

Formulation of Lipid-Based Nanoparticles for Simultaneous Delivery of Lapatinib and Anti-Survivin siRNA for HER2+ Breast Cancer Treatment
 ,
,
Abstract
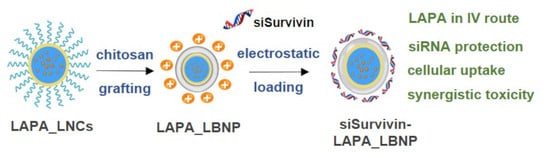
1. Introduction
2. Results and Discussion
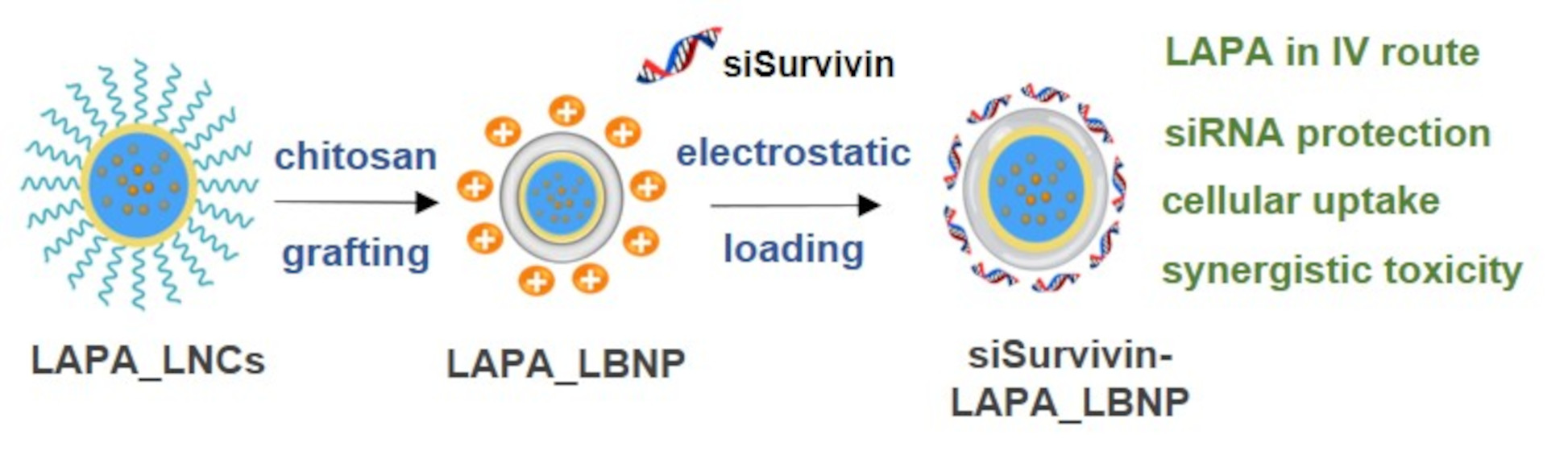
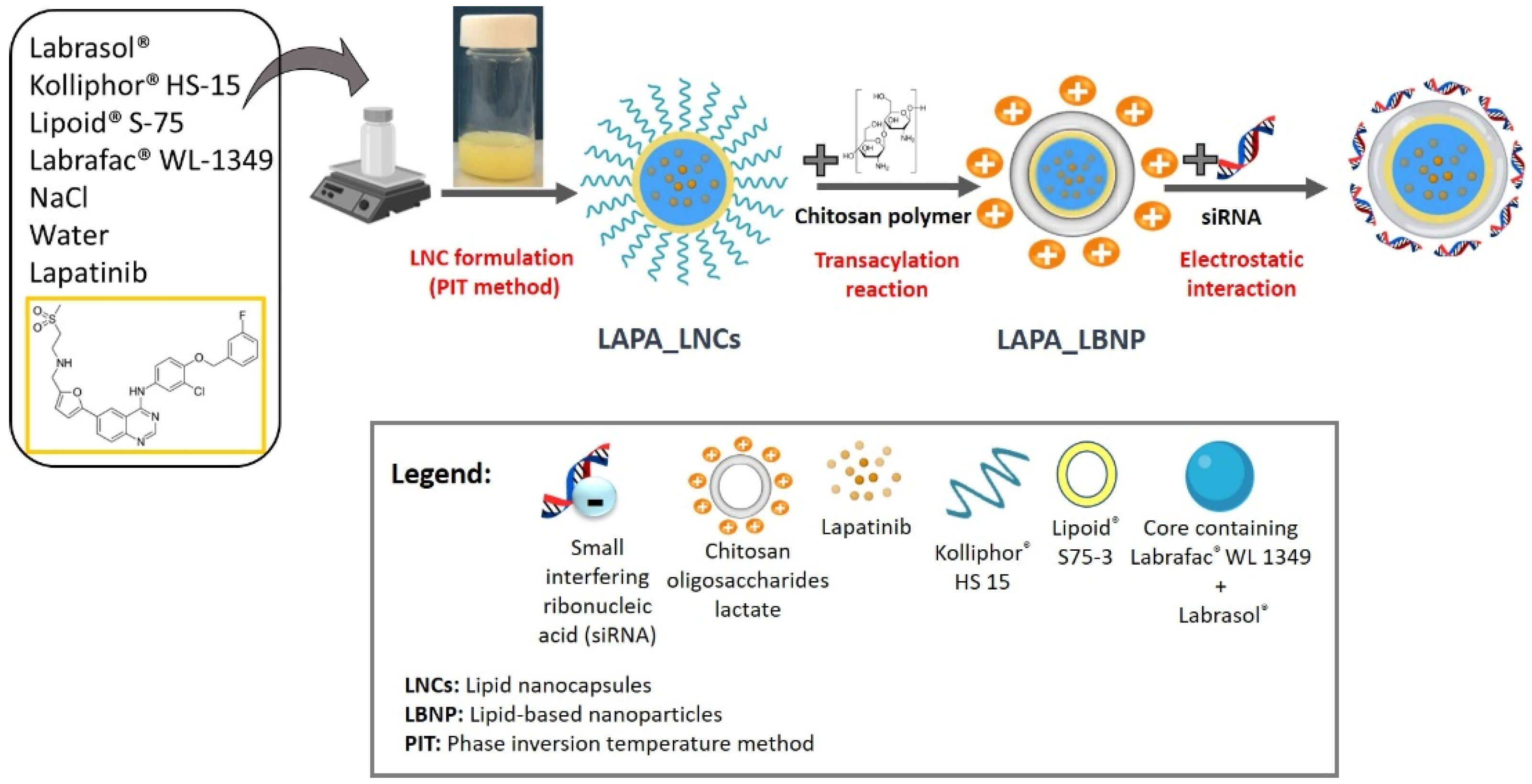
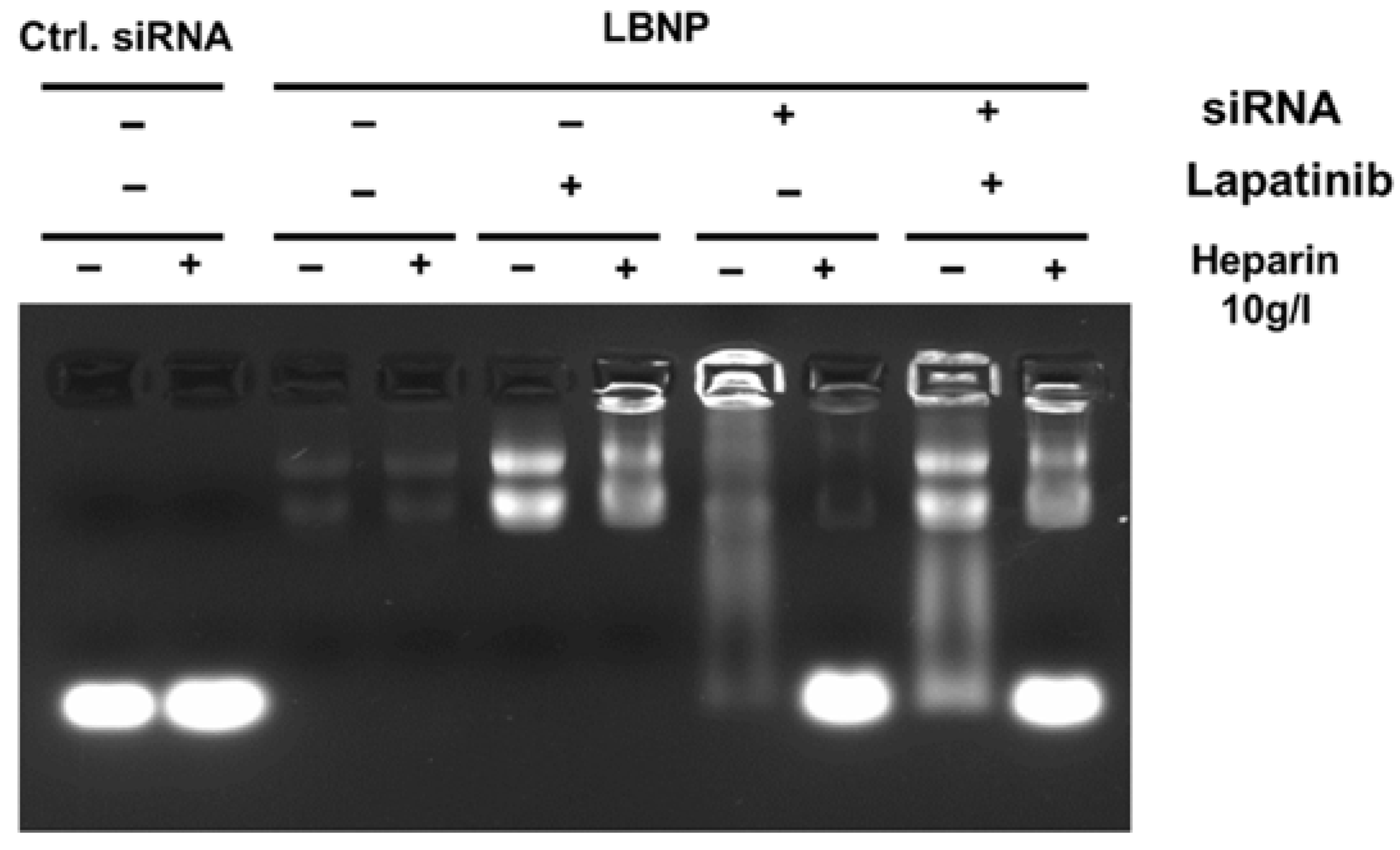
2.1. Formulation of LAPA_LBNP
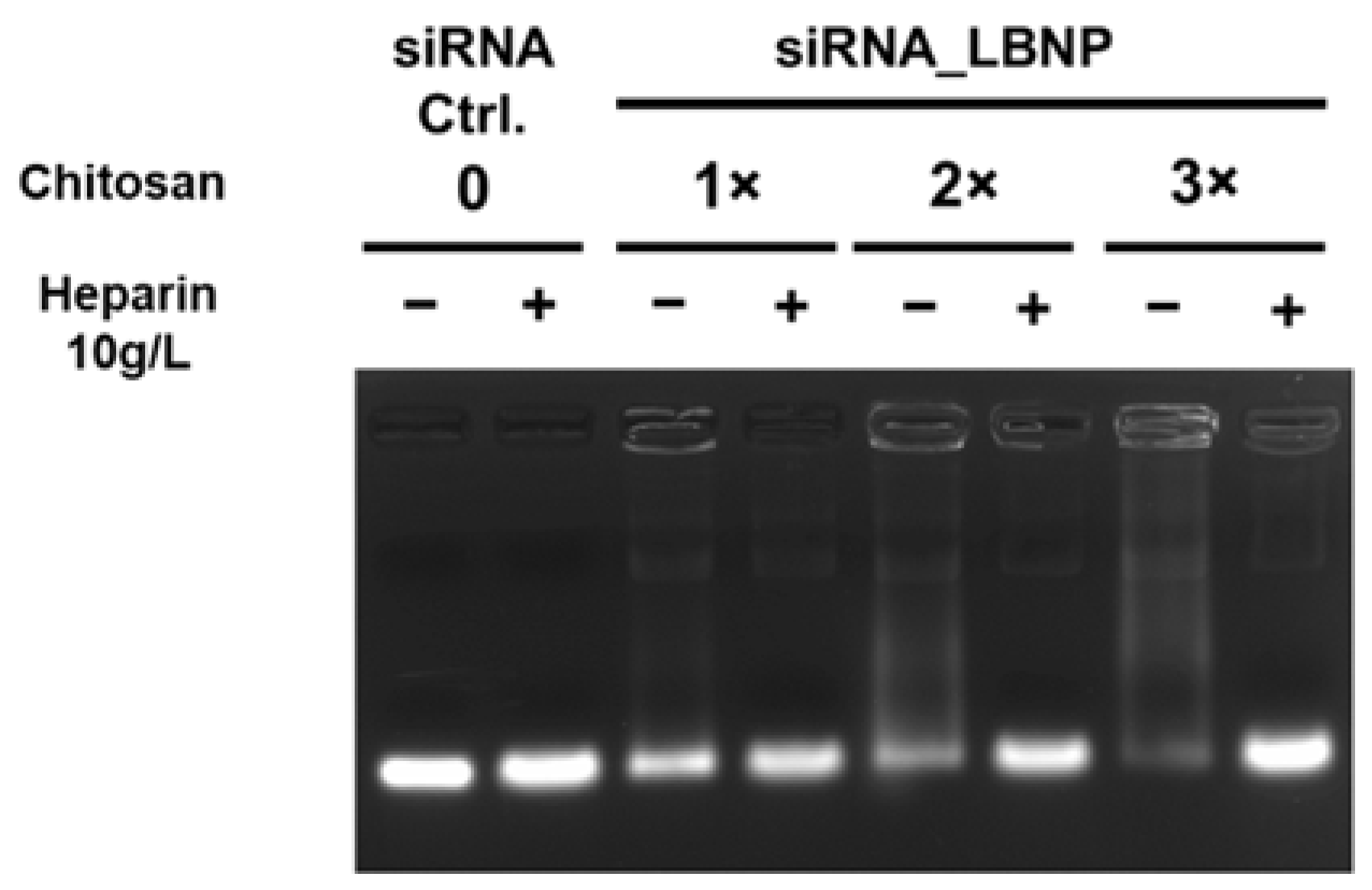
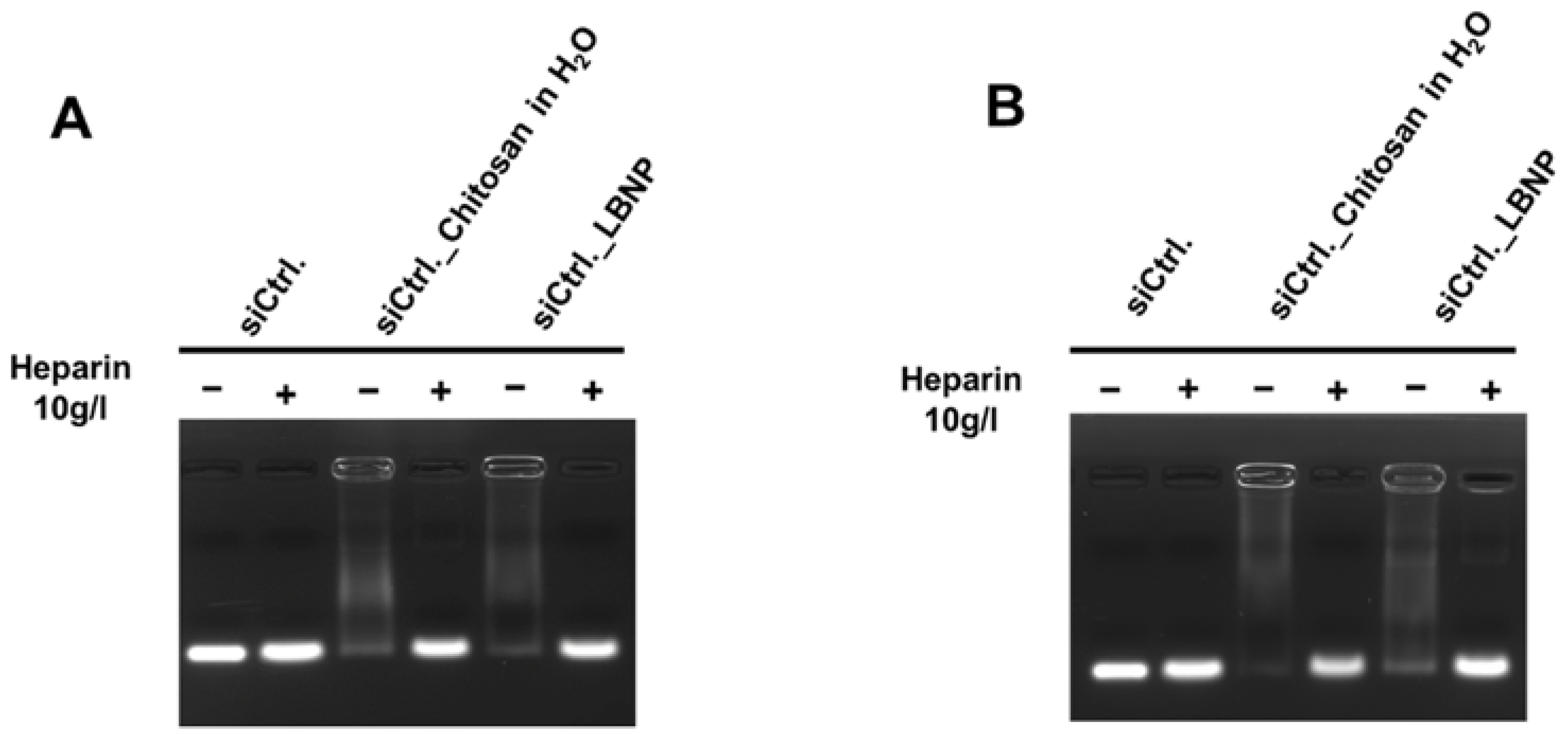
2.2. Optimization of Chitosan LBNP
2.3. Characterization of LBNP
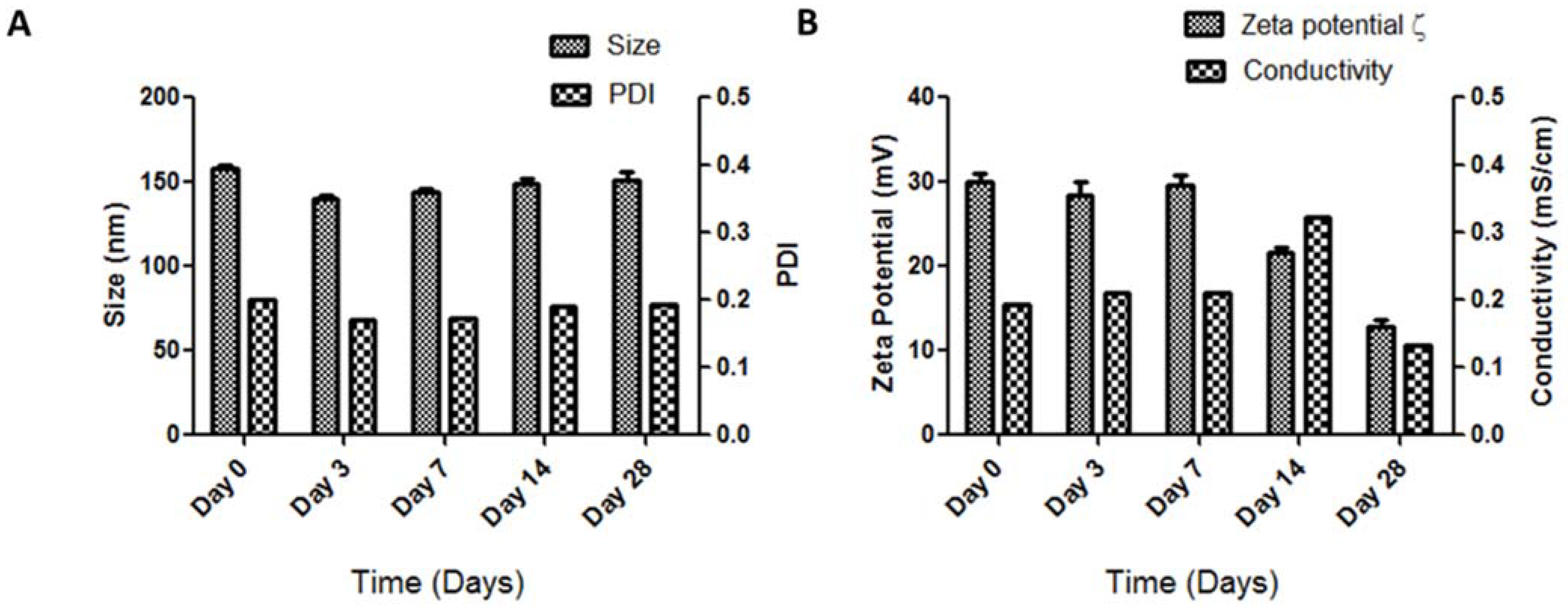
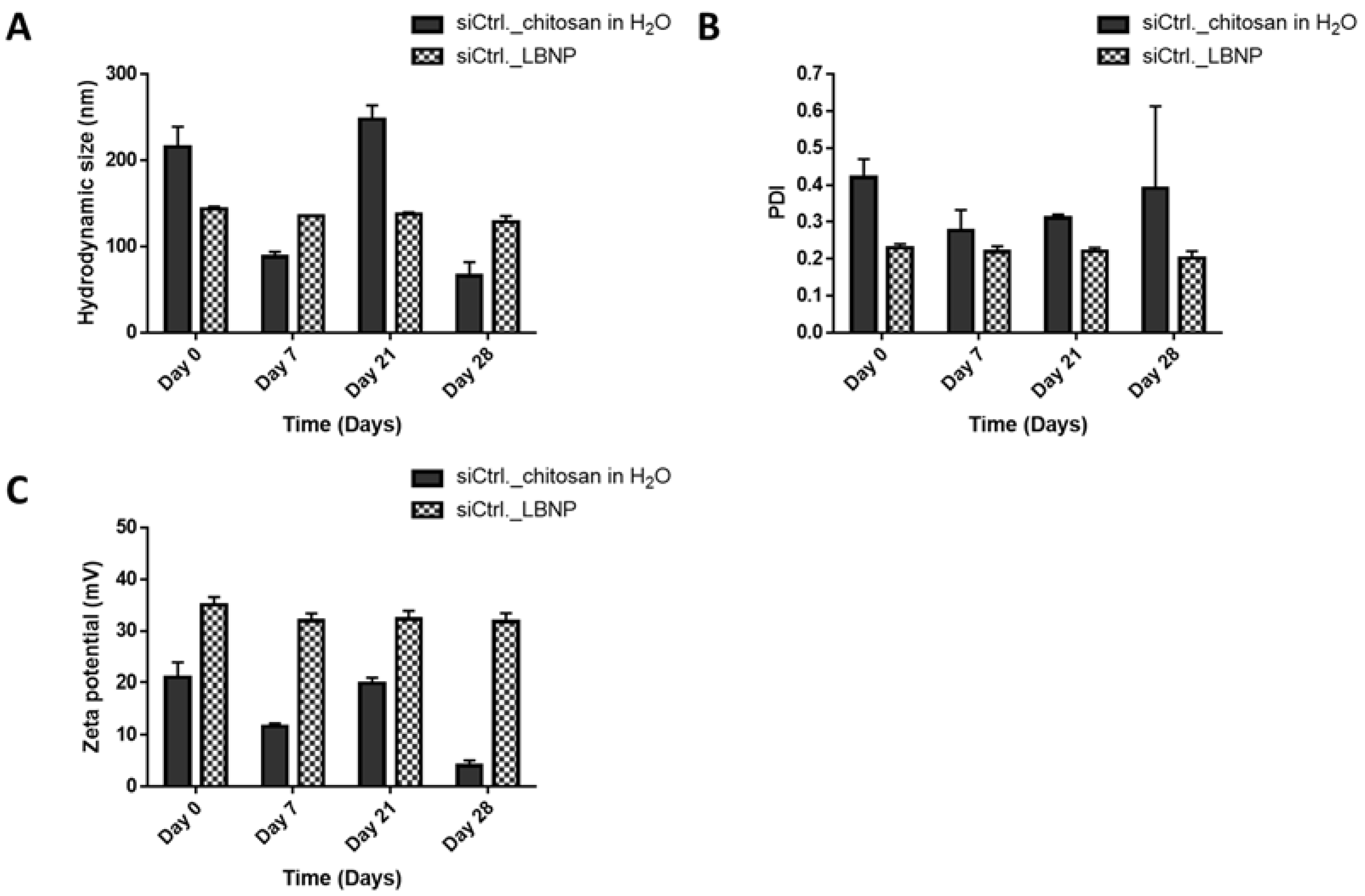
2.4. Storage Stability of siRNA_LBNP
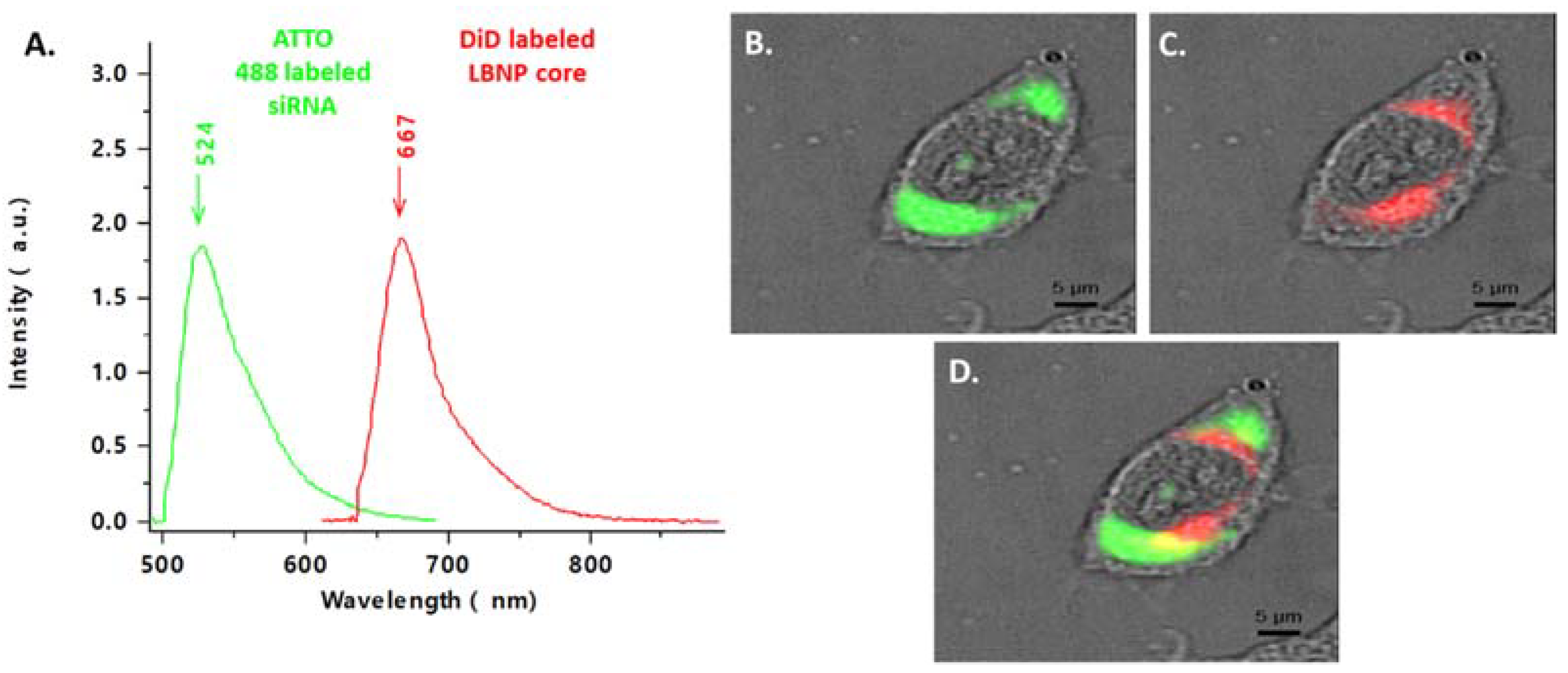
2.5. Cellular Uptake of Fluorescent siRNA_LBNP
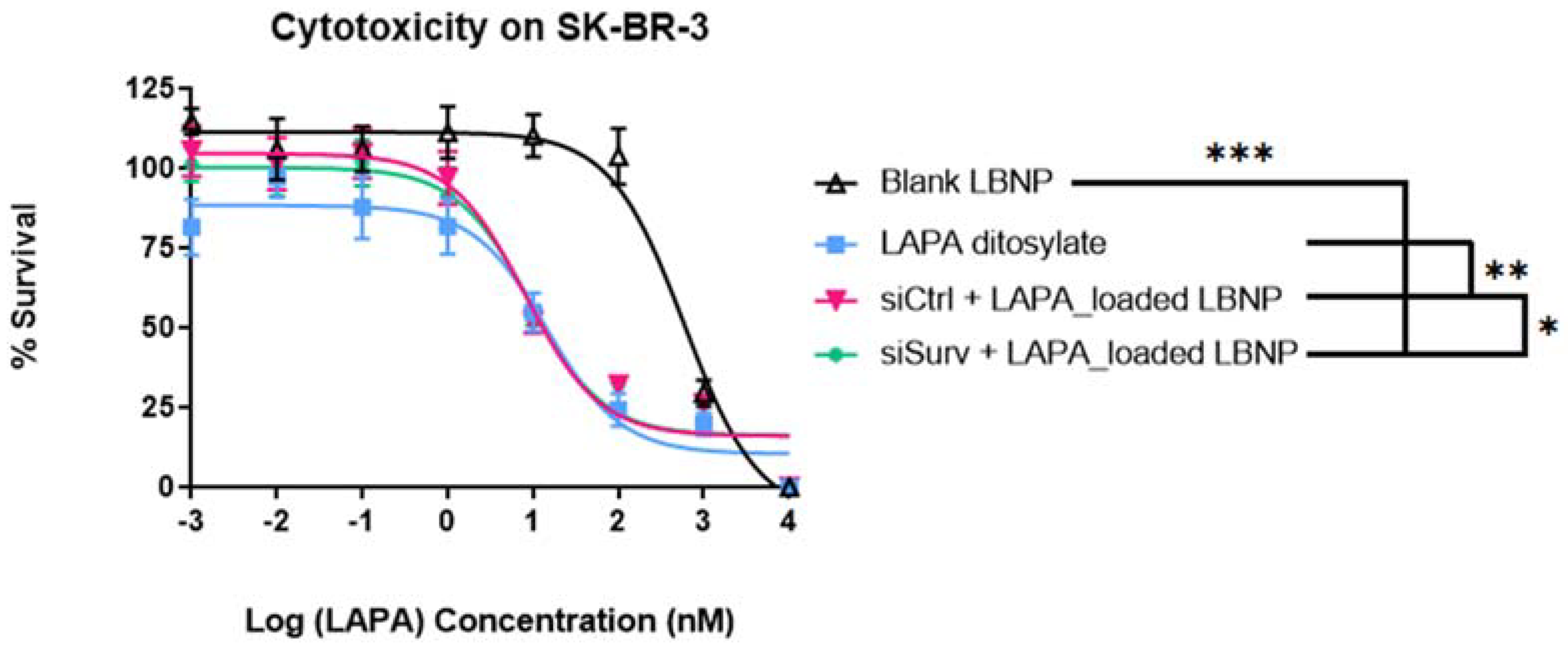
2.6. Cell Viability Analysis of LAPA_LBNP and siSurvivin-LAPA_LBNP
3. Materials and Methods
3.1. Materials
3.2. Nanocarrier Preparation
3.2.1. Formulation of LAPA-Loaded Lipid Nanocapsules
3.2.2. Formulation of siRNA (Co-Loaded) Lipid-Based Nanoparticles
3.3. Physicochemical Characteristics of the Nanocarrier
3.3.1. Particle Size and Zeta Potential
3.3.2. Encapsulation Efficiency
3.3.3. Agarose Gel Electrophoresis
3.3.4. Storage Stability of the Nanoparticles
3.4. Nanocarrier Cellular Evaluation
3.4.1. Cell Line and Culture
3.4.2. Confocal Spectral Imaging (CSI)
3.4.3. In Vitro Cytotoxicity
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nuciforo, P.; Radosevic-Robin, N.; Ng, T.; Scaltriti, M. Quantification of HER Family Receptors in Breast Cancer. Breast Cancer Res. 2015, 17, 53. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-Positive Breast Cancer and Tyrosine Kinase Inhibitors: The Time Is Now. Npj Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Czerniecki, B.J. Clinical Development of Immunotherapies for HER2+ Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. Npj Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Drago, J.Z.; Modi, S. Implementing Antibody-Drug Conjugates (ADCs) in HER2-Positive Breast Cancer: State of the Art and Future Directions. Breast Cancer Res. 2021, 23, 84. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Tiwari, A.K.; Wu, C.-P.; Su, X.; Wang, S.-R.; Liu, D.; Ashby, C.R.; Huang, Y.; Robey, R.W.; Liang, Y.; et al. Lapatinib (Tykerb, GW572016) Reverses Multidrug Resistance in Cancer Cells by Inhibiting the Activity of ATP-Binding Cassette Subfamily B Member 1 and G Member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef]
- Ratain, M.J.; Cohen, E.E. The Value Meal: How to Save $1,700 Per Month or More on Lapatinib. J. Clin. Oncol. 2007, 25, 3397–3398. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Sadeghi, S.; Finn, R.S. Lapatinib, a Dual-Targeted Small Molecule Inhibitor of EGFR and HER2, in HER2-Amplified Breast Cancer: From Bench to Bedside. Clin. Med. Insights Ther. 2011, 3, CMT.S3783. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Chen, C.; Chen, J.; Wei, Y.; Cao, S.; Jiang, X. Incorporation of Lapatinib into Core–Shell Nanoparticles Improves Both the Solubility and Anti-Glioma Effects of the Drug. Int. J. Pharm. 2014, 461, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.; Goodin, S. Lapatinib: A Dual Inhibitor of Human Epidermal Growth Factor Receptor Tyrosine Kinases. Clin. Ther. 2008, 30, 1426–1447. [Google Scholar] [CrossRef] [PubMed]
- Eljack, S.; David, S.; Faggad, A.; Chourpa, I.; Allard-Vannier, E. Nanoparticles Design Considerations to Co-Deliver Nucleic Acids and Anti-Cancer Drugs for Chemoresistance Reversal. Int. J. Pharm. X 2022, 4, 100126. [Google Scholar] [CrossRef]
- Greco, F.; Vicent, M.J. Combination Therapy: Opportunities and Challenges for Polymer–Drug Conjugates as Anticancer Nanomedicines. Adv. Drug Deliv. Rev. 2009, 61, 1203–1213. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Fumita, S.; Okamoto, W.; Nishio, K.; Nakagawa, K. Roles of BIM Induction and Survivin Downregulation in Lapatinib-Induced Apoptosis in Breast Cancer Cells with HER2 Amplification. Oncogene 2011, 30, 4097–4106. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, Versatile Modulation of Cell Division and Apoptosis in Cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar] [CrossRef]
- Altieri, D.C. Validating Survivin as a Cancer Therapeutic Target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A Novel Anti-Apoptosis Gene, Survivin, Expressed in Cancer and Lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of Apoptosis and Mitotic Spindle Checkpoint by Survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Sun, W.; Chen, X.; Xie, C.; Wang, Y.; Lin, L.; Zhu, K.; Shuai, X. Co-Delivery of Doxorubicin and Anti-BCL-2 SiRNA by PH-Responsive Polymeric Vector to Overcome Drug Resistance in In Vitro and In Vivo HepG2 Hepatoma Model. Biomacromolecules 2018, 19, 2248–2256. [Google Scholar] [CrossRef]
- Majumder, J.; Minko, T. Multifunctional Lipid-Based Nanoparticles for Codelivery of Anticancer Drugs and SiRNA for Treatment of Non-Small Cell Lung Cancer with Different Level of Resistance and EGFR Mutations. Pharmaceutics 2021, 13, 1063. [Google Scholar] [CrossRef]
- Babaei, M.; Abnous, K.; Taghdisi, S.M.; Taghavi, S.; Saljooghi, A.S.; Ramezani, M.; Alibolandi, M. Targeted Rod-Shaped Mesoporous Silica Nanoparticles for the Co-Delivery of Camptothecin and Survivin ShRNA in to Colon Adenocarcinoma In Vitro and In Vivo. Eur. J. Pharm. Biopharm. 2020, 156, 84–96. [Google Scholar] [CrossRef]
- Heurtault, B.; Saulnier, P.; Pech, B.; Proust, J.; Benoit, J. A Novel Phase Inversion-Based Process for the Preparation of Lipid Nanocarriers. Pharm. Res. 2002, 19, 875–880. [Google Scholar] [CrossRef]
- Allard, E.; Passirani, C.; Garcion, E.; Pigeon, P.; Vessières, A.; Jaouen, G.; Benoit, J.-P. Lipid Nanocapsules Loaded with an Organometallic Tamoxifen Derivative as a Novel Drug-Carrier System for Experimental Malignant Gliomas. J. Control. Release 2008, 130, 146–153. [Google Scholar] [CrossRef]
- Malzert-Fréon, A.; Saint-Lorant, G.; Hennequin, D.; Gauduchon, P.; Poulain, L.; Rault, S. Influence of the Introduction of a Solubility Enhancer on the Formulation of Lipidic Nanoparticles with Improved Drug Loading Rates. Eur. J. Pharm. Biopharm. 2010, 75, 117–127. [Google Scholar] [CrossRef]
- David, S.; Resnier, P.; Guillot, A.; Pitard, B.; Benoit, J.-P.; Passirani, C. SiRNA LNCs–A Novel Platform of Lipid Nanocapsules for Systemic SiRNA Administration. Eur. J. Pharm. Biopharm. 2012, 81, 448–452. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Resnier, P.; Galopin, N.; Sibiril, Y.; Clavreul, A.; Cayon, J.; Briganti, A.; Legras, P.; Vessières, A.; Montier, T.; Jaouen, G.; et al. Efficient Ferrocifen Anticancer Drug and Bcl-2 Gene Therapy Using Lipid Nanocapsules on Human Melanoma Xenograft in Mouse. Pharmacol. Res. 2017, 126, 54–65. [Google Scholar] [CrossRef]
- Resnier, P.; Lepeltier, E.; Emina, A.L.; Galopin, N.; Bejaud, J.; David, S.; Ballet, C.; Benvegnu, T.; Pecorari, F.; Chourpa, I.; et al. Model Affitin and PEG Modifications onto SiRNA Lipid Nanocapsules: Cell Uptake and in Vivo Biodistribution Improvements. RSC Adv. 2019, 9, 27264–27278. [Google Scholar] [CrossRef]
- Bastiancich, C.; Bozzato, E.; Luyten, U.; Danhier, F.; Bastiat, G.; Préat, V. Drug Combination Using an Injectable Nanomedicine Hydrogel for Glioblastoma Treatment. Int. J. Pharm. 2019, 559, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Labrak, Y.; Heurtault, B.; Frisch, B.; Saulnier, P.; Lepeltier, E.; Miron, V.E.; Muccioli, G.G.; des Rieux, A. Impact of Anti-PDGFRα Antibody Surface Functionalization on LNC Uptake by Oligodendrocyte Progenitor Cells. Int. J. Pharm. 2022, 618, 121623. [Google Scholar] [CrossRef] [PubMed]
- Lollo, G.; Matha, K.; Bocchiardo, M.; Bejaud, J.; Marigo, I.; Virgone-Carlotta, A.; Dehoux, T.; Rivière, C.; Rieu, J.-P.; Briançon, S.; et al. Drug Delivery to Tumours Using a Novel 5-FU Derivative Encapsulated into Lipid Nanocapsules. J. Drug Target. 2019, 27, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.-C.; Edwards-Levy, F. Coating Alginate Beads with Cross-Linked Biopolymers: A Novel Method Based on a Transacylation Reaction. J. Microencapsul. 1996, 13, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, K.; Saulnier, P.; Boesen, K.; Benoit, J.-P. Lagarce, F. Anti-Epidermal Growth Factor Receptor SiRNA Carried by Chitosan-Transacylated Lipid Nanocapsules Increases Sensitivity of Glioblastoma Cells to Temozolomide. Int. J. Nanomed. 2014, 9, 1479. [Google Scholar] [CrossRef]
- Djekic, L.; Primorac, M. The Influence of Cosurfactants and Oils on the Formation of Pharmaceutical Microemulsions Based on PEG-8 Caprylic/Capric Glycerides. Int. J. Pharm. 2008, 352, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Tawa, R.; Konishi, T.; Shibata, N.; Takada, K. A Novel Emulsifier, Labrasol, Enhances Gastrointestinal Absorption of Gentamicin. Life Sci. 2001, 69, 2899–2910. [Google Scholar] [CrossRef] [PubMed]
- Libster, D.; Aserin, A.; Wachtel, E.; Shoham, G.; Garti, N. An HII Liquid Crystal-Based Delivery System for Cyclosporin A: Physical Characterization. J. Colloid Interface Sci. 2007, 308, 514–524. [Google Scholar] [CrossRef]
- Katas, H.; Alpar, H.O. Development and Characterisation of Chitosan Nanoparticles for SiRNA Delivery. J. Control. Release 2006, 115, 216–225. [Google Scholar] [CrossRef]
- Li, J.; Cai, C.; Li, J.; Li, J.; Li, J.; Sun, T.; Wang, L.; Wu, H.; Yu, G. Chitosan-Based Nanomaterials for Drug Delivery. Molecules 2018, 23, 2661. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Atyabi, F.; Rastegari, A.; Kheshtchin, N.; Arab, S.; Hassannia, H.; Ajami, M.; Mirsanei, Z.; Habibi, S.; Masoumi, F.; et al. CD73 Specific SiRNA Loaded Chitosan Lactate Nanoparticles Potentiate the Antitumor Effect of a Dendritic Cell Vaccine in 4T1 Breast Cancer Bearing Mice. J. Control. Release 2017, 246, 46–59. [Google Scholar] [CrossRef]
- Benchamas, G.; Huang, G.; Huang, S.; Huang, H. Preparation and Biological Activities of Chitosan Oligosaccharides. Trends Food Sci. Technol. 2021, 107, 38–44. [Google Scholar] [CrossRef]
- Abrica-González, P.; Zamora-Justo, J.A.; Sotelo-López, A.; Vázquez-Martínez, G.R.; Balderas-López, J.A.; Muñoz-Diosdado, A.; Ibáñez-Hernández, M. Gold Nanoparticles with Chitosan, N-Acylated Chitosan, and Chitosan Oligosaccharide as DNA Carriers. Nanoscale Res. Lett. 2019, 14, 258. [Google Scholar] [CrossRef] [PubMed]
- Bellich, B.; D’Agostino, I.; Semeraro, S.; Gamini, A.; Cesàro, A. “The Good, the Bad and the Ugly” of Chitosans. Mar. Drugs 2016, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Syeda, J.; Wasan, K.; Wasan, E. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef]
- Zargar, V.; Asghari, M.; Dashti, A. A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Buss, J.H.; Begnini, K.R.; Bruinsmann, F.A.; Ceolin, T.; Sonego, M.S.; Pohlmann, A.R.; Guterres, S.S.; Collares, T.; Seixas, F.K. Lapatinib-Loaded Nanocapsules Enhances Antitumoral Effect in Human Bladder Cancer Cell. Front. Oncol. 2019, 9, 203. [Google Scholar] [CrossRef]
- Wilson, J.N.; Liu, W.; Brown, A.S.; Landgraf, R. Binding-Induced, Turn-on Fluorescence of the EGFR/ERBB Kinase Inhibitor, Lapatinib. Org. Biomol. Chem. 2015, 13, 5006–5011. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Miranda, O.R.; Thompson, M.A.; Pabelick, C.M.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Prakash, Y.S.; Mukherjee, P. Effect of Nanoparticle Surface Charge at the Plasma Membrane and Beyond. Nano Lett. 2010, 10, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.; Hussain, S.M.; Schrand, A.M.; Braydich-Stolle, L.K.; Hess, K.L.; Jones, R.L.; Schlager, J.J. Unique Cellular Interaction of Silver Nanoparticles: Size-Dependent Generation of Reactive Oxygen Species. J. Phys. Chem. B 2008, 112, 13608–13619. [Google Scholar] [CrossRef]
- Jiang, J.; Oberdörster, G.; Elder, A.; Gelein, R.; Mercer, P.; Biswas, P. Does Nanoparticle Activity Depend upon Size and Crystal Phase? Nanotoxicology 2008, 2, 33–42. [Google Scholar] [CrossRef]
- Thorek, D.L.J.; Tsourkas, A. Size, Charge and Concentration Dependent Uptake of Iron Oxide Particles by Non-Phagocytic Cells. Biomaterials 2008, 29, 3583–3590. [Google Scholar] [CrossRef]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.-H.; Qoronfleh, M.W. Therapeutic Efficacy of Nanoparticles and Routes of Administration. Biomater. Res. 2019, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.T.; Haes, A.J. What Does Nanoparticle Stability Mean? J. Phys. Chem. C 2019, 123, 16495–16507. [Google Scholar] [CrossRef] [PubMed]
- Večeř, M.; Pospíšil, J. Stability and Rheology of Aqueous Suspensions. Procedia Eng. 2012, 42, 1720–1725. [Google Scholar] [CrossRef]
- Mouzouvi, C.R.A.; Umerska, A.; Bigot, A.K.; Saulnier, P. Surface Active Properties of Lipid Nanocapsules. PLoS ONE 2017, 12, e0179211. [Google Scholar] [CrossRef]
- Dunne, G.; Breen, L.; Collins, D.M.; Roche, S.; Clynes, M.; O’Connor, R. Modulation of P-Gp Expression by Lapatinib. Investig. New Drugs 2011, 29, 1284–1293. [Google Scholar] [CrossRef]
- Polli, J.W.; Olson, K.L.; Chism, J.P.; St John-Williams, L.; Yeager, R.L.; Woodard, S.M.; Otto, V.; Castellino, S.; Demby, V.E. An Unexpected Synergist Role of P-Glycoprotein and Breast Cancer Resistance Protein on the Central Nervous System Penetration of the Tyrosine Kinase Inhibitor Lapatinib (N-{3-Chloro-4-[(3-Fluorobenzyl)Oxy]Phenyl}-6-[5-({[2-(Methylsulfonyl)Ethyl]Amino}methyl)-2-Furyl]-4-Quinazolinamine; GW572016): TABLE 1. Drug Metab. Dispos. 2009, 37, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse Drug-Resistance Mechanisms Can Emerge from Drug-Tolerant Cancer Persister Cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Meng, F.; Deng, C.; Klok, H.-A.; Zhong, Z. Dual and Multi-Stimuli Responsive Polymeric Nanoparticles for Programmed Site-Specific Drug Delivery. Biomaterials 2013, 34, 3647–3657. [Google Scholar] [CrossRef]
- Yamashita, F.; Hashida, M. Pharmacokinetic Considerations for Targeted Drug Delivery. Adv. Drug Deliv. Rev. 2013, 65, 139–147. [Google Scholar] [CrossRef]
- Xia, W.; Bisi, J.; Strum, J.; Liu, L.; Carrick, K.; Graham, K.M.; Treece, A.L.; Hardwicke, M.A.; Dush, M.; Liao, Q.; et al. Regulation of Survivin by ErbB2 Signaling: Therapeutic Implications for ErbB2-Overexpressing Breast Cancers. Cancer Res. 2006, 66, 1640–1647. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | DH (nm) | PDI | Zeta Potential (mV) | EE (%) |
|---|---|---|---|---|
| LNCs | 86.9 ± 12.9 | 0.116 ± 0.02 | −4.15 ± 4.35 | - |
| LAPA_LBNP | 126.9 ± 20.60 | 0.14 ± 0.08 | +28.42 ± 6.69 | 94.51 ± 6.63 |
| siRNA-LAPA_LBNP | 123.9 ± 17.10 | 0.09 ± 0.05 | +20.84 ± 8.67 |
| Tested Formulations | IC50 (nM) |
|---|---|
| LBNP | 6481 ± 1486 |
| LAPA_ditosylate | 159.0 ± 12.4 |
| siCtrl.-LAPA_LBNP | 99.7 ± 12.8 |
| siSurv.-LAPA_LBNP | 76.8 ± 12.3 |
| Formulations | Statistical Significance | |
|---|---|---|
| Control | Evaluated/Compared | p-Value |
| LBNP | siCtrl.-LAPA_LBNP | 0.0001 *** |
| siSurv.-LAPA_LBNP | ||
| LAPA Ditosylate | ||
| siCtrl.-LAPA_LBNP | LAPA Ditosylate | 0.0006 *** |
| siCtrl.-LAPA_LBNP | siSurv.-LAPA_LBNP | 0.0418 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eljack, S.; David, S.; Chourpa, I.; Faggad, A.; Allard-Vannier, E. Formulation of Lipid-Based Nanoparticles for Simultaneous Delivery of Lapatinib and Anti-Survivin siRNA for HER2+ Breast Cancer Treatment. Pharmaceuticals 2022, 15, 1452. https://doi.org/10.3390/ph15121452
Eljack S, David S, Chourpa I, Faggad A, Allard-Vannier E. Formulation of Lipid-Based Nanoparticles for Simultaneous Delivery of Lapatinib and Anti-Survivin siRNA for HER2+ Breast Cancer Treatment. Pharmaceuticals. 2022; 15(12):1452. https://doi.org/10.3390/ph15121452
Chicago/Turabian StyleEljack, Sahar, Stephanie David, Igor Chourpa, Areeg Faggad, and Emilie Allard-Vannier. 2022. "Formulation of Lipid-Based Nanoparticles for Simultaneous Delivery of Lapatinib and Anti-Survivin siRNA for HER2+ Breast Cancer Treatment" Pharmaceuticals 15, no. 12: 1452. https://doi.org/10.3390/ph15121452
APA StyleEljack, S., David, S., Chourpa, I., Faggad, A., & Allard-Vannier, E. (2022). Formulation of Lipid-Based Nanoparticles for Simultaneous Delivery of Lapatinib and Anti-Survivin siRNA for HER2+ Breast Cancer Treatment. Pharmaceuticals, 15(12), 1452. https://doi.org/10.3390/ph15121452