
Facile Entry to Pharmaceutically Important 3-Difluoromethyl-quinoxalin-2-ones Enabled by Visible-Light-Driven Difluoromethylation of Quinoxalin-2-ones


Abstract
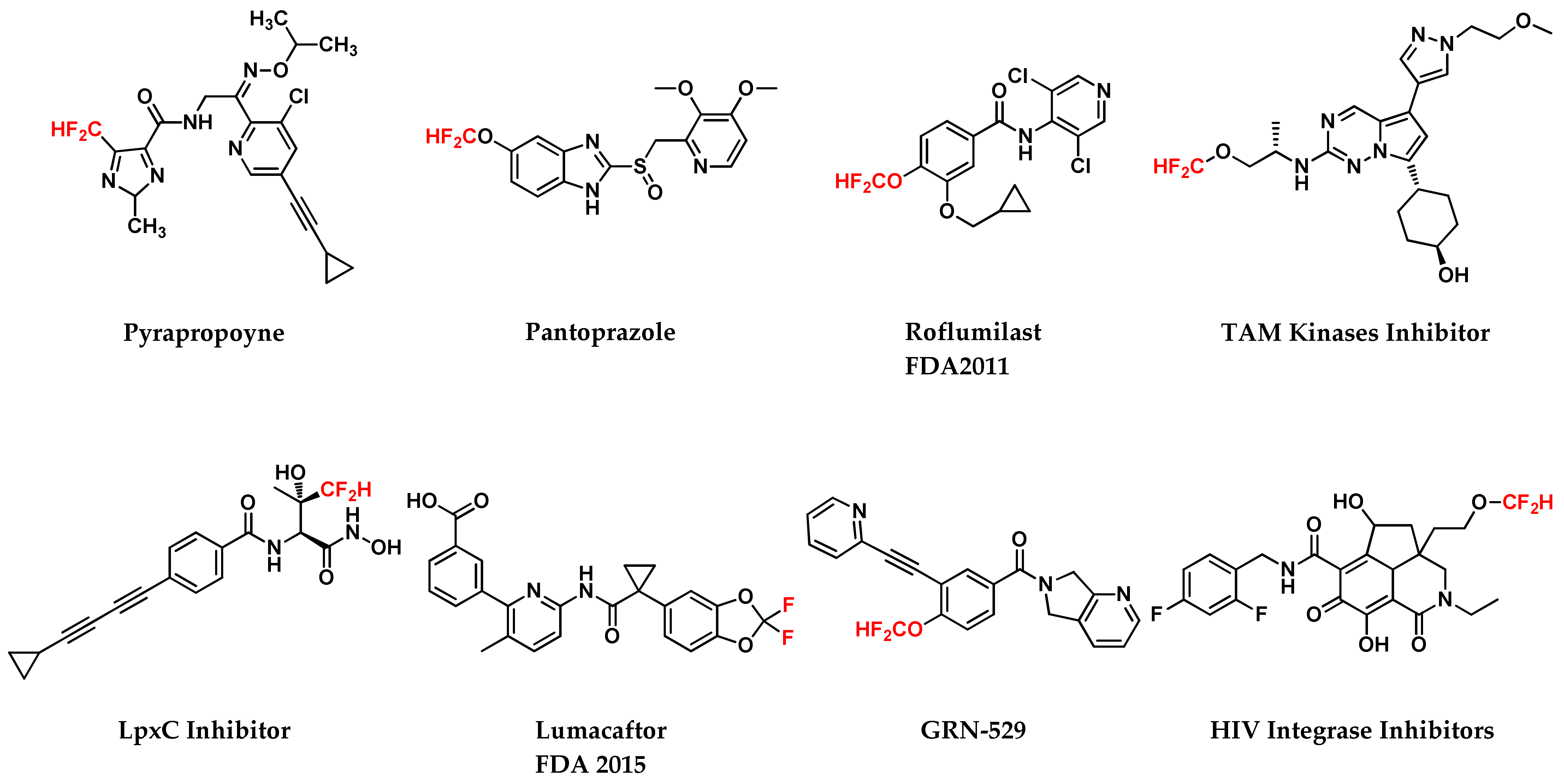
1. Introduction
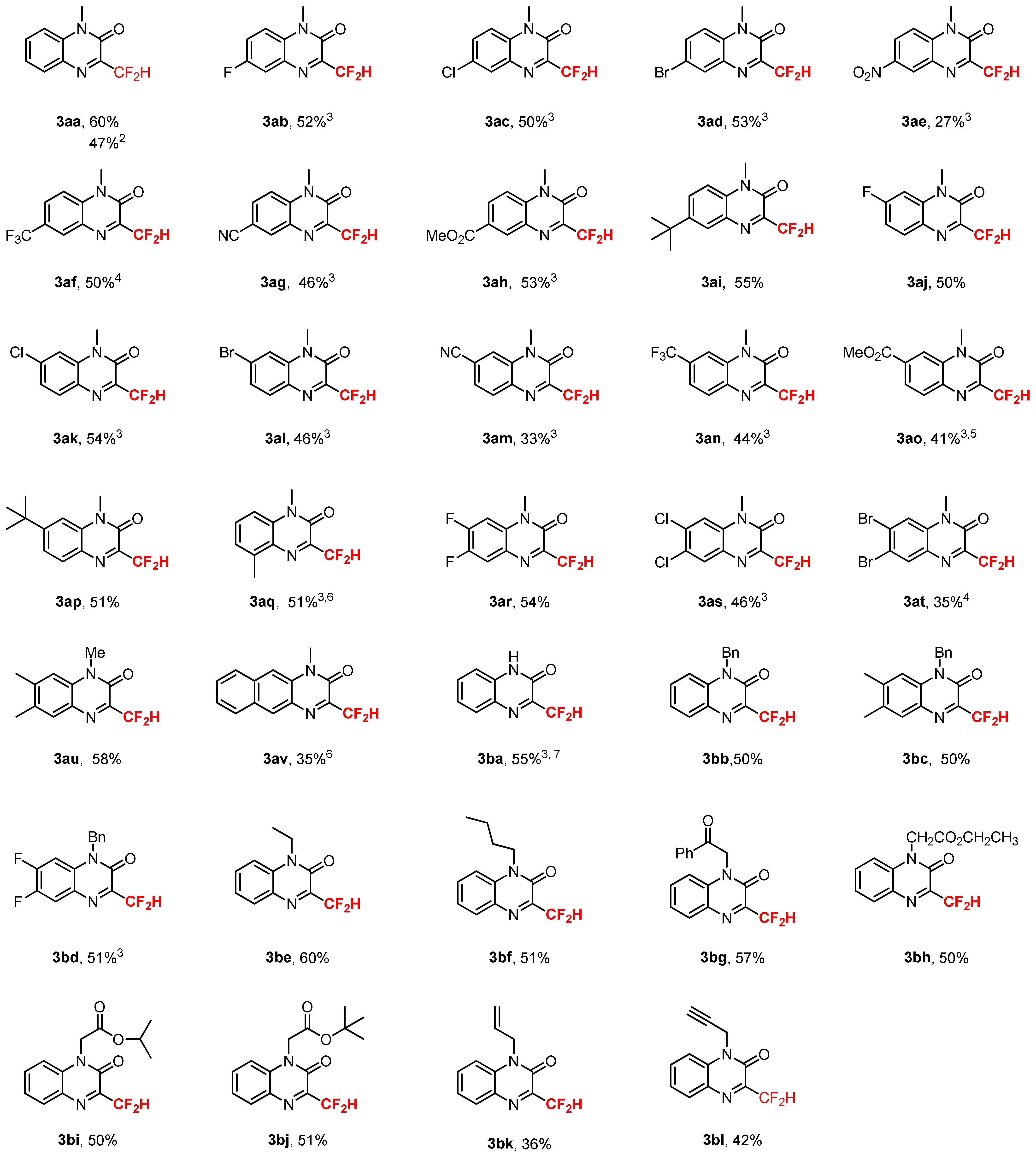
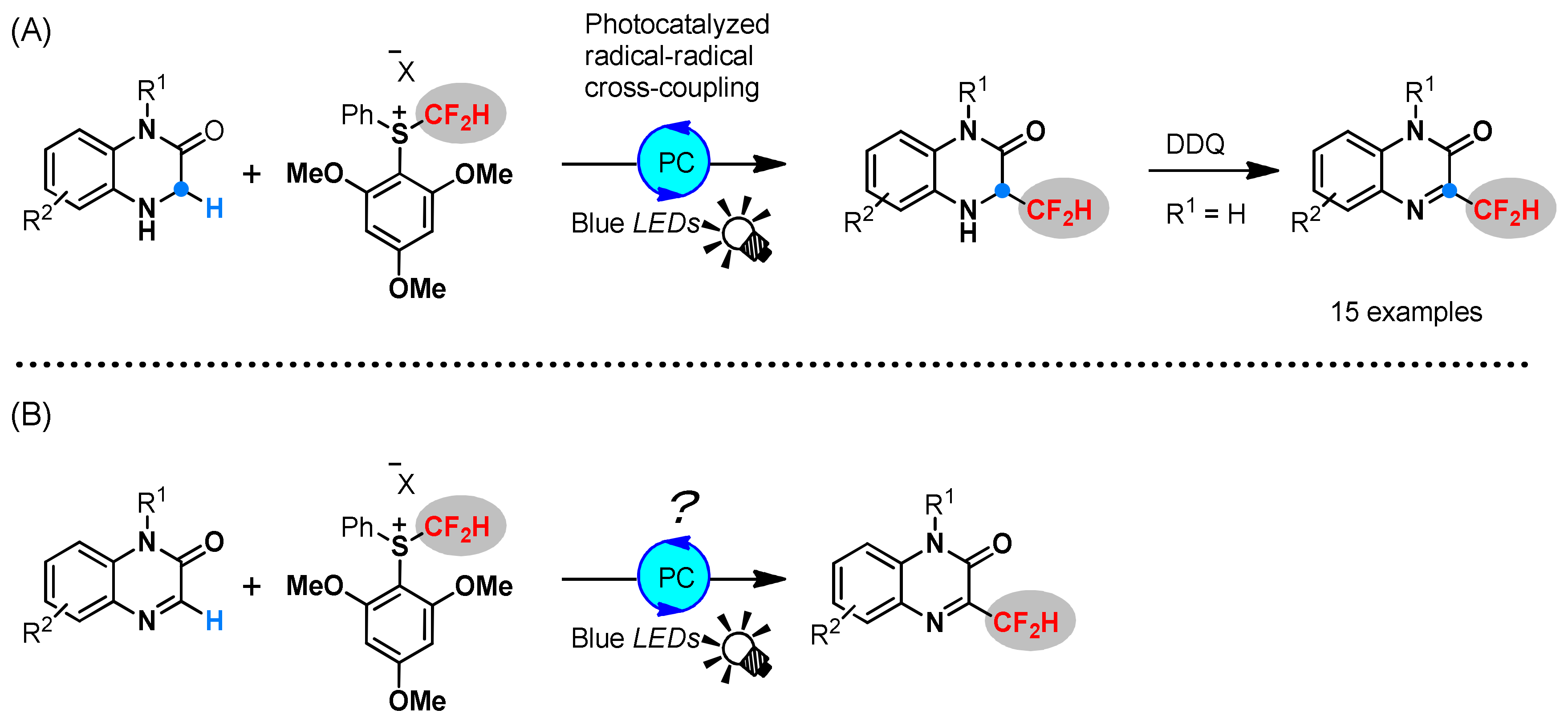
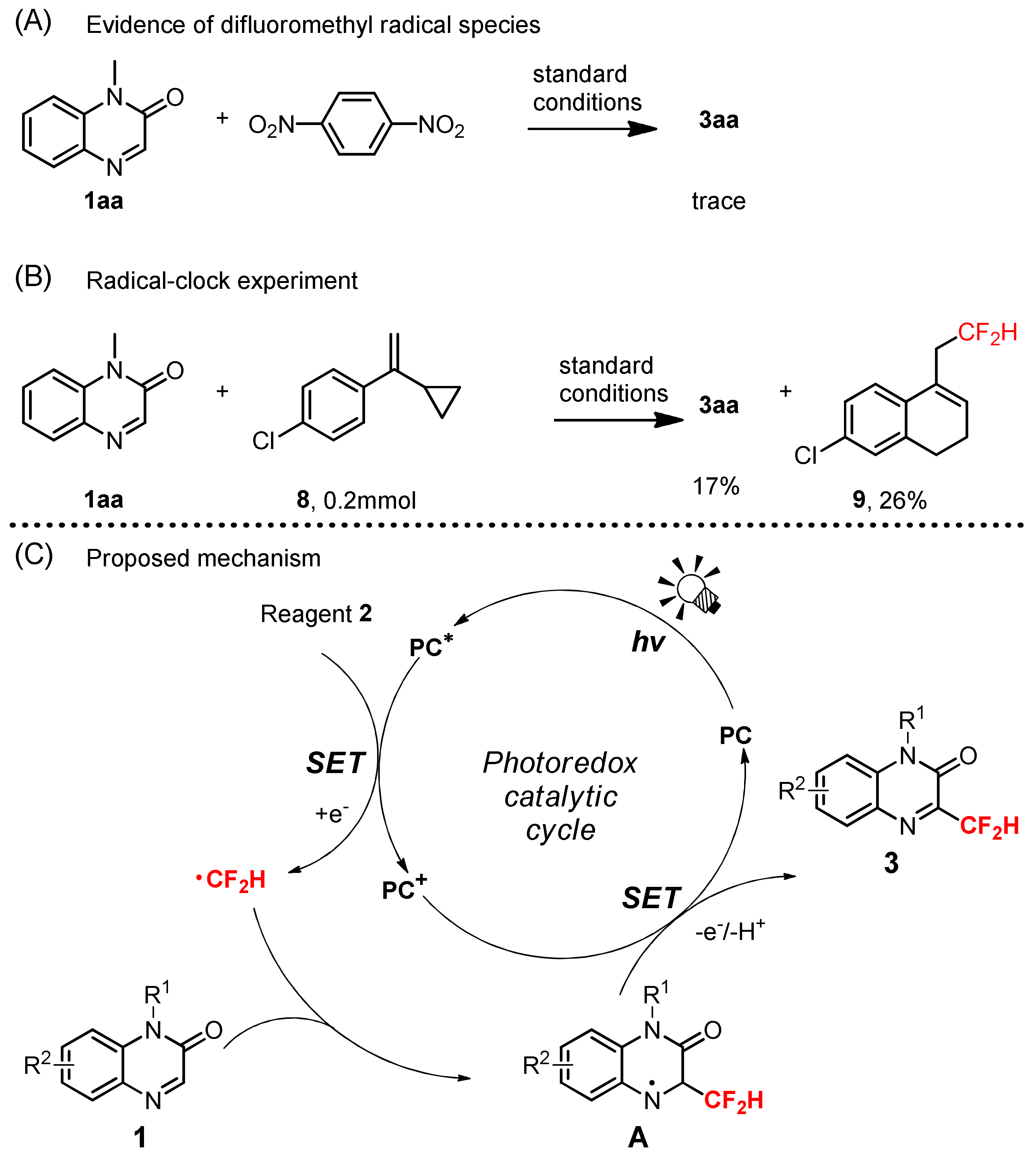
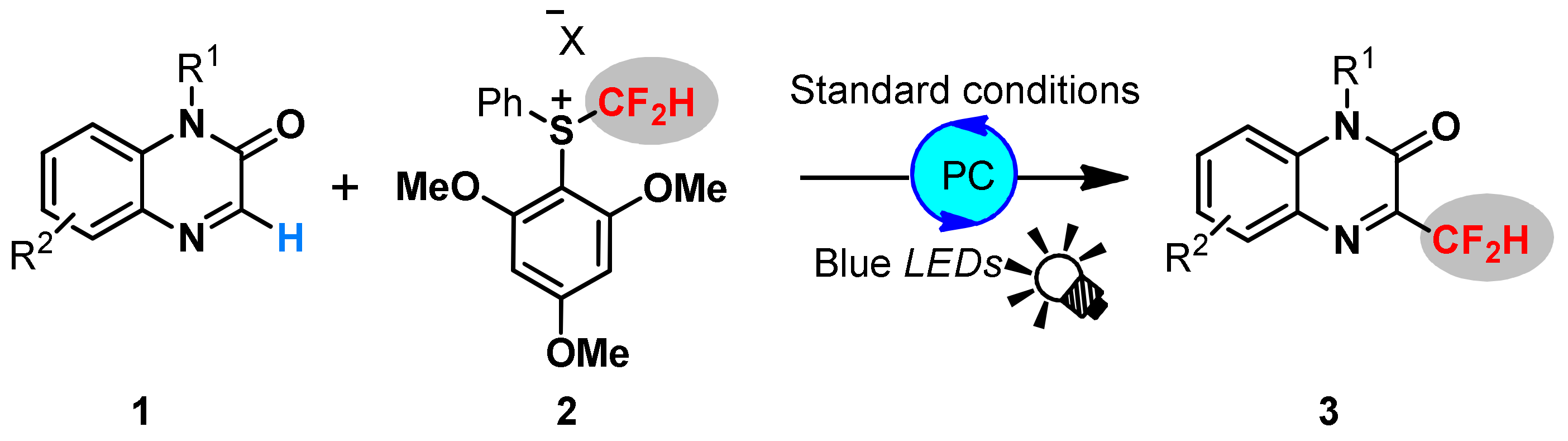
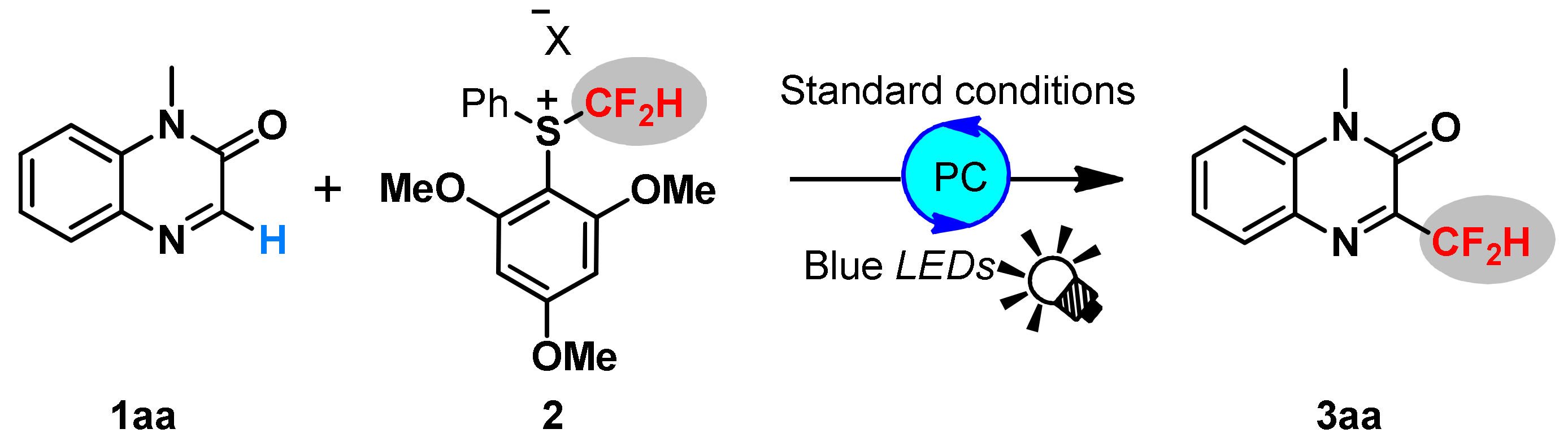
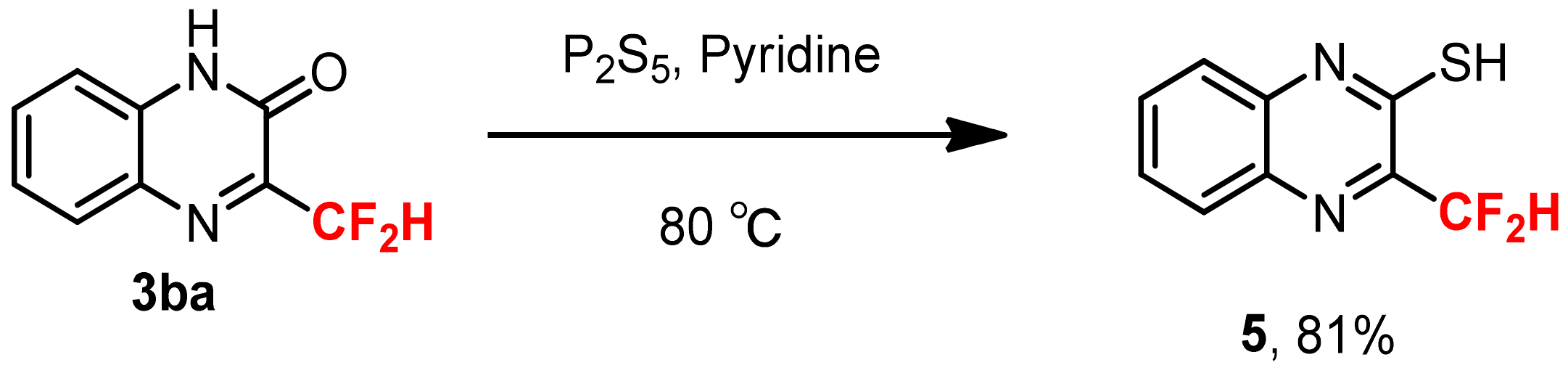
2. Results and Discussions
3. Materials and Methods
3.1. General Experimental Information
3.2. Reaction Apparatus
3.3. Synthesis of Quinoxalin-2-Ones
3.4. General Procedure for Visible-Light Redox-Catalyzed Difluoromethylation of Quinoxalin-2 ones
3.5. Larger-Scale Experiment
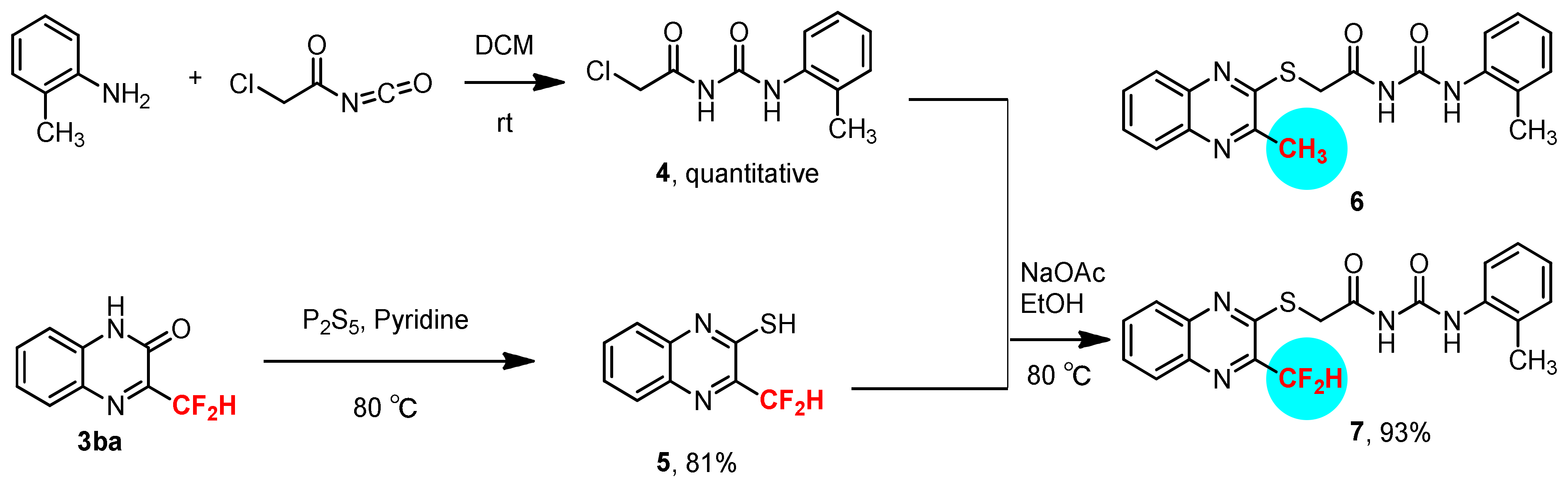
3.6. Synthesis of 3-Difluoroemthyl-quinoxalin-2-thiol 5
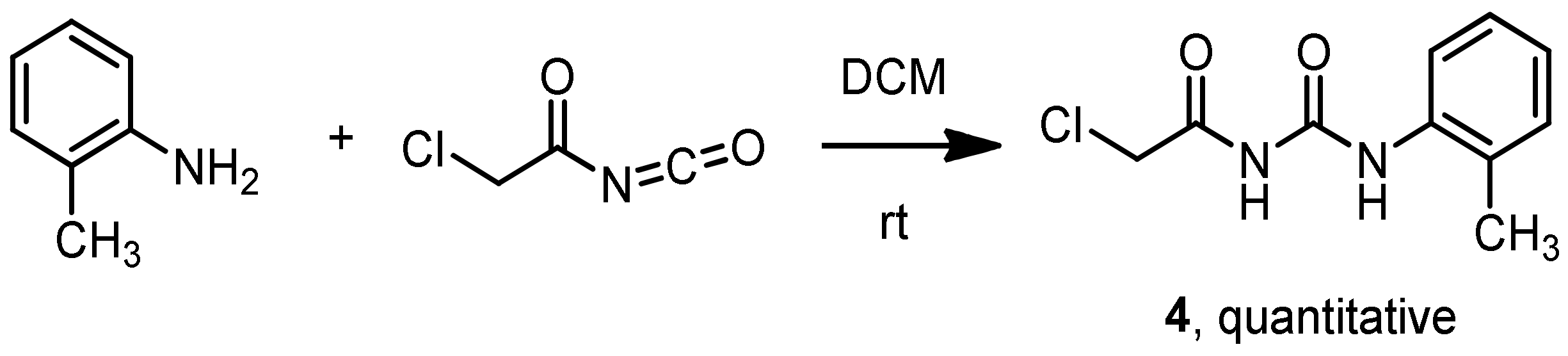
3.7. Synthesis of 1-(2-Chloroacetyl)-3-(2-methylphenyl)-urea 4
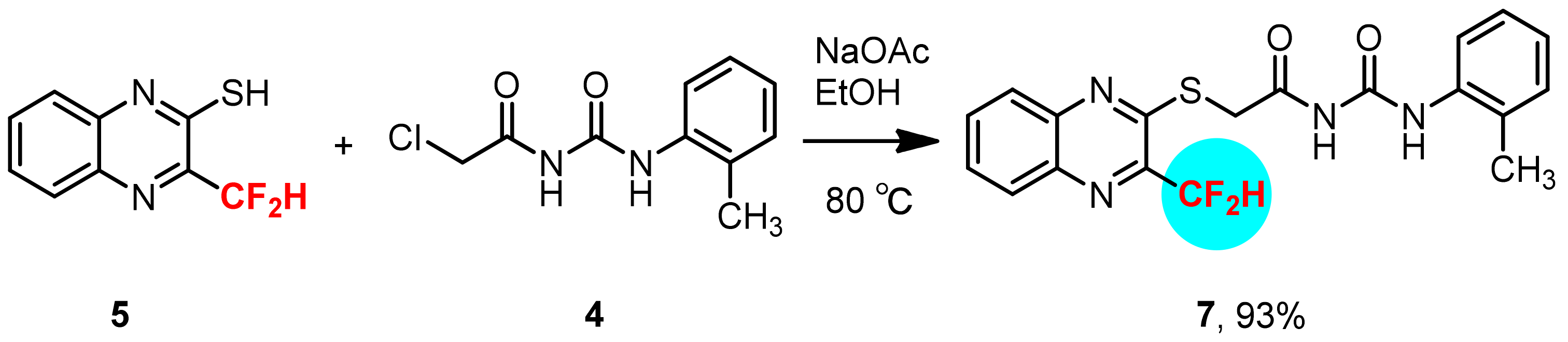
3.8. Synthesis of 1-(2-Methylphenyl)-3-[2-(3-difluoromethyl-quinoxalin-2-ylsulfanyl)-acetyl]-urea 7
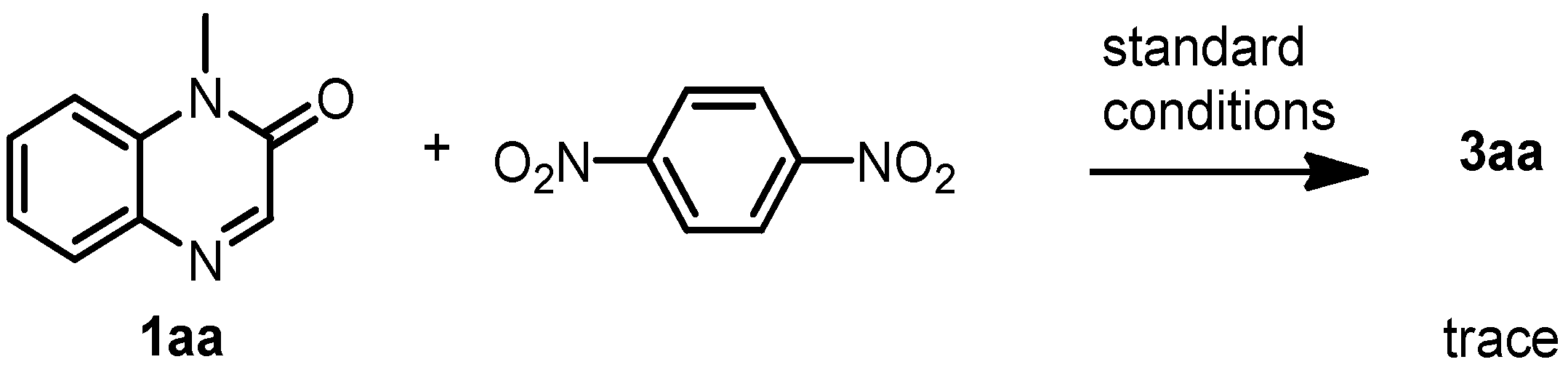
3.9. Radical Trapping Experiment
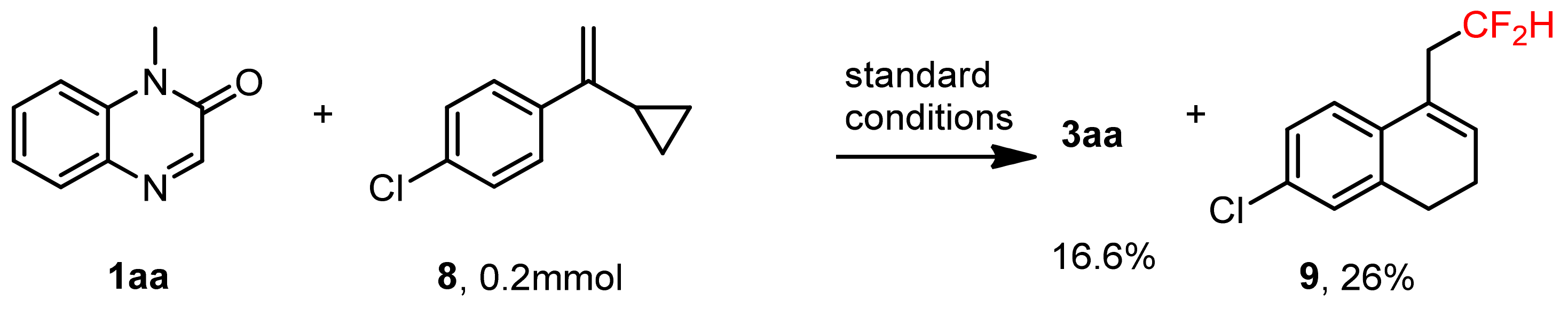
3.10. Radical-Clock Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Swallow, S. Fluorine in medicinal chemistry. Prog. Med. Chem. 2015, 54, 65–133. [Google Scholar] [PubMed]
- Goure, W.F.; Leschinsky, K.L.; Wratten, S.J.; Chupp, J.P. Synthesis and herbicidal activity of N-substituted 2,6-bis(polyfluoromethyl)dihydropyridine-3,5-dicarboxylates. J. Agric. Food Chem. 1991, 39, 981–986. [Google Scholar] [CrossRef]
- Jeschke, P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef]
- Isanbor, C.; O’Hagan, D. Fluorine in Medicinal Chemistry: A Review of Anti-Cancer Agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- MÜller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Gouverneur, V.; Muller, K. Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications; Imperial College Press: London, UK, 2012. [Google Scholar]
- Wang, J.; Sanchez-Rosello, M.; Acena, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: New structural trends and therapeutic areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Bremer, M. Nematic liquid crystals for active matrix displays: Molecular design and synthesis. Angew. Chem. Int. Ed. 2000, 39, 4216–4235. [Google Scholar] [CrossRef]
- Tasaka, T.; Takenaka, S.; Kabu, K.; Morita, Y.; Okamoto, H. Smectic phase exhibited by dissymmetric liquid crystals: Effect of a terminal fluoromethyl group on mesomorphic properties. Ferroelectronics 2002, 276, 83–92. [Google Scholar] [CrossRef]
- Boltalina, O.V.; Nakajima, T. New Fluorinated Carbons: Fundamentals and Applications; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Banks, R.E.; Smart, B.E.; Tatlow, J.C. (Eds.) Organofluorine Chemistry, Principles and Applications; Plenum: New York, NY, USA, 1994. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (accessed on 1 December 2022).
- Erickson, J.A.; McLoughlin, J.I. Hydrogen Bond Donor Properties of the Difluoromethyl Group. J. Org. Chem. 1995, 60, 1626–1631. [Google Scholar] [CrossRef]
- Sessler, C.D.; Rahm, M.; Becker, S.; Goldberg, J.M.; Wang, F.; Lippard, S.J. CF2H, a Hydrogen Bond Donor. J. Am. Chem. Soc. 2017, 139, 9325–9332. [Google Scholar] [CrossRef]
- Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem. 2017, 60, 797–804. [Google Scholar] [CrossRef]
- Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; et al. CF2H, a Functional Group-Dependent Hydrogen-Bond Donor: Is It a More or Less Lipophilic Bioisostere of OH, SH, and CH3? J. Med. Chem. 2019, 62, 5628–5637. [Google Scholar] [CrossRef]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef]
- Martínez, M.D.; Luna, L.; Tesio, A.Y.; Feresin, G.E.; Durán, F.J.; Burton, G. Antioxidant properties in a nonpolar environment of difluoromethyl bioisosteres of methylhydroxycinnamates. J. Pharm. Pharmacol. 2016, 68, 233–244. [Google Scholar] [CrossRef]
- Rodriguez, C.R.; del Fueyo, M.C.; Santillán, V.J.; Virginia, D.M.; Veleiro, A.S.; Castro, O.A.; Burton, G. Synthesis and biological activity of fluorinated analogues of the DAF-12 receptor antagonist 24-hydroxy-4-cholen-3-one. Steroids 2019, 151, 108469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiang, X.-X.; Chen, J.; Yang, C.; Pan, Y.-L.; Cheng, J.-P.; Meng, Q.; Li, X. Direct C-H difluoromethylation of hetercycles via organic photoredox catalysis. Nat. Commun. 2020, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, S.; Wang, F.; Qing, F.-L.; Chu, L. Silver-Enabled General Radical Difluoromethylation Reaction with TMSCF2H. Angew. Chem. Int. Ed. 2021, 60, 4300–4306. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Li, C.; Li, Y.; Zhu, Y.; Teng, P.; Gu, Y.-C.; Zhang, W.-H. Direct Difluoromethylation of Heterocycles through Photosensitized Electron Transfer. Eur. J. Org. Chem. 2022, 2022, e202100896. [Google Scholar] [CrossRef]
- Lu, S.-L.; Li, X.; Qin, W.-B.; Liu, J.-J.; Huang, Y.-Y.; Wong, H.N.C.; Liu, G.-K. Air- and Light-Stable S-(Difluoromethyl)Sulfonium Salts: C-Selective Electrophilic Difluoromethylation of β-Ketoesters and Malonates. Org. Lett. 2018, 20, 6925–6929. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-K.; Li, X.; Qin, W.-B.; Peng, X.-S.; Wong, H.N.C.; Zhang, L.; Zhang, X. Facile Difluoromethylation of Aliphatic Alcohols with an S-(Difluoro-methyl)Sulfonium Salt: Reaction, Scope and Mechanistic Study. Chem. Commun. 2019, 55, 7446–7449. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-K.; Qin, W.-B.; Li, X.; Lin, L.-T.; Wong, H.N.C. Difluoromethylation of Phenols and Thiophenols with the S-(Difluoromethyl)Sulfonium Salt: Reaction, Scope, and Mechanistic Study. J. Org. Chem. 2019, 84, 15948–15957. [Google Scholar] [CrossRef]
- Liu, G.-K.; Li, X.; Qin, W.-B.; Lin, W.-F.; Lin, L.-T.; Chen, J.-Y.; Liu, J.-J. Selective O-Difluoromethylation of 1,3-Diones Using S-(Difluoromethyl)-Sulfonium Salt. Chin. Chem. Lett. 2019, 30, 1515–1518. [Google Scholar] [CrossRef]
- Qin, W.-B.; Xiong, W.; Li, X.; Chen, J.-Y.; Lin, L.-T.; Wong, H.N.C.; Liu, G.-K. Visible-Light-Driven Difluoromethylation of Isocyanides with S-(Difluoromethyl)Diarylsulfonium Salt: Access to a Wide Variety of Difluoromethylated Phenanthridines and Isoquinolines. J. Org. Chem. 2020, 85, 10479–10487. [Google Scholar] [CrossRef]
- Qin, W.-B.; Xiong, W.; Zhao, Y.-S.; Fu, K.-Z.; Su, L.; Liu, G.-K. Difluoromethyl radical triggered tandem reaction of N-Allylamides to difluoromethylated β-amino alcohols by photoredox catalysis. Org. Lett. 2021, 23, 8482–8487. [Google Scholar] [CrossRef]
- Xiong, W.; Qin, W.-B.; Zhao, Y.-S.; Fu, K.-Z.; Liu, G.-K. Direct C(sp3)–H difluoromethylation via radical–radical cross-coupling by visible-light photoredox catalysis. Org. Chem. Front. 2022, 9, 2141–2148. [Google Scholar] [CrossRef]
- Chen, D.; Wang, Z.J.; Bao, W. Copper-catalyzed cascade syntheses of 2H-benzo[b][1,4]thiazin-3(4H)-ones and quinoxalin-2(1H)-ones through capturing S and N atom respectively from AcSH and TsNH2. J. Org. Chem. 2010, 75, 5768–5771. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Obata, T.; Yamazaki, Y.; Mori, Y.; Hirokawa, H.; Koseki, J.-I.; Hattori, T.; Niitsu, K.; Takeda, S.; Aburada, M.; et al. Potent Platelet-Derived Growth Factor-b Receptor (PDGF-bR) Inhibitors: Synthesis and Structure–Activity Relationships of 7-[3-(Cyclohexylmethyl)ureido]-3-{1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl}quinoxalin-2(1H)-one Derivatives. Chem. Pharm. Bull. 2007, 55, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.J.; TenBrink, R.E.; Stelzer, L.S.; Belonga, K.L.; Carter, D.B.; Im, H.K.; Im, W.B.; Sethy, V.H.; Tang, A.H.; VonVoigtlander, P.F.; et al. High-Affinity Partial Agonist Imidazo[1,5-a]quinoxaline Amides, Carbamates, and Ureas at the γ-Aminobutyric Acid A/Benzodiazepine Receptor Complex. J. Med. Chem. 1996, 39, 158–175. [Google Scholar] [CrossRef]
- Marie Loughran, H.; Han, Z.; Wrobel, J.E.; Decker, S.E.; Ruthel, G.; Freedman, B.D.; Harty, R.N.; Reitz Allen, B. Quinoxaline-based inhibitors of Ebola and Marburg VP40 egress. Bioorg. Med. Chem. Lett. 2016, 26, 3429–3435. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | PC | Base | Temp. (℃) | Solvent | Yield (%) |
| 1 | PC I | LiOH | 25 °C | AcOEt | 53 |
| 2 | 4CzIPN | LiOH | 25 °C | AcOEt | 35 |
| 3 | Perylene | LiOH | 25 °C | AcOEt | 52 |
| 4 | Rose Bengal | LiOH | 25 °C | AcOEt | NR |
| 5 | Eosin Y | LiOH | 25 °C | AcOEt | trace |
| 6 | fac-Ir(ppy)3 | LiOH | 25 °C | AcOEt | 39 |
| 7 | Ir(dFppy)3 | LiOH | 25 °C | AcOEt | 41 |
| 8 | PC II | LiOH | 25 °C | AcOEt | 60 |
| 9 | PC II | LiOAc | 25 °C | AcOEt | 58 |
| 10 | PC II | tBuOLi | 25 °C | AcOEt | 46 |
| 11 | PC II | K2CO3 | 25 °C | AcOEt | 36 |
| 12 | PC II | Na2CO3 | 25 °C | AcOEt | 24 |
| 13 | PC II | DIPEA | 25 °C | AcOEt | NR |
| 14 | PC II | LiOH | 25 °C | DCM | 56 |
| 15 | PC II | LiOH | 25 °C | CHCl3 | 45 |
| 16 | PC II | LiOH | 25 °C | CH3CN | 45 |
| 17 | PC II | LiOH | 25 °C | THF | 24 |
| 18 | PC II | LiOH | 40 °C | AcOEt | 51 |
| 19 | PC II | LiOH | 0 °C | AcOEt | 43 |
| 20 2 | PC II | LiOH | 25 °C | AcOEt | 60 |
| 21 3 | PC II (5 mol%) | LiOH | 25 °C | AcOEt | 57 |
| 22 4 | PC II/no light | LiOH | 25 °C | AcOEt | NR |
| 23 5 | no PC II | LiOH | 25 °C | AcOEt | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, K.-Z.; Chen, X.-X.; Zhao, Y.-S.; Gu, Y.-Q.; Liu, G.-K. Facile Entry to Pharmaceutically Important 3-Difluoromethyl-quinoxalin-2-ones Enabled by Visible-Light-Driven Difluoromethylation of Quinoxalin-2-ones. Pharmaceuticals 2022, 15, 1552. https://doi.org/10.3390/ph15121552
Fu K-Z, Chen X-X, Zhao Y-S, Gu Y-Q, Liu G-K. Facile Entry to Pharmaceutically Important 3-Difluoromethyl-quinoxalin-2-ones Enabled by Visible-Light-Driven Difluoromethylation of Quinoxalin-2-ones. Pharmaceuticals. 2022; 15(12):1552. https://doi.org/10.3390/ph15121552
Chicago/Turabian StyleFu, Kai-Zhong, Xu-Xin Chen, Ya-Shi Zhao, Yuan-Qing Gu, and Guo-Kai Liu. 2022. "Facile Entry to Pharmaceutically Important 3-Difluoromethyl-quinoxalin-2-ones Enabled by Visible-Light-Driven Difluoromethylation of Quinoxalin-2-ones" Pharmaceuticals 15, no. 12: 1552. https://doi.org/10.3390/ph15121552
APA StyleFu, K.-Z., Chen, X.-X., Zhao, Y.-S., Gu, Y.-Q., & Liu, G.-K. (2022). Facile Entry to Pharmaceutically Important 3-Difluoromethyl-quinoxalin-2-ones Enabled by Visible-Light-Driven Difluoromethylation of Quinoxalin-2-ones. Pharmaceuticals, 15(12), 1552. https://doi.org/10.3390/ph15121552