
Bezafibrate Exerts Neuroprotective Effects in a Rat Model of Sporadic Alzheimer’s Disease

 ,
,  , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
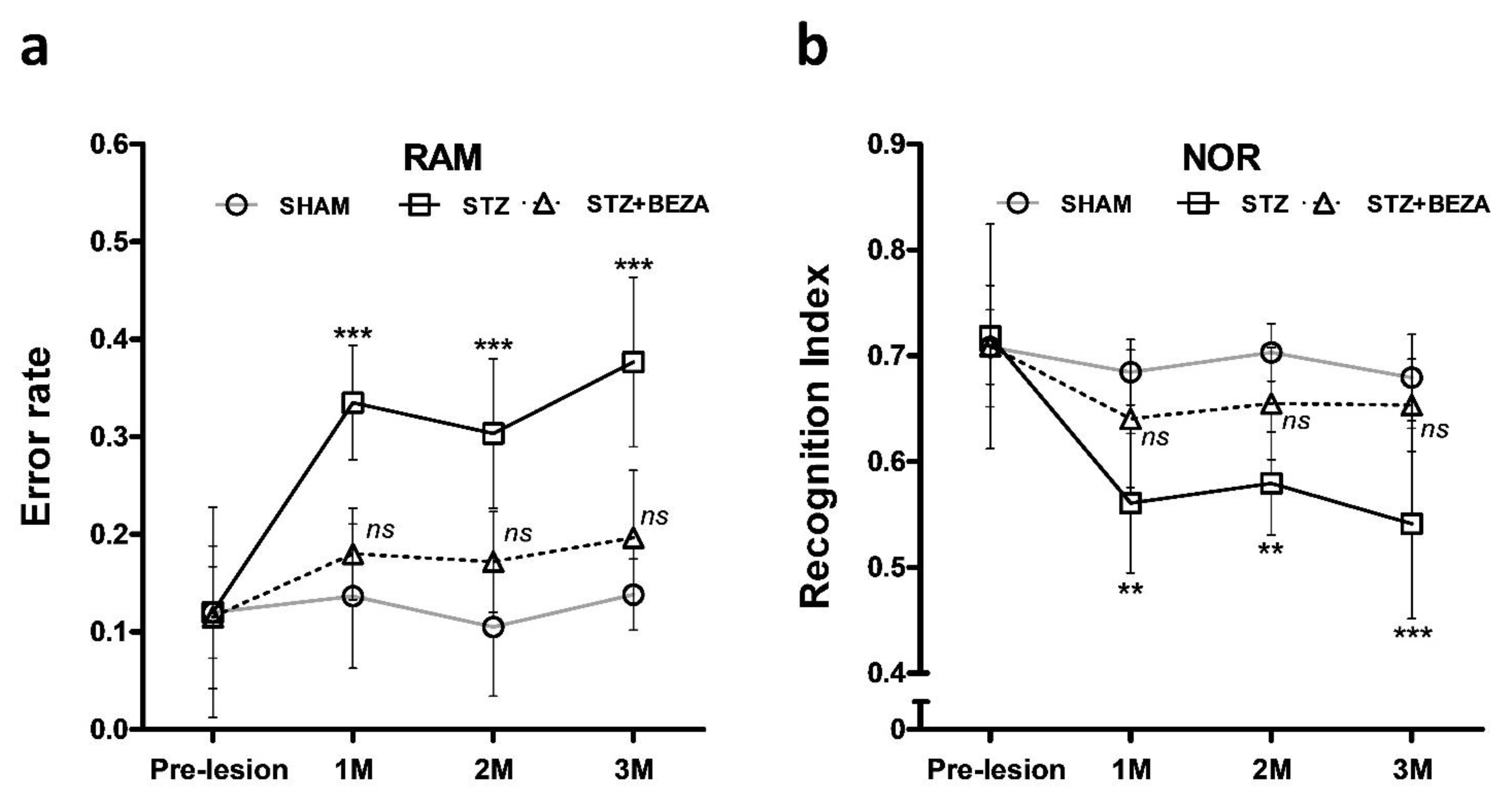
2.1. Bezafibrate Rescued the STZ-ICV-Induced Behavioral Deficits
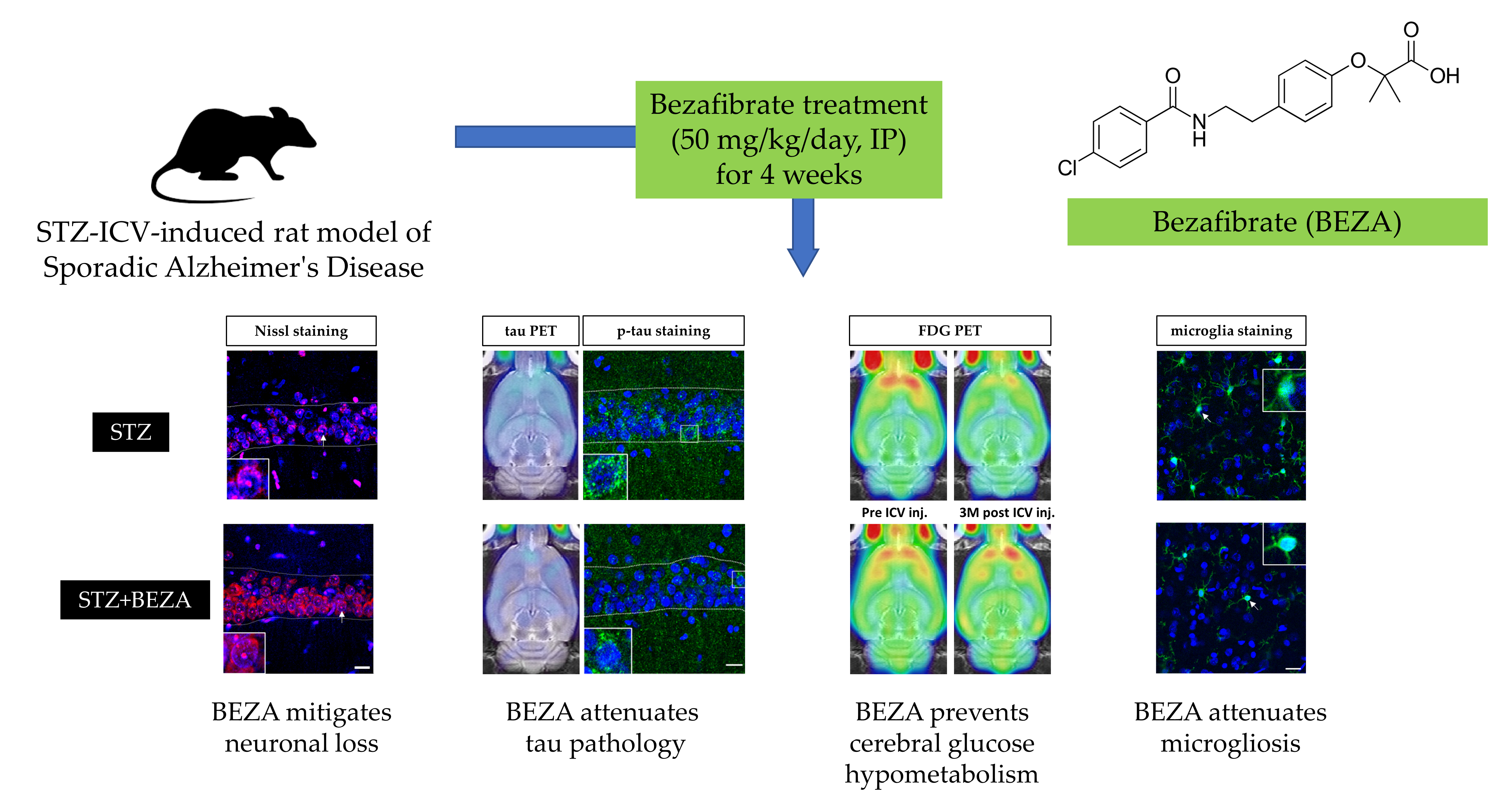
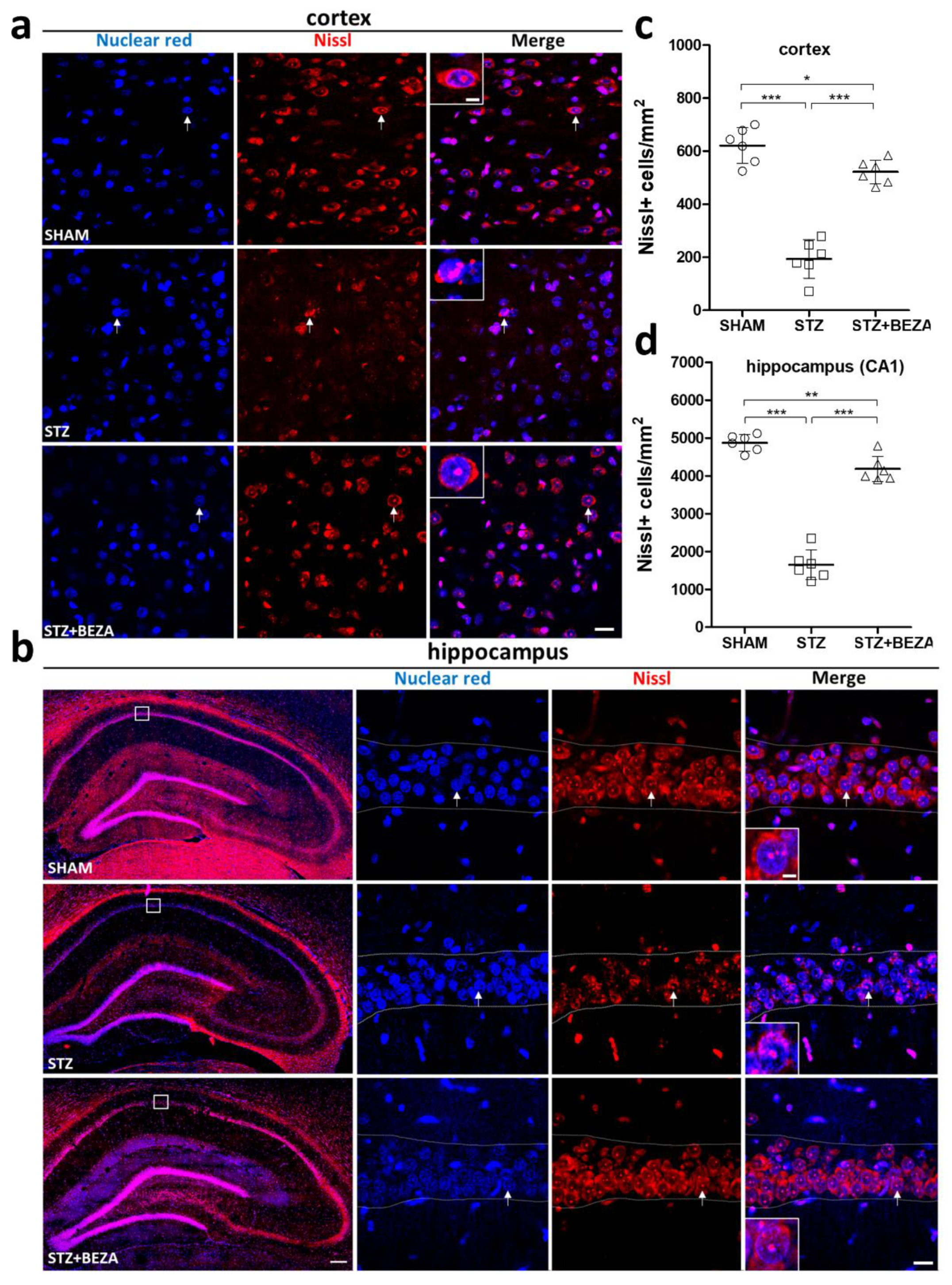
2.2. Bezafibrate Mitigated the STZ-ICV-Induced Brain Neuronal Loss
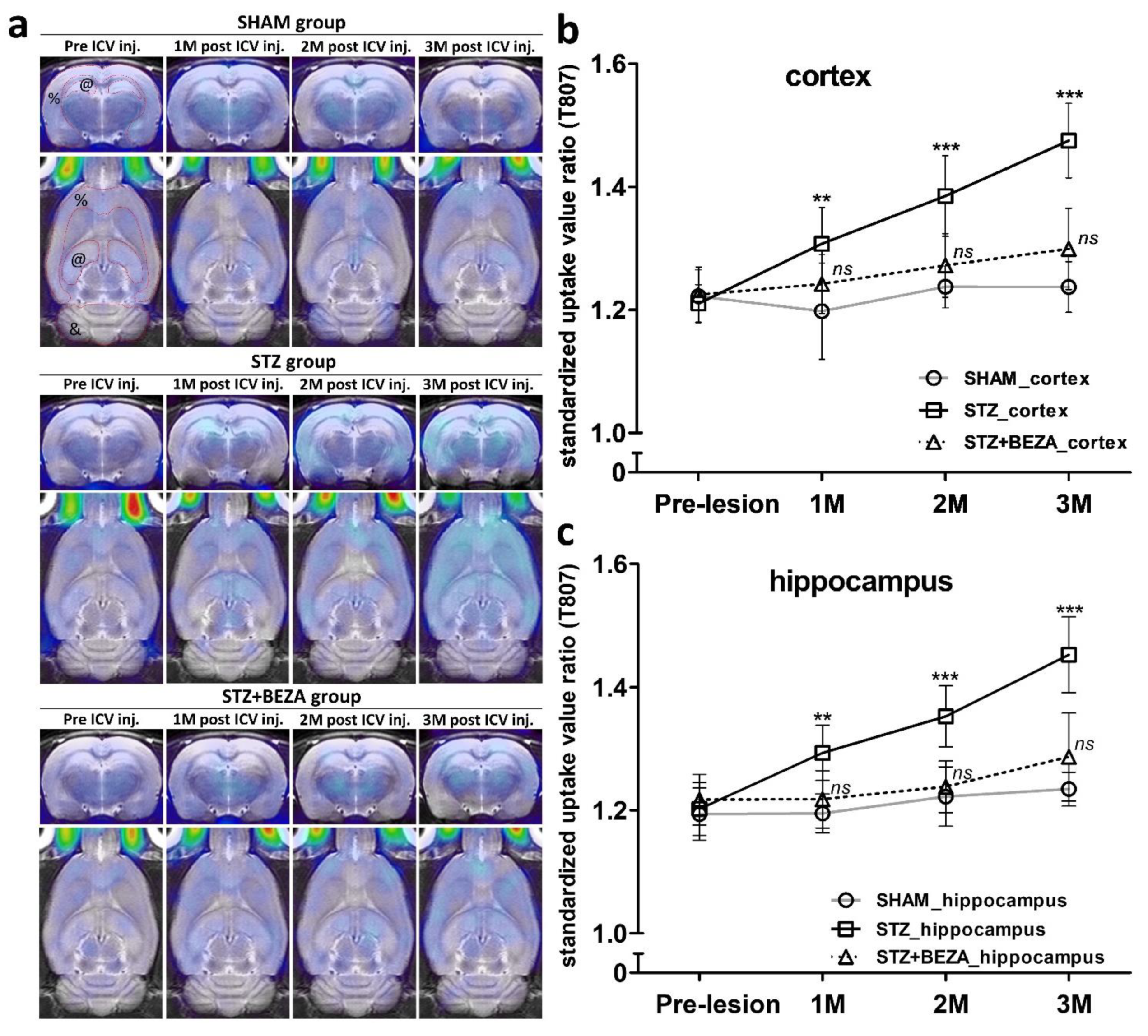
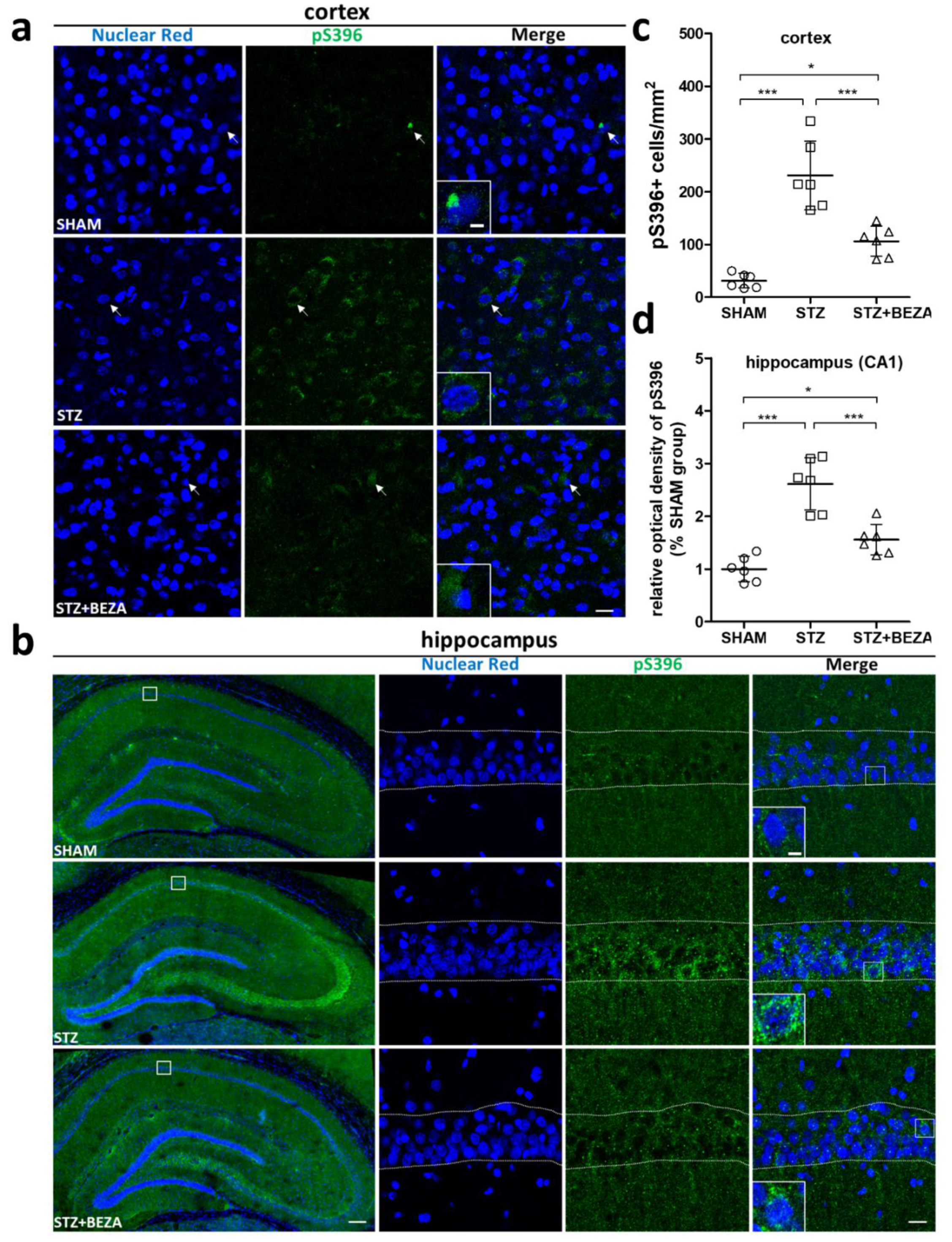
2.3. Bezafibrate Alleviated the Severity of Tau Pathology in STZ-ICV-Injected Rats
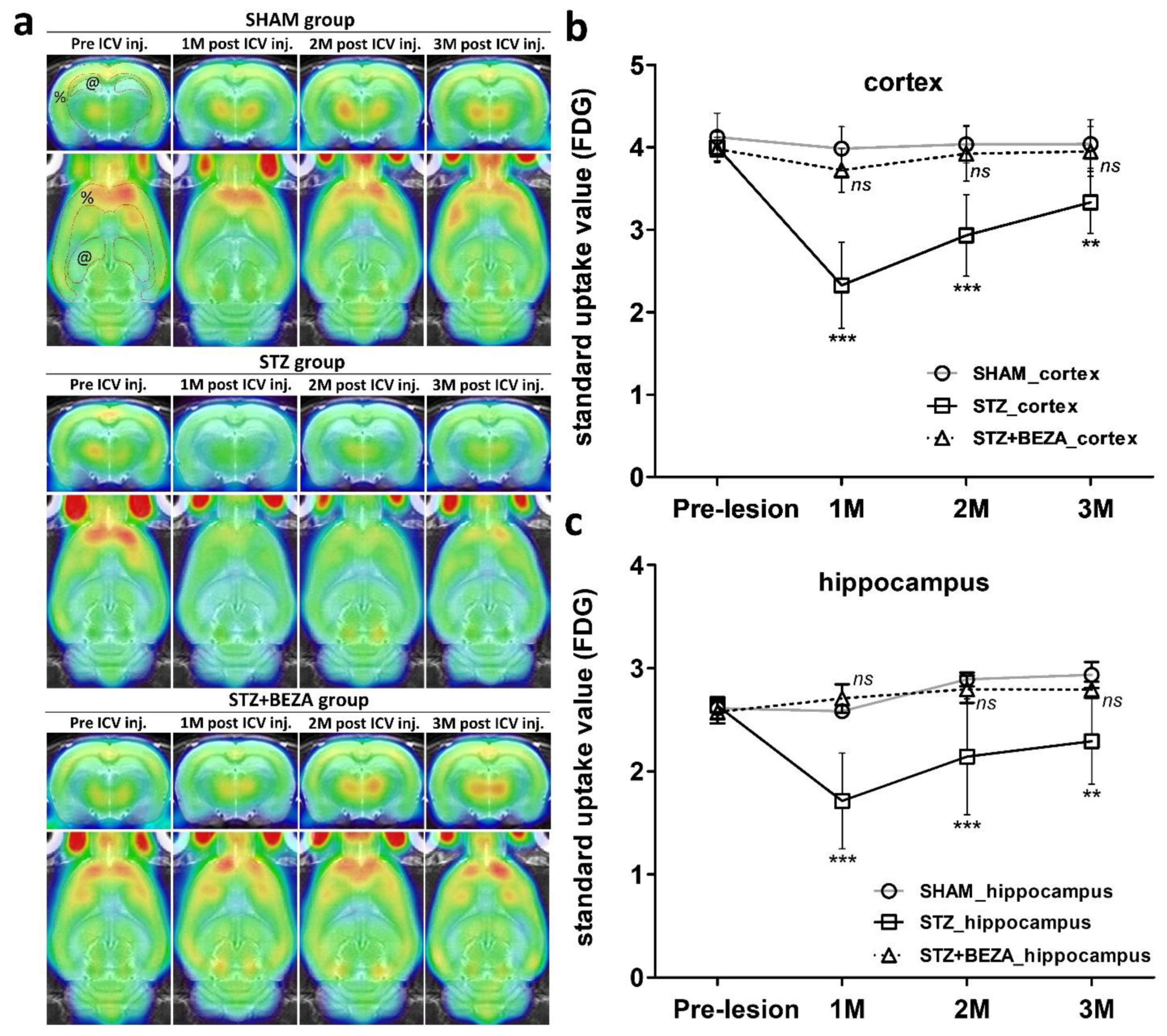
2.4. Bezafibrate Prevented STZ-ICV-Induced Cerebral Glucose Hypometabolism
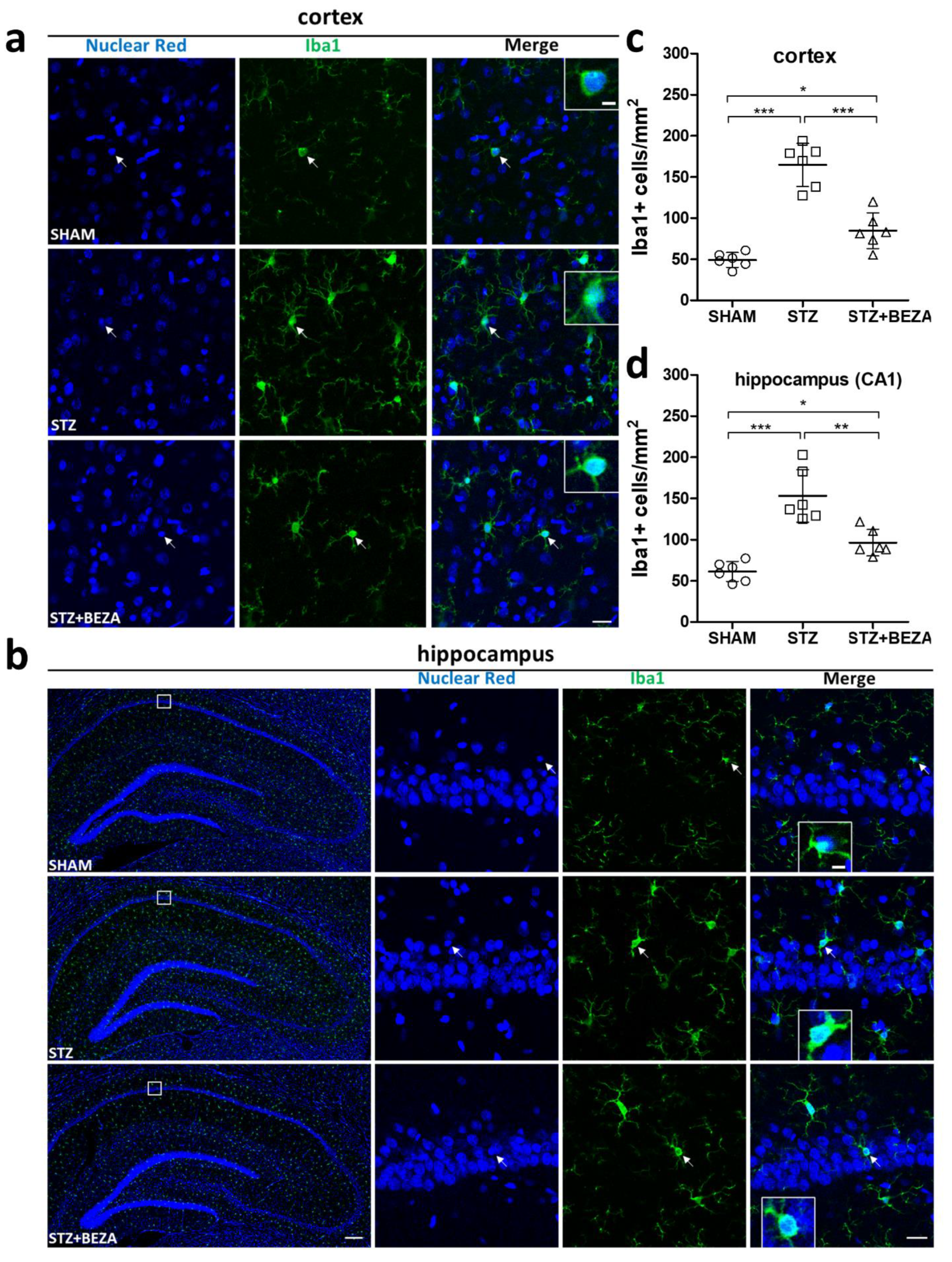
2.5. Bezafibrate Reduced Microgliosis in STZ-ICV-Injected Rats
3. Discussion
4. Materials and Methods
4.1. Animals
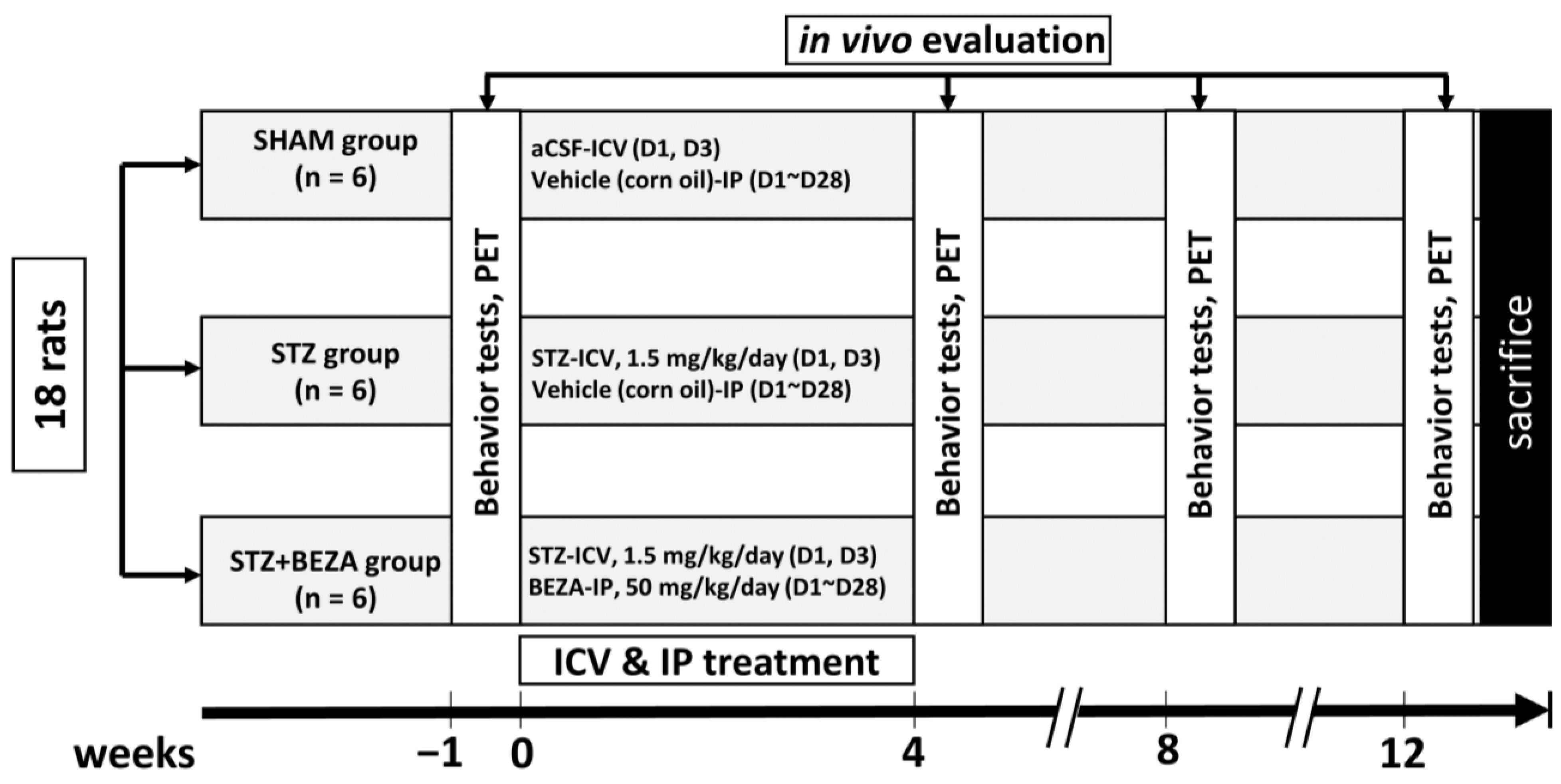
4.2. Animal Groups and Drugs Treatments
4.2.1. Animal Group Assignments and Experimental Procedures
4.2.2. Procedures of STZ-ICV Administration and Bezafibrate Treatment
4.3. Animal Behavior Tests
4.3.1. Radial Arm Maze Test
4.3.2. Novel Object Recognition Test
4.4. Animal PET and Radiopharmaceuticals
4.5. Immunofluorescence Staining
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimer’s Dis. 2018, 62, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef]
- Kumar, M.; Bansal, N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2021. advance online publication. [Google Scholar] [CrossRef]
- Franko, A.; Huypens, P.; Neschen, S.; Irmler, M.; Rozman, J.; Rathkolb, B.; Neff, F.; Prehn, C.; Dubois, G.; Baumann, M.; et al. Bezafibrate Improves Insulin Sensitivity and Metabolic Flexibility in STZ-Induced Diabetic Mice. Diabetes 2016, 65, 2540–2552. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, G.; Burckhardt, B.C. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. In Drug Transporters; Springer: Berlin/Heidelberg, Germany, 2011; pp. 29–104. [Google Scholar]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurother. 2008, 5, 481–489. [Google Scholar] [CrossRef]
- Dumont, M.; Stack, C.; Elipenahli, C.; Jainuddin, S.; Gerges, M.; Starkova, N.; Calingasan, N.Y.; Yang, L.; Tampellini, D.; Starkov, A.A.; et al. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum. Mol. Genet. 2012, 21, 5091–5105. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, B.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Dysregulation of Insulin Signaling, Glucose Transporters, O-GlcNAcylation, and Phosphorylation of Tau and Neurofilaments in the Brain: Implication for Alzheimer’s Disease. Am. J. Pathol. 2009, 175, 2089–2098. [Google Scholar] [CrossRef]
- Knezovic, A.; Osmanovic-Barilar, J.; Curlin, M.; Hof, P.R.; Šimić, G.; Riederer, P.; Salkovic-Petrisic, M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J. Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef]
- Knezovic, A.; Barilar, J.O.; Babic, A.; Bagaric, R.; Farkas, V.; Riederer, P.; Salkovic-Petrisic, M. Glucagon-like peptide-1 mediates effects of oral galactose in streptozotocin-induced rat model of sporadic Alzheimer’s disease. Neuropharmacol. 2018, 135, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Silva, D.; Carrettiero, D.C.; Oliveira, A.S.A.; Rodrigues, S.; Dos Santos-Lopes, J.; Canas, P.; Cunha, R.; Almeida, M.C.; Ferreira, T.L. Anandamide Effects in a Streptozotocin-Induced Alzheimer’s Disease-Like Sporadic Dementia in Rats. Front. Neurosci. 2018, 12, 653. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Silva, D.; Vizin, R.C.L.; Martins, T.; Ferreira, T.; Almeida, M.; Carrettiero, D. Intracerebral Injection of Streptozotocin to Model Alzheimer Disease in Rats. Bio-Protocol 2019, 9, e3397. [Google Scholar] [CrossRef]
- Wang, T.; Zheng, W.; Xu, H.; Zhou, J.-M.; Wang, Z.-Y. Clioquinol Inhibits Zinc-Triggered Caspase Activation in the Hippocampal CA1 Region of a Global Ischemic Gerbil Model. PLoS ONE 2010, 5, e11888. [Google Scholar] [CrossRef]
- Xia, C.-F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [18F] T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D.; Mintun, M.S. Regional profiles of the candidate tau PET ligand18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139, 1539–1550. [Google Scholar] [CrossRef]
- Pontecorvo, M.J.; Keene, C.D.; Beach, T.G.; Montine, T.J.; Arora, A.K.; Sr, M.D.D.; Navitsky, M.; Kennedy, I.; Joshi, A.D.; Lu, M.; et al. Comparison of regional flortaucipir PET with quantitative tau immunohistochemistry in three subjects with Alzheimer’s disease pathology: A clinicopathological study. EJNMMI Res. 2020, 10, 65. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, N.; Wang, S.; Yao, Z.; Xiao, F.; Lu, J.; Chen, B. Neuronal loss and microgliosis are restricted to the core of Aβ deposits in mouse models of Alzheimer’s disease. Aging Cell 2021, 20, e13380. [Google Scholar] [CrossRef]
- Ghosh, R.; Sil, S.; Gupta, P.; Ghosh, T. Optimization of intracerebroventricular streptozotocin dose for the induction of neuroinflammation and memory impairments in rats. Metab. Brain Dis. 2020, 35, 1279–1286. [Google Scholar] [CrossRef]
- Lithfous, S.; Dufour, A.; Després, O. Spatial navigation in normal aging and the prodromal stage of Alzheimer’s disease: Insights from imaging and behavioral studies. Ageing Res. Rev. 2013, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Laatu, S.; Revonsuo, A.; Jäykkä, H.; Portin, R.; Rinne, J.O. Visual object recognition in early Alzheimer’s disease: Deficits in semantic processing. Acta Neurol. Scand. 2003, 108, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Floresco, S.; Seamans, J.K.; Phillips, A.G. Selective Roles for Hippocampal, Prefrontal Cortical, and Ventral Striatal Circuits in Radial-Arm Maze Tasks with or Without a Delay. J. Neurosci. 1997, 17, 1880–1890. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2011, 13, 93–110. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Spreading of Tau Pathology in Sporadic Alzheimer’s Disease Along Cortico-cortical Top-Down Connections. Cereb. Cortex 2018, 28, 3372–3384. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Li, C.-H.; Chen, T.-F.; Chiu, M.-J.; Yen, R.-F.; Shih, M.-C.; Lin, C.-H. Integrated 18F-T807 Tau PET, Structural MRI, and Plasma Tau in Tauopathy Neurodegenerative Disorders. Front. Aging Neurosci. 2021, 13, 646440. [Google Scholar] [CrossRef] [PubMed]
- Nestor, P.J.; Altomare, D.; Festari, C.; Drzezga, A.; Rivolta, J.; Walker, Z.; Bouwman, F.; Orini, S.; Law, I.; Agosta, F.; et al. Clinical utility of FDG-PET for the differential diagnosis among the main forms of dementia. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1509–1525. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 707268. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Lester-Coll, N.; Plater, J.; Wands, J.R. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 89–109. [Google Scholar] [CrossRef]
- de la Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Van Giau, V.; Vo, V.G.; Nguyen, T.; Nguyen, T. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.; Gong, C.-X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Z.; Mao, Y.-F.; Zheng, T.; Zhang, B. Intranasal Insulin Ameliorates Cerebral Hypometabolism, Neuronal Loss, and Astrogliosis in Streptozotocin-Induced Alzheimer’s Rat Model. Neurotox. Res. 2018, 33, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Neschen, S.; Rozman, J.; Rathkolb, B.; Aichler, M.; Feuchtinger, A.; Brachthäuser, L.; Neff, F.; Kovarova, M.; Wolf, E.; et al. Bezafibrate ameliorates diabetes via reduced steatosis and improved hepatic insulin sensitivity in diabetic TallyHo mice. Mol. Metab. 2017, 6, 256–266. [Google Scholar] [CrossRef]
- Shiochi, H.; Ohkura, T.; Fujioka, Y.; Sumi, K.; Yamamoto, N.; Nakanishi, R.; Matsuzawa, K.; Izawa, S.; Ohkura, H.; Inoue, K.; et al. Bezafibrate improves insulin resistance evaluated using the glucose clamp technique in patients with type 2 diabetes mellitus: A small-scale clinical study. Diabetol. Metab. Syndr. 2014, 6, 113. [Google Scholar] [CrossRef][Green Version]
- Piemontese, L. An innovative approach for the treatment of Alzheimer’s disease: The role of peroxisome proliferator-activated receptors and their ligands in development of alternative therapeutic interventions. Neural Regen. Res. 2019, 14, 43–45. [Google Scholar] [CrossRef]
- Kummer, M.P.; Schwarzenberger, R.; Sayah-Jeanne, S.; Dubernet, M.; Walczak, R.; Hum, D.W.; Schwartz, S.; Axt, D.; Heneka, M.T. Pan-PPAR Modulation Effectively Protects APP/PS1 Mice from Amyloid Deposition and Cognitive Deficits. Mol. Neurobiol. 2014, 51, 661–671. [Google Scholar] [CrossRef]
- Malm, T.; Mariani, M.; Donovan, L.J.; Neilson, L.; Landreth, E.G. Activation of the nuclear receptor PPARδ is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. J. Neuroinflamm. 2015, 12, 1–15. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, J.B.; Jeżyna, M. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Guzman-Martinez, L.; Troncoso, C.C.; Faias, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef]
- Heppner, F.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2017, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Ginhoux, F.; Shimohama, S. Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem. Pharmacol. 2021, 192, 114754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Z.; Blanchard, J.; Dai, C.-L.; Sun, S.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; Gong, C.-X. A Non-transgenic Mouse Model (icv-STZ Mouse) of Alzheimer’s Disease: Similarities to and Differences from the Transgenic Model (3xTg-AD Mouse). Mol. Neurobiol. 2013, 47, 711–725. [Google Scholar] [CrossRef]
- Shoham, S.; Bejar, C.; Kovalev, E.; Schorer-Apelbaum, D.; Weinstock, M. Ladostigil prevents gliosis, oxidative–nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacol. 2007, 52, 836–843. [Google Scholar] [CrossRef]
- D’Orio, B.; Fracassi, A.; Cerù, M.P.; Moreno, S. Targeting PPARalpha in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 345–354. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Ozpinar, A. PPAR-δ and erucic acid in multiple sclerosis and Alzheimer’s Disease. Likely benefits in terms of immunity and metabolism. Int. Immunopharmacol. 2019, 69, 245–256. [Google Scholar] [CrossRef]
- Landreth, G. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 937–944. [Google Scholar] [CrossRef]
- Ponce-Lopez, T.; Liy-Salmeron, G.; Hong, E.; Meneses, A. Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho-GSK3β decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Res. 2011, 1426, 73–85. [Google Scholar] [CrossRef]
- Foster, J.E.; Gott, K.; Schuyler, M.R.; Kozak, W.; Tesfaigzi, Y. LPS-induced neutrophilic inflammation and Bcl-2 expression in metaplastic mucous cells. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L405–L414. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.E.; Zrull, M.C.; Luine, V.N. Chronic restraint stress enhances radial arm maze performance in female rats. Brain Res. 2001, 904, 279–289. [Google Scholar] [CrossRef]
- Bhuvanendran, S.; Kumari, Y.; Othman, I.; Shaikh, M.F. Amelioration of Cognitive Deficit by Embelin in a Scopolamine-Induced Alzheimer’s Disease-Like Condition in a Rat Model. Front. Pharmacol. 2018, 9, 665. [Google Scholar] [CrossRef]
- de Cristóbal, J.; García-García, L.; Delgado, M.; Pérez, M.; Pozo, M.A.; Medina, M. Longitudinal Assessment of a Transgenic Animal Model of Tauopathy by FDG-PET Imaging. J. Alzheimer’s Dis. 2014, 40, S79–S89. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Chiu, M.-J.; Yen, R.-F.; Tsai, C.-L.; Hsieh, H.-Y.; Chiu, C.-H.; Wu, C.-H.; Hsin, L.-W.; Tzen, K.-Y.; Cheng, C.-Y.; et al. An one-pot two-step automated synthesis of [18F] T807 injection, its biodistribution in mice and monkeys, and a preliminary study in humans. PLoS ONE 2019, 14, e0217384. [Google Scholar] [CrossRef]
- Jhao, Y.-T.; Chiu, C.-H.; Chen, C.-F.F.; Chou, T.-K.; Lin, Y.-W.; Ju, Y.-T.; Wu, S.-C.; Yan, R.-F.; Shiue, C.-Y.; Chueh, S.-H.; et al. The Effect of Sertoli Cells on Xenotransplantation and Allotransplantation of Ventral Mesencephalic Tissue in a Rat Model of Parkinson’s Disease. Cells 2019, 8, 1420. [Google Scholar] [CrossRef]
- O’Brien, F.E.; O’Connor, R.M.; Clarke, G.; Dinan, T.G.; Griffin, B.T.; Cryan, J.F. P-glycoprotein Inhibition Increases the Brain Distribution and Antidepressant-Like Activity of Escitalopram in Rodents. Neuropsychopharmacology 2013, 38, 2209–2219. [Google Scholar] [CrossRef]
- Tournier, N.; Bauer, M.; Pichler, V.; Nics, L.; Klebermass, E.-M.; Bamminger, K.; Matzneller, P.; Weber, M.; Karch, R.; Caille, F.; et al. Impact of P-Glycoprotein Function on the Brain Kinetics of the Weak Substrate 11C-Metoclopramide Assessed with PET Imaging in Humans. J. Nucl. Med. 2019, 60, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: London, UK, 2009; pp. 35–63. [Google Scholar]
- Vemuri, P.; Lowe, V.J.; Knopman, D.S.; Senjem, M.L.; Kemp, B.J.; Schwarz, C.; Przybelski, S.A.; Machulda, M.M.; Petersen, R.C.; Jack, C.R., Jr. Tau-PET uptake: Regional variation in average SUVR and impact of amyloid deposition. Alzheimer’s Dementia Diagn. Assess. Dis. Monit. 2017, 6, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Regalado-Reyes, M.; Furcila, D.; Hernández, F.; Ávila, J.; De Felipe, J.; León-Espinosa, G. Phospho-Tau Changes in the Human CA1 During Alzheimer’s Disease Progression. J. Alzheimer’s Dis. 2019, 69, 277–288. [Google Scholar] [CrossRef]
- Moloney, C.M.; Lowe, V.J.; Murray, M.E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimer’s Dement. 2021, 17, 1554–1574. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Alonso-Gil, S.; Santos, Á.; Perez-Castillo, A. Phosphodiesterase 7 Regulation in Cellular and Rodent Models of Parkinson’s Disease. Mol. Neurobiol. 2019, 57, 806–822. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, L.-F.; Jhao, Y.-T.; Chiu, C.-H.; Sun, L.-H.; Chou, T.-K.; Shiue, C.-Y.; Cheng, C.-Y.; Ma, K.-H. Bezafibrate Exerts Neuroprotective Effects in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals 2022, 15, 109. https://doi.org/10.3390/ph15020109
Lin L-F, Jhao Y-T, Chiu C-H, Sun L-H, Chou T-K, Shiue C-Y, Cheng C-Y, Ma K-H. Bezafibrate Exerts Neuroprotective Effects in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals. 2022; 15(2):109. https://doi.org/10.3390/ph15020109
Chicago/Turabian StyleLin, Li-Fan, Yun-Ting Jhao, Chuang-Hsin Chiu, Lu-Han Sun, Ta-Kai Chou, Chyng-Yann Shiue, Cheng-Yi Cheng, and Kuo-Hsing Ma. 2022. "Bezafibrate Exerts Neuroprotective Effects in a Rat Model of Sporadic Alzheimer’s Disease" Pharmaceuticals 15, no. 2: 109. https://doi.org/10.3390/ph15020109
APA StyleLin, L.-F., Jhao, Y.-T., Chiu, C.-H., Sun, L.-H., Chou, T.-K., Shiue, C.-Y., Cheng, C.-Y., & Ma, K.-H. (2022). Bezafibrate Exerts Neuroprotective Effects in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals, 15(2), 109. https://doi.org/10.3390/ph15020109