossRef]
- Nyawade, E.A.; Onani, M.O.; Meyer, S.; Dube, P. Synthesis, characterization and antibacterial activity studies of new 2-pyrral-L-amino acid Schiff base palladium (II) complexes. Chem. Pap. 2020, 74, 3705–3715. [Google Scholar] [CrossRef]
- Boubakri, L.; Mansour, L.; Harrath, A.H.; Ozdemir, I.; Yasar, S.; Hamdi, N. N-Heterocyclic carbene-Pd(II)-PPh3 complexes as a new highly efficient catalyst system for the Sonogashira cross-coupling reaction: Synthesis, characterization and biological activities. J. Coord. Chem. 2018, 71, 183–199. [Google Scholar] [CrossRef]
- Sumar Ristovic, M.; Zianna, A.; Psomas, G.; Hatzidimitriou, A.; Coutouli-Argyropoulou, E.; Lalia-Kantouri, M. Interaction of dinuclear cadmium(II) 5-Cl-salicylaldehyde complexes with calf-thymus DNA. Mater. Sci. Eng. C 2016, 61, 579–590. [Google Scholar] [CrossRef]
- Zianna, A.; Geromichalou, E.; Geromichalos, G.; Fiotaki, A.-M.; Hatzidimitriou, A.G.; Kalogiannis, S.; Psomas, G. Zinc(II) complexes of 3,5–dibromo–salicylaldehyde and α–diimines: Synthesis, characterization and in vitro and in silico biological profile. J. Inorg. Biochem. 2022, 226, 111659. [Google Scholar] [CrossRef] [PubMed]
- Zianna, A.; Geromichalos, G.; Psoma, E.; Kalogiannis, S.; Hatzidimitriou, A.G.; Psomas, G. Structure and in vitro and in silico biological activity of zinc(II) complexes with 3,5–dichloro–salicylaldehyde. J. Inorg. Biochem. 2022, 229, 111727. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ji, J.; Wang, C.; Jia, A.; Zhang, Q. Coordination modes of salicylaldehyde derivatives in the ruthenium(II) nitrosyl and bis(2,2′-bipyridine) ruthenium(II) complexes. J. Coord. Chem. 2020, 73, 1306–1313. [Google Scholar] [CrossRef]
- Sahajpal, A.; Robinson, S.D.; Mazid, M.A.; Motevalli, M.; Hursthouse, M.B. Complexes of the platinum metals. Part 39. Salicylaldehydato(2–) derivatives of ruthenium, osmium, and iridium. X-ray crystal structures of [Ru(OC6H4CO)(HOC6H4CH2OH) (CO)(PPh3)2]·CH2Cl2 and [Ru(OC6H4CO)(MeOH)(CO)(PPh3)2]·MeOH. J. Chem. Soc. Dalton Trans. 1990, 7, 2119–2124. [Google Scholar] [CrossRef]
- Kordestani, N.; Rudbari, H.A.; Fernandes, A.R.; Raposo, L.R.; Baptista, P.V.; Ferreira, D.; Bruno, G.; Bella, G.; Scopelliti, R.; Braun, J.D.; et al. Antiproliferative Activities of Diimine-Based Mixed Ligand Copper(II) Complexes. ACS Comb. Sci. 2020, 22, 89–99. [Google Scholar] [CrossRef]
- Jayamani, A.; Bellam, R.; Gopu, G.; Ojwach, S.O.; Sengottuvelan, N. Copper(II) complexes of bidentate mixed ligands as artificial nucleases: Synthesis, crystal structure, characterization and evaluation of biological properties. Polyhedron 2018, 156, 138–149. [Google Scholar] [CrossRef]
- Han, Y.; Wang, L.; Li, Q.; Xue, L. Synthesis, Structures, and Antimicrobial Activities of Two Cobalt(II) Complexes [Co(L1)2(OH2)2] and [Co(L2)2]. Acta Chim. Slov. 2017, 64, 179–185. [Google Scholar] [CrossRef][Green Version]
- Lalia-Kantouri, M.; Gdaniec, M.; Choli-Papadopoulou, T.; Badounas, A.; Papadopoulos, C.D.; Czapik, A.; Geromichalos, G.D.; Sahpazidou, D.; Tsitouroudi, F. Effect of cobalt(II) complexes with dipyridylamine and salicylaldehydes on cultured tumor and non-tumor cells: Synthesis, crystal structure investigations and biological activity. J. Inorg. Biochem. 2012, 117, 25–34. [Google Scholar] [CrossRef]
- Zianna, A.; Psomas, G.; Hatzidimitriou, A.; Coutouli-Argyropoulou, E.; Lalia-Kantouri, M. Zinc complexes of salicylaldehydes: Synthesis, characterization and DNA-binding properties. J. Inorg. Biochem. 2013, 127, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Zianna, A.; Geromichalos, G.D.; Hatzidimitriou, A.G.; Coutouli-Argyropoulou, E.; Lalia-Kantouri, M.; Psomas, G. Palladium(II) complexes with salicylaldehyde ligands: Synthesis, characterization, structure, in vitro and in silico study of the interaction with calf-thymus DNA and albumins. J. Inorg. Biochem. 2019, 194, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ntanatsidis, S.; Perontsis, S.; Konstantopoulou, S.; Kalogiannis, S.; Hatzidimitriou, A.G.; Papadopoulos, A.N.; Psomas, G. Manganese(II) complexes of substituted salicylaldehydes and α-diimines: Synthesis, characterization and biological activity. J. Inorg. Biochem. 2022, 227, 111693. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 6th ed.; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Ahmadi, M.; Fasihizad, A.; Machura, B.; Kruszynski, R.; Akbari, A.; Barak, T. Palladium(II) complex with salicylaldehyde: Synthesis, characterization, crystal structure and thermal decomposition into PdO nano-particle. Can. J. Basic Appl. Sci. 2015, 3, 155–163. [Google Scholar]
- Marchi, R.C.; Campos, I.A.S.; Santana, V.T.; Carlos, R.M. Chemical implications and considerations on techniques used to assess the in vitro antioxidant activity of coordination compounds. Coord. Chem. Rev. 2022, 451, 2142752. [Google Scholar] [CrossRef]
- Slavova-Kazakova, A.; Karamac, M.; Kancheva, V.; Amarowicz, R. Antioxidant Activity of Flaxseed Extracts in Lipid Systems. Molecules 2016, 21, 17. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Ntella, M.; Mpompou, L.; Karallaki, F.; Papadopoulos, A.; Hadjipavlou-Litina, D.; Lazari, D. Study of the antioxidant activity of Thymus sibthorpii Bentham (Lamiaceae). J. Enz. Inhib. Med. Chem. 2016, 31, 154–159. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Hadjipavlou-Litina, D. Biological evaluation of several coumarin derivatives designed as possible anti-inflammatory/antioxidants agents. J. Enz. Inhib. Med. Chem. 2003, 180, 63–69. [Google Scholar] [CrossRef]
- Gulcin, I.; Beydemır, S.; Alici, H.A.; Elmasta, M.; Buyukokuroglu, M.E. In vitro antioxidant properties of morphine. Pharmacol. Res. 2004, 49, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakasha, G.K.; Rao, L.; Sakariah, K. Antioxidant activities of flavidin in different in vitro model systems. Bioorg. Med. Chem. 2004, 12, 5141–5146. [Google Scholar] [CrossRef]
- Elo, H.; Kuure, M.; Pelttari, E. Correlation of the antimicrobial activity of salicylaldehydes with broadening of the NMR signal of the hydroxyl proton. Possible involvement of proton exchange processes in the antimicrobial activity. Eur. J. Med. Chem. 2015, 92, 750–753. [Google Scholar] [CrossRef]
- Pelttari, E.; Karhumaki, E.; Langshaw, J.; Perakyla, H.; Elo, H. Antimicrobial Properties of Substituted Salicylaldehydes and Related Compounds. Naturforsch. 2007, 62, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009, 5, 1685–1704. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Pierre, V.C.; Barton, J.K. Metallo-intercalators and metallo-insertors. Chem. Commun. 2007, 44, 4565–4576. [Google Scholar] [CrossRef]
- Pratviel, G.; Bernadou, J.; Meunier, B. DNA and RNA cleavage by metal complexes. Adv. Inorg. Chem. 1998, 45, 251–262. [Google Scholar]
- Pyle, A.M.; Rehmann, J.P.; Meshoyrer, R.; Kumar, C.V.; Turro, N.J.; Barton, J.K. Mixed-Ligand Complexes of Ruthenium(II): Factors Governing Binding to DNA. J. Am. Chem. Soc. 1989, 111, 3053–3063. [Google Scholar] [CrossRef]
- Wolfe, A.; Shimer, G.; Meehan, T. Polycyclic Aromatic Hydrocarbons Physically Intercalate into Duplex Regions of Denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef]
- Dimitrakopoulou, A.; Dendrinou-Samara, C.; Pantazaki, A.A.; Alexiou, M.; Nordlander, E.; Kessissoglou, D.P. Synthesis, structure and interactions with DNA of novel tetranuclear, [Mn4(II/II/II/IV)] mixed valence complexes. J. Inorg. Biochem. 2008, 102, 618–628. [Google Scholar] [CrossRef]
- Garcia-Gimenez, J.L.; Gonzalez-Alvarez, M.; Liu-Gonzalez, M.; Macias, B.; Borras, J.; Alzuet, G. Toward the development of metal-based synthetic nucleases: DNA binding and oxidative DNA cleavage of a mixed copper(II) complex with N-(9H-purin-6-yl)benzenesulfonamide and 1,10-phenantroline. Antitumor activity in human Caco-2 cells and Jurkat T lymphocytes. Evaluation of p53 and Bcl-2 proteins in the apoptotic mechanism. J. Inorg. Biochem. 2009, 103, 923–934. [Google Scholar] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Plenum Press: New York, NY, USA, 2006. [Google Scholar]
- Zhao, G.; Lin, H.; Zhu, S.; Sun, H.; Chen, Y. Dinuclear palladium (II) complexes containing two monofunctional [Pd(en)(pyridine) Cl]+ units bridged by Se or S. Synthesis, characterization, cytotoxicity and kinetic studies of DNA-binding. J. Inorg. Biochem. 1998, 70, 219–226. [Google Scholar] [CrossRef]
- Heller, D.P.; Greenstock, C.L. Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye. Biophys. Chem. 1994, 50, 305–312. [Google Scholar] [CrossRef]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.E.; Christ, D.D. Chapter 33. Plasma Protein Binding of Drugs. Ann. Rep. Med. Chem. 1996, 31, 327–336. [Google Scholar]
- Tan, C.; Liu, J.; Li, H.; Zheng, W.; Shi, S.; Chen, L.; Ji, L. Differences in structure, physiological stability, electrochemistry, cytotoxicity, DNA and protein binding properties between two Ru(III) complexes. J. Inorg. Biochem. 2008, 102, 347–358. [Google Scholar] [CrossRef]
- Stella, L.; Capodilupo, A.L.; Bietti, M. A reassessment of the association between azulene and [60]fullerene. Possible pitfalls in the determination of binding constants through fluorescence spectroscopy. Chem. Commun. 2008, 39, 4744–4746. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Hytönen, V.P.; Nordlund, H.R.; Kulomaa, M.S. Genetically engineered avidins and streptavidins. Cell. Mol. Life Sci. 2006, 63, 2992–3017. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Lazou, M.; Tarushi, A.; Gritzapis, P.; Psomas, G. Transition metal complexes with a novel guanine-based (E)-2-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazolin-4(3H)-one: Synthesis, characterization, interaction with DNA and albumins and antioxidant activity. J. Inorg. Biochem. 2020, 206, 111019. [Google Scholar] [CrossRef]
- Kakoulidou, C.; Kosmas, V.-R.; Hatzidimitriou, A.G.; Fylaktakidou, K.C.; Psomas, G. Structure and biological profile of transition metal complexes with (E)-4-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazoline. J. Inorg. Biochem. 2021, 219, 111448. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Mizuuchi, K.; O’Dea, M.H.; Nash, H.A. DNA gyrase: An enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci. USA 1976, 73, 3872–3876. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Chu, D.T.W. Type II DNA Topoisomerases as antibacterial targets. Curr. Pharm. Des. 1996, 2, 195–208. [Google Scholar]
- Gentry, A.C.; Osheroff, N. DNA Topoisomerases: Type II. In Encyclopedia of Biological Chemistry, 2nd ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Ali, J.A.; Orphanides, G.; Maxwell, A. Nucleotide Binding to the 43-Kilodalton N-Terminal Fragment of the DNA Gyrase B Protein. Biochemistry 1995, 34, 9801–9808. [Google Scholar] [CrossRef] [PubMed]
- Lafitte, D.; Lamour, V.; Tsvetkov, P.O.; Makarov, A.A.; Klich, M.; Deprez, P.; Moras, D.; Briand, C.; Gilli, R. DNA Gyrase Interaction with Coumarin-Based Inhibitors: The Role of the Hydroxybenzoate Isopentenyl Moiety and the 5‘-Methyl Group of the Noviose. Biochemistry 2002, 41, 7217–7223. [Google Scholar] [CrossRef]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta 2015, 1851, 331–339. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, F.d.C.P.; Mendes, A.N. Computational analysis of eugenol inhibitory activity in lipoxygenase and cyclooxygenase pathways. Sci. Rep. 2020, 10, 16204. [Google Scholar] [CrossRef]
- Tong, W.G.; Ding, X.Z.; Adrian, T.E. The mechanisms of lipoxygenase inhibitor-induced apoptosis in human breast cancer cells. Biochem. Bioph. Res. Comm. 2002, 296, 942–948. [Google Scholar] [CrossRef]
- Rackova, L.; Oblozinsky, M.; Kostalova, D.; Kettmann, V.; Bezakova, L. radical scavenging activity and lipoxygenase inhibition of Mahonia aquifolium extract and isoquinoline alkaloids. J. Inflamm. 2007, 4, 15. [Google Scholar] [CrossRef]
- Koukoulitsa, C.; Hadjipavlou-Litina, D.; Geromichalos, G.; Skaltsa, H.; Enz, J. Inhibitory effect on soybean lipoxygenase and docking studies of some secondary metabolites, isolated from Origanum vulgare L. ssp. Hirtum. Inhib. Med. Chem. 2007, 22, 99–104. [Google Scholar] [CrossRef]
- Kaur, G.; Silakari, O. Multiple target-centric strategy to tame inflammation. Fut. Med. Chem. 2017, 9, 1361–1376. [Google Scholar] [CrossRef]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef]
- Frisch, A.E.; Nielsen, A.B.; Holder, A.J. Gaussview; Gaussian Inc.: Pittsburgh, PE, USA, 2003. [Google Scholar]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems (Electronics); John Wiley Sons Inc.: New York, NY, USA, 2001. [Google Scholar]
- Gunasekaran, S.; Balaji, R.A.; Kumeresan, S.; Anand, G.; Srinivasan, S. Experimental and theoretical investigations of spectroscopic properties of N-acetyl-5-methoxytryptamine. Can. J. Anal. Sci. Spectrosc. 2008, 53, 149–162. [Google Scholar]
- Govindarajan, M.; Karabacak, M.; Periandy, S.; Tanuja, D. Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation and NLO, HOMO-LUMO, NBO analysis of organic 2,4,5-trichloroaniline. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 97, 231–245. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1043. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Arjunan, V.; Devi, L.; Subbalakshmi, R.; Rani, T.; Mohan, S. Synthesis, vibrational, NMR, quantum chemical and structure-activity relation studies of 2-hydroxy-4-methoxyacetophenone. Spectrochim. Acta A 2014, 130, 164–177. [Google Scholar] [CrossRef]
- Zumdahl, S.S. Chemical Principles, 5th ed.; Chapter 13.2 “Electronegativity”; Houghton Mifflin Company: Boston, MA, USA, 2005; pp. 587–590. [Google Scholar]
- Parthasarathi, R.; Padmanabhan, J.; Sarkar, U.; Maiti, B.; Subramanian, V.; Chattaraj, P.K. Toxicity Analysis of Benzidine Through Chemical Reactivity and Selectivity Profiles: A DFT Approac. J. Mol. Des. 2003, 2, 798–813. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Bulat, F.A. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1731–1742. [Google Scholar] [CrossRef]
- Mok, S.W.F.; Zeng, W.; Niu, Y.; Coghi, P.; Wu, Y.; Sin, W.M.; Ng, S.I.; Gordillo-Martínez, F.; Gao, J.Y.; Law, B.Y.K.; et al. Method for Rapid Screening of Anilide-Containing AMPK Modulators Based on Computational Docking and Biological Validation. Front. Pharmacol. 2018, 9, 710. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Tewari, M.; Shukla, H.S.; Roy, B.K. In Silico Profiling of the Potentiality of Curcumin and Conventional Drugs for CagA Oncoprotein Inactivation. Arch. Pharm. 2015, 348, 548–555. [Google Scholar] [CrossRef]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-Drug Interactions for UDP-Glucuronosyltransferase Substrates: A Pharmacokinetic Explanation for Typically Observed Low Exposure (AUCI/AUC) Ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef]
- Cheng, F.; Yu, Y.; Zhou, Y.; Shen, Z.; Xiao, W.; Liu, G.; Li, W.; Lee, P.W.; Tang, Y. Insights into Molecular Basis of Cytochrome P450 Inhibitory Promiscuity of Compounds. J. Chem. Inf. Model 2001, 51, 2482–2495. [Google Scholar] [CrossRef]
- Van Booven, D.; Marsh, S.; McLeod, H.; Carrillo, M.W.; Sangkuhl, K.; Klein, T.E.; Altman, R.B. Cytochrome P450 2C9-CYP2C9. Pharmacogenet. Genom. 2010, 20, 277–281. [Google Scholar] [CrossRef]
- Bertilsson, L.; Dahl, M.; Dal’en, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef]
- Dai, D.; Tang, J.; Rose, R.; Hodgson, E.; Bienstock, R.J.; Mohrenweiser, H.W.; Goldstein, J.A. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J. Pharmacol. Exp. Ther. 2001, 299, 825–831. [Google Scholar]
- Lin, T.E.; HuangFu, W.C.; Chao, M.W.; Sung, T.Y.; Chang, C.D.; Chen, Y.Y.; Hsieh, J.H.; Tu, H.J.; Huang, H.L.; Pan, S.L.; et al. A Novel Selective JAK2 Inhibitor Identified Using Pharmacological Interactions. Front Pharmacol. 2018, 9, 1379. [Google Scholar] [CrossRef]
- Elsafadi, M.; Manikandan, M.; Almalki, S.; Mahmood, A.; Shinwari, T.; Vishnubalaji, R.; Mobarak, M.; Alfayez, M.; Aldahmash, A.; Kassem, M.; et al. Transgelin is a poor prognostic factor associated with advanced colorectal cancer (CRC) stage promoting tumor growth and migration in a TGF β-dependent manner. Cell Death Dis. 2020, 11, 341. [Google Scholar] [CrossRef]
- Chen, Z.; He, S.; Zhan, Y.; He, A.; Fang, D.; Gong, Y.; Li, X.; Zhou, L. TGF-β-induced transgelin promotes bladder cancer metastasis by regulating epithelial-mesenchymal transition and invadopodia formation. EBioMedicine 2019, 47, 208–220. [Google Scholar] [CrossRef]
- Choi, C.; Cho, W.K.; Park, S.; Shin, S.-W.; Park, W.; Kim, H.; Choi, D.H. Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation. Int. J. Mol. Sci. 2020, 21, 2691. [Google Scholar] [CrossRef]
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 1961, 3, 208–211. [Google Scholar] [CrossRef]
- Reichmann, M.F.; Rice, S.A.; Thomas, C.A.; Doty, P. A Further Examination of the Molecular Weight and Size of Desoxypentose Nucleic Acid. J. Am. Chem. Soc. 1954, 76, 3047–3053. [Google Scholar] [CrossRef]
- M86–E01078; User Manual, Apex2, Version 2. Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2006.
- Siemens Industrial Automation, Inc. SADABS: Area-Detector Absorption Correction; Siemens Industrial Automation, Inc.: Madison, WI, USA, 1996. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Watkin, D.J.; Prout, C.K.; Pearce, L.J. CAMERON Program; Chemical Crystallographic Laboratory, Oxford University: Oxford, UK, 1996. [Google Scholar]
- Ruch, R.J.; Cheng, C.; Klaunig, J.E. Prevention of cytotoxicity and inhibition of intercellular communication by antioxidant catechins isolated from Chinese green tea. Carcinogenesis 1989, 10, 1003–1008. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef]
- Irgi, E.P.; Geromichalos, G.D.; Balala, S.; Kljun, J.; Kalogiannis, S.; Papadopoulos, A.; Turel, I.; Psomas, G. Cobalt(II) complexes with the quinolone antimicrobial drug oxolinic acid: Structure and biological perspectives. RSC Adv. 2015, 5, 36353–36367. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Zhang, G.; Tao, W.; Tang, S. Interaction of the flavonoid hesperidin with bovine serum albumin: A fluorescence quenching study. J. Lumin. 2007, 126, 211–218. [Google Scholar] [CrossRef]
- Drew, H.R.; Wing, R.M.; Takano, T.; Broka, C.; Tanaka, S.; Itakura, K.; Dickerson, R.E. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. USA 1981, 78, 2179–2183. [Google Scholar] [CrossRef]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.C.; Gerstmeier, J.; Schexnaydre, E.E.; Borner, F.; Garscha, U.; Neau, D.B.; Werz, O.; Newcomer, M.E. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat. Chem. Biol. 2020, 16, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; McKeever, B.M.; Xu, S.; Wisniewski, D.; Miller, D.K.; Yamin, T.T.; Spencer, R.H.; Chu, L.; Ujjainwalla, F.; Cunningham, B.R.; et al. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science 2007, 317, 510–512. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.E.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W.J. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Halgren, T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Halgren, T. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Geromichalos, G.D.; Alifieris, C.E.; Geromichalou, E.G.; Trafalis, D.T. Overview on the current status on virtual high-through put screening and combinatorial chemistry approaches inmulti-target anticancer drug discovery; Part II. J. BUON 2016, 21, 764–779. [Google Scholar]
- Bashford, D.; Case, D.A. Generalized born models ofmacromolecular solvation effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System 0.99; DeLano Scientific: San Carlos, CA, USA, 2006. [Google Scholar]
- Pyka, A.; Babuska, M.; Zachariasz, M. A comparison of theoretical methods of calculation of partition coefficients for selected drugs. Acta Pol. Pharm. 2006, 63, 159–167. [Google Scholar] [PubMed]
- Leung, S.S.F.; Mijalkovic, J.; Borrelli, K.; Jacobson, M.P. Testing physical models of passive membrane permeation. J. Chem. Inf. Model. 2012, 52, 1621–1636. [Google Scholar] [CrossRef]
- Sharma, R.; Majer, F.; Peta, V.K.; Wang, J.; Keaveney, R.; Kelleher, D.; Long, A.; Gilmer, J.F. Bile acid toxicity structure–activity relationships: Correlations between cell viability and lipophilicity in a panel of new and known bile acids using an oesophageal cell line (HET-1A). Bioorg. Med. Chem. 2010, 18, 6886–6895. [Google Scholar] [CrossRef]
- Pliska, V.; Testa, B.; van De Waterbed, H. Lipophilicity in Drug Action and Toxicology; John Wiley & Sons Ltd.: New York, NY, USA, 1996; p. 439. ISBN 9783527293834. [Google Scholar]
- Darmostuk, M.; Jurášek, M.; Lengyel, K.; Zelenka, J.; Rumlová, M.; Drašar, P.; Ruml, T. Conjugation of chlorins with spermine enhances phototoxicity to cancer cells in vitro. J. Photochem. Photobiol. 2017, 168, 175–184. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef]
- Refsgaard, H.H.F.; Jensen, B.F.; Brockhoff, P.B.; Padkjaer, S.B.; Guldbrandt, M.; Christensen, M.S. In silico prediction of membrane permeability from calculated molecular parameters. J. Med. Chem. 2005, 48, 805–811. [Google Scholar] [CrossRef]
- Li, D.; Kerns, E.H. Lipophilicity Methods in Drug-Like Properties, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Bakht, M.A.; Yar, M.S.; Abdel-Hamid, S.G.; Qasoumi, S.I.; Samad, A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5862–5869. [Google Scholar] [CrossRef] [PubMed]
- Hagan, O.S.; Swainston, N.; Handl, J.; Kell, D.B. A ‘rule of 0.5’ for the metabolite-likeness of approved pharmaceutical drugs. Metabolomics 2015, 11, 323–339. [Google Scholar]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Zakharov, A.V.; Lagunin, A.A.; Poroikov, V.V. QNA-based ‘Star Track’ QSAR approach. SAR QSAR Environ. Res. 2009, 20, 679–709. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Borodina, Y.V.; Lagunin, A.A.; Kos, A.J. Robustness of biological activity spectra predicting by computer program PASS for noncongeneric sets of chemical compounds. Chem. Inf. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Zakharov, A.V.; Filimonov, D.A.; Poroikov, V.V. A new approach to QSAR modelling of acute toxicity. SAR QSAR Environ. Res. 2007, 18, 285–298. [Google Scholar] [CrossRef]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of rat acute toxicity on the basis of PASS prediction. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Sadym, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. Prediction of Biological Activity Spectra via The Internet. SAR QSAR Environ. Res. 2003, 14, 339–347. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Ihlenfeld, W.; Gloriozova, T.A.; Lagunin, A.A.; Borodina, Y.V.; Stepanchikova, A.V.; Nicklaus, M. CPASS Biological Activity Spectrum Predictions in the Enhanced Open NCI Database Browser. J. Chem. Inf. Comput. Sci. 2003, 43, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [PubMed]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. SOMP: Web server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics 2015, 31, 2046–2048. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Rudik, A.; Filimonov, D.; Druzhilovsky, D.; Poroikov, V. ROSC-Pred: Web-service for rodent organ-specific carcinogenicity prediction. Bioinformatics 2017, 34, 710–712. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Quantitative prediction of antitarget interaction profiles for chemical compounds. Chem. Res. Toxicol. 2012, 25, 2378–2385. [Google Scholar] [CrossRef]
- Poroikov, V.; Akimov, D.; Shabelnikova, E.; Filimonov, D. Top 200 Medicines: Can New Actions be Discovered Through Computer-aided Prediction? SAR QSAR Environ. Res. 2001, 12, 327–344. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Lagunin, A.; Ivanov, S.; Rudik, A.; Filimonov, D.; Poroikov, V. DIGEP-Pred: Web service for in silico prediction of drug-induced gene expression profiles based on structural formula. Bioinformatics 2013, 29, 2062–2063. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | DPPH% (30 min) | DPPH% (60 min) | ABTS% | H2O2% |
|---|---|---|---|---|
| 4-Et2N-saloH | 2.47 ± 0.09 | 3.20 ± 0.08 | 25.84 ± 0.34 | 76.57 ± 0.71 |
| 3,5-diBr-saloH [13] | 7.97 ± 0.53 | 10.22 ± 0.30 | 16.55 ± 0.31 | 76.05 ± 1.51 |
| 3,5-diCl-saloH [14] | 15.68 ± 0.15 | 18.80 ± 0.52 | 84.89 ± 0.16 | 79.92 ± 0.34 |
| 5-F-saloH | 3.96 ± 1.16 | 5.56 ± 1.06 | 19.57 ± 0.58 | 71.84 ± 0.95 |
| 4-OMe-saloH | 4.16 ± 0.10 | 6.27 ± 0.25 | 40.83 ± 0.44 | 94.24 ± 0.67 |
| [Pd(4-Et2N-salo)2], 1 | 2.45 ± 0.09 | 4.00 ± 0.53 | 97.05 ± 0.48 | 18.19 ± 0.29 |
| [Pd(3,5-diBr-salo)2], 2 | 7.99 ± 0.24 | 14.63 ± 0.53 | 50.46 ± 0.91 | 49.21 ± 0.32 |
| [Pd(3,5-diCl-salo)2], 3 | 4.60 ± 0.20 | 2.91 ± 0.11 | 63.45 ± 0.38 | 51.26 ± 0.70 |
| [Pd(5-F-salo)2], 4 | 14.82 ± 0.45 | 13.61 ± 0.48 | 63.30 ± 0.53 | 48.31 ± 0.67 |
| [Pd(4-OMe-salo)2], 5 | 2.29 ± 0.36 | 3.00 ± 0.25 | 96.66 ± 0.46 | 58.38 ± 0.54 |
| NDGA | 87.08 ± 0.12 | 87.47 ± 0.12 | Not tested | Not tested |
| BHT | 61.30 ± 1.16 | 79.78 ± 1.12 | Not tested | Not tested |
| Trolox | Not tested | Not tested | 98.10 ± 0.48 | Not tested |
| L-ascorbic acid | Not tested | Not tested | Not tested | 60.80 ± 0.20 |
| Compound | S. aureus | B. subtilis | E. coli | X. campestris |
|---|---|---|---|---|
| 4-Et2N-saloH | >200 (>1035) | >200 (>1035) | >200 (>1035) | >200 (>1035) |
| 3,5-diBr-saloH | 25 (89) | 25 (89) | 50 (179) | 25 (89) |
| 3,5-diCl-saloH | 50 (262) | 50 (262) | 50 (262) | 50 (262) |
| 5-F-saloH | 200 (1427) | 200 (1427) | 200 (1427) | 200 (1427) |
| 4-OMe-saloH | >200 (>1314) | >200 (>1314) | >200 (>1314) | >200 (>1314) |
| [Pd(4-Et2N-salo)2], 1 | 100 (204) | 100 (204) | 100 (204) | 200 (407) |
| [Pd(3,5-diBr-salo)2], 2 | 25 (38) | 25 (38) | 25 (38) | 25 (38) |
| [Pd(3,5-diCl-salo)2], 3 | 25 (51) | 50 (103) | 25 (51) | 25 (51) |
| [Pd(5-F-salo)2], 4 | 100 (259) | 100 (259) | 100 (259) | 100 (259) |
| [Pd(4-OMe-salo)2], 5 | 100 (245) | 100 (245) | 100 (245) | 100 (245) |
| Compound | Band (ΔA/A0 a (%), Δλ b (nm)) | Kb (M−1) |
|---|---|---|
| 4-Et2N-saloH | 349 (−1 a, 0 b) | 5.06 (±0.14) × 105 |
| 3,5-dibromo-saloH [13] | 337 (<−50, elim c), 427 (>+50, 0) | 3.71 (±0.14) × 105 |
| 3,5-dichloro-saloH [14] | 335 (>−50, +2); 426 (>+50, +9) | 5.36 (±0.15) × 105 |
| 5-fluoro-saloH | 334 (−30, +1); 421 (>+50, 0) | 8.37 (±0.47) × 104 |
| 4-OMe-saloH [23] | 315 (−44, 1) | 9.25 (±0.12) × 105 |
| [Pd(4-Et2N-salo)2], 1 | 339 (+3, 0); 350 (+2, 0); 388 (−10, 0) | 3.66 (±0.22) × 105 |
| [Pd(3,5-diBr-salo)2], 2 | 310 (−10, 0); 426 (+20, 0) | 3.18 (±0.21) × 105 |
| [Pd(3,5-diCl-salo)2], 3 | 314 (−14, +2); 425 (+24, 0) | 1.90 (±0.12) × 106 |
| [Pd(5-F-salo)2], 4 | 308 (−2, 0); 426 (+15, −10) | 2.84 (±0.14) × 105 |
| [Pd(4-OMe-salo)2], 5 | 310 (sh) (−12.5, +3); 386 (+16, −3) | 1.27 (±0.10) × 106 |
| Compound | ΔΙ/Ι0 (%) | KSV (M−1) | kq (M−1s−1) |
|---|---|---|---|
| 4-Et2N-saloH | 43.4 | 2.89 (±0.07) × 104 | 1.25 (±0.03) × 1012 |
| 3,5-diBr-saloH [13] | 46.6 | 3.95 (±0.10) × 104 | 1.72 (±0.04) × 1012 |
| 3,5-diCl-saloH [14] | 43.2 | 3.56 (±0.09) × 104 | 1.55 (±0.04) × 1012 |
| 5-F-saloH | 51.3 | 3.79 (±0.11) × 104 | 1.73 (±0.05) × 1012 |
| 4-OMe-saloH [23] | 62.5 | 5.19 (±0.17) × 104 | 2.26 (±0.07) × 1012 |
| [Pd(4-Et2N-salo)2], 1 | 51.2 | 1.03 (±0.02) × 105 | 4.49 (±0.09) × 1012 |
| [Pd(3,5-diBr-salo)2], 2 | 64.8 | 1.00 (±0.02) × 105 | 4.35 (±0.10) × 1012 |
| [Pd(3,5-diCl-salo)2], 3 | 60.6 | 7.53 (±0.18) × 104 | 3.27 (±0.08) × 1012 |
| [Pd(5-F-salo)2], 4 | 51.4 | 5.71 (±0.10) × 104 | 2.48 (±0.04) × 1012 |
| [Pd(4-OMe-salo)2], 5 | 44.2 | 4.26 (±0.08) × 104 | 1.85 (±0.04) × 1012 |
| Compound | ΔΙ/Ιο (%) | kq (M−1 s−1) | K (M−1) |
|---|---|---|---|
| BSA | |||
| 4-Et2N-saloH | 1.41 (±0.06) × 1013 | 4.16 (±0.23) × 105 | |
| 3,5-diBr-saloH [13] | 3.65 (±0.25) × 1013 | 2.97 (±0.16) × 106 | |
| 3,5-diCl-saloH [14] | 1.47 (±0.09) × 1013 | 1.64 (±0.10) × 106 | |
| 5-F-saloH | 1.98 (±0.08) × 1012 | 4.31 (±0.31) × 104 | |
| 4-OMe-saloH [23] | 1.16 (±0.14) × 1013 | 4.11 (±0.23) × 105 | |
| [Pd(4-Et2N-salo)2], 1 | 92.9 | 5.72 (±0.44) × 1013 | 1.08 (±0.06) × 105 |
| [Pd(3,5-diBr-salo)2], 2 | 97.5 | 2.11 (±0.08) × 1014 | 1.50 (±0.06) × 106 |
| [Pd(3,5-diCl-salo)2], 3 | 97.2 | 1.11 (±0.04) × 1014 | 7.82 (±0.24) × 105 |
| [Pd(5-F-salo)2], 4 | 85.7 | 2.24 (±0.14) × 1013 | 8.79 (±0.13) × 104 |
| [Pd(4-OMe-salo)2], 5 | 97.2 | 2.17 (±0.09) × 1014 | 9.18 (±0.27) × 105 |
| HSA | |||
| 4-Et2N-saloH | 2.10 (±0.22) × 1013 | 4.03 (±0.17) × 105 | |
| 3,5-diBr-saloH [13] | 1.72 (±0.06) × 1013 | 4.04 (±0.30) × 105 | |
| 3,5-diCl-saloH [14] | 7.11 (±0.32) × 1012 | 6.33 (±0.14) × 105 | |
| 5-F-saloH | 1.96 (±0.07) × 1012 | 4.65 (±0.35) × 104 | |
| 4-OMe-saloH | 2.28 (±0.13) × 1012 | 9.67 (±0.48) × 104 | |
| [Pd(4-Et2N-salo)2], 1 | 91.8 | 3.48 (±0.14) × 1013 | 1.25 (±0.06) × 105 |
| [Pd(3,5-diBr-salo)2], 2 | 88.8 | 4.22 (±0.13) × 1013 | 4.12 (±0.15) × 105 |
| [Pd(3,5-diCl-salo)2], 3 | 90.4 | 5.00 (±0.12) × 1013 | 4.10 (±0.17) × 105 |
| [Pd(5-F-salo)2], 4 | 75.7 | 1.82 (±0.06) × 1013 | 1.38 (±0.05) × 105 |
| [Pd(4-OMe-salo)2], 5 | 92.2 | 6.69 (±0.23) × 1013 | 3.56 (±0.12) × 105 |
| Compound | No Marker | Marker: Warfarin | Marker: Ibuprofen |
|---|---|---|---|
| BSA | |||
| 4-Et2N-saloH | 4.16 (±0.23) × 105 | 1.33 (±0.07) × 105 | 4.86 (±0.55) × 104 |
| 3,5-diBr-saloH [13] | 2.97 (±0.16) × 106 | 2.84 (±0.10) × 105 | 7.11 (±0.29) × 105 |
| 3,5-diCl-saloH [14] | 1.64 (±0.10) × 106 | 5.09 (±0.09) × 105 | 1.35 (±0.55) × 105 |
| 5-F-saloH | 4.31 (±0.31) × 104 | 3.02 (±0.31) × 104 | 4.60 (±0.44) × 104 |
| 4-OMe-saloH | 4.11 (±0.23) × 105 | 1.46 (±0.33) × 104 | 7.90 (±0.21) × 104 |
| [Pd(4-Et2N-salo)2], 1 | 1.08 (±0.06) × 105 | 9.34 (±0.60) × 104 | 8.94 (±0.63) × 104 |
| [Pd(3,5-diBr-salo)2], 2 | 1.50 (±0.06) × 106 | 9.64 (±0.70) × 104 | 1.05 (±0.09) × 105 |
| [Pd(3,5-diCl-salo)2], 3 | 7.82 (±0.24) × 105 | 3.26 (±0.34) × 105 | 2.89 (±0.10) × 105 |
| [Pd(5-F-salo)2], 4 | 8.79 (±0.13) × 104 | 6.05 (±0.27) × 104 | 9.27 (±0.48) × 104 |
| [Pd(4-OMe-salo)2], 5 | 9.18 (±0.27) × 105 | 1.46 (±0.14) × 105 | 6.52 (±0.46) × 104 |
| HSA | |||
| 4-Et2N-saloH | 4.03 (±0.17) × 105 | 2.28 (±0.09) × 105 | 4.21 (±0.40) × 104 |
| 3,5-diBr-saloH [13] | 4.04 (±0.30) × 105 | 1.23 (±0.05) × 105 | 4.56 (±0.37) × 104 |
| 3,5-diCl-saloH [14] | 6.33 (±0.14) × 105 | 1.52 (±0.07) × 105 | 1.87 (±0.08) × 105 |
| 5-F-saloH | 4.65 (±0.35) × 104 | 3.02 (±0.31) × 104 | 6.22 (±0.21) × 104 |
| 4-OMe-saloH | 9.67 (±0.48) × 104 | 1.66 (±0.21) × 104 | 4.69 (±0.40) × 104 |
| [Pd(4-Et2N-salo)2], 1 | 1.25 (±0.06) × 105 | 6.89 (±0.28) × 104 | 1.86 (±0.12) × 105 |
| [Pd(3,5-diBr-salo)2], 2 | 4.12 (±0.15) × 105 | 1.43 (±0.09) × 105 | 3.23 (±0.13) × 105 |
| [Pd(3,5-diCl-salo)2], 3 | 4.10 (±0.17) × 105 | 2.47 (±0.09) × 105 | 3.78 (±0.14) × 105 |
| [Pd(5-F-salo)2], 4 | 1.38 (±0.05) × 105 | 8.98 (±0.82) × 104 | 1.10 (±0.08) × 105 |
| [Pd(4-OMe-salo)2], 5 | 3.56 (±0.12) × 105 | 1.00 (±0.18) × 105 | 1.45 (±0.17) × 104 |
| Compound | CT DNA | E. coli DNA-Gyrase | S. aureus DNA-Gyrase | 5-LOX | FLAP |
|---|---|---|---|---|---|
| [Pd(4-Et2N-salo)2], 1 | −44.86 | −14.12 | −38.00 | −30.87 | −33.20 |
| [Pd(3,5-diBr-salo)2], 2 | −34.91 | −20.56 | −28.71 | −21.78 | −32.58 |
| [Pd(3,5-diCl-salo)2], 3 | −44.21 | −24.87 | −38.26 | −23.25 | −38.29 |
| [Pd(5-F-salo)2], 4 | −32.92 | −24.07 | −27.92 | −17.21 | −28.82 |
| [Pd(4-OMe-salo)2], 5 | −36.83 | −14.23 | −35.66 | −22.74 | −26.18 |
| EB | −40.47 | - | - | - | - |
| Chlorobiocin (CBN) | - | −18.26 | - | - | - |
| Moxifloxacin (MFX) | - | - | −42.18 | - | - |
| QPT-1 | - | - | −45.66 | - | - |
| Trolox | - | - | - | −22.40 | - |
| Complex | DFT Energy (Hartrees) | EHOMO (eV) | ELUMO (eV) | η (eV) | s (eV−1) | χ (eV) | μ (eV) | ω (eV) | |
|---|---|---|---|---|---|---|---|---|---|
| 6-31G*(d,p)/LANL2DZ Double-ζ | ωB97X-D/cc-pVTZ Triple-ζ | ||||||||
| 1 | 2.94 | ||||||||
| X-ray | −1392.40893 | −1383.65149 | −5.09 | −1.45 | 1.82 | 0.274 | 3.27 | −3.27 | |
| 2 | 6.05 | ||||||||
| cis | −11260.3055 | −6.11 | −2.77 | ||||||
| trans | −11260.3069 | −11247.40728 | −6.12 | −2.82 | 1.65 | 0.303 | 4.47 | −4.47 | |
| 3 | 5.97 | ||||||||
| cis | −2805.56390 | −6.10 | −2.72 | ||||||
| trans | −2805.56547 | −2796.91806 | −6.10 | −2.79 | 1.655 | 0.302 | 4.445 | −4.445 | |
| 4 | 4.86 | ||||||||
| cis | −1165.65626 | −5.75 | −2.33 | ||||||
| trans | −1165.65708 | −1159.02699 | −5.72 | −2.36 | 1.68 | 0.297 | 4.04 | −4.04 | |
| 5 | 3.86 | ||||||||
| cis | −1196.25448 | −5.52 | −1.90 | ||||||
| trans | −1195.47476 | −1189.11065 | −5.57 | −1.93 | 1.82 | 0.274 | 3.75 | −3.75 |
| Comp. | ZPE (kJ/mol/kcal/mol) a | Vibrational (v) Corrections (kJ/mol) | S° (J/mol·K) | H° (au) | G° (au) | Cv (J/mol·K) | |
|---|---|---|---|---|---|---|---|
| Hv | Hv-TSv | ||||||
| 1 | 1242.63/296.99 | 1308.7656 | 1110.2507 | 665.82 | −1391.91074 | −1391.98635 | 333.41 |
| 2 | 445.12/106.39 | 494.7563 | 321.2041 | 582.10 | −11260.1184 | −11260.1845 | 227.80 |
| 3 | 450.30/107.62 | 498.7245 | 331.2835 | 561.60 | −2805.37550 | −2805.43927 | 227.03 |
| 4 | 509.41/121.75 | 551.4305 | 399.8988 | 508.24 | −1165.44706 | −1165.50478 | 208.31 |
| 5 | 725.91/173.49 | 775.1167 | 609.3427 | 556.01 | −1195.95943 | −1196.02257 | 242.02 |
| Complex | CPK Area (Å2) | CPK Volume (Å3) | PSA (Å2) | CPK Ovality | Polarizability |
|---|---|---|---|---|---|
| 1 | 458.89 | 426.90 | 45.789 | 1.67 | 75.15 |
| 2 | 357.82 | 326.54 | 43.555 | 1.56 | 67.08 |
| 3 | 339.68 | 308.59 | 43.792 | 1.54 | 65.62 |
| 4 | 289.42 | 263.24 | 44.710 | 1.46 | 61.93 |
| 5 | 337.02 | 307.98 | 58.305 | 1.53 | 65.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zianna, A.; Geromichalos, G.; Fiotaki, A.-M.; Hatzidimitriou, A.G.; Kalogiannis, S.; Psomas, G. Palladium(II) Complexes of Substituted Salicylaldehydes: Synthesis, Characterization and Investigation of Their Biological Profile. Pharmaceuticals 2022, 15, 886. https://doi.org/10.3390/ph15070886
Zianna A, Geromichalos G, Fiotaki A-M, Hatzidimitriou AG, Kalogiannis S, Psomas G. Palladium(II) Complexes of Substituted Salicylaldehydes: Synthesis, Characterization and Investigation of Their Biological Profile. Pharmaceuticals. 2022; 15(7):886. https://doi.org/10.3390/ph15070886
Chicago/Turabian StyleZianna, Ariadni, George Geromichalos, Augusta-Maria Fiotaki, Antonios G. Hatzidimitriou, Stavros Kalogiannis, and George Psomas. 2022. "Palladium(II) Complexes of Substituted Salicylaldehydes: Synthesis, Characterization and Investigation of Their Biological Profile" Pharmaceuticals 15, no. 7: 886. https://doi.org/10.3390/ph15070886
APA StyleZianna, A., Geromichalos, G., Fiotaki, A.-M., Hatzidimitriou, A. G., Kalogiannis, S., & Psomas, G. (2022). Palladium(II) Complexes of Substituted Salicylaldehydes: Synthesis, Characterization and Investigation of Their Biological Profile. Pharmaceuticals, 15(7), 886. https://doi.org/10.3390/ph15070886