
Exploring Novel Pyridine Carboxamide Derivatives as Urease Inhibitors: Synthesis, Molecular Docking, Kinetic Studies and ADME Profile

 , , , ,
, , , ,
Abstract
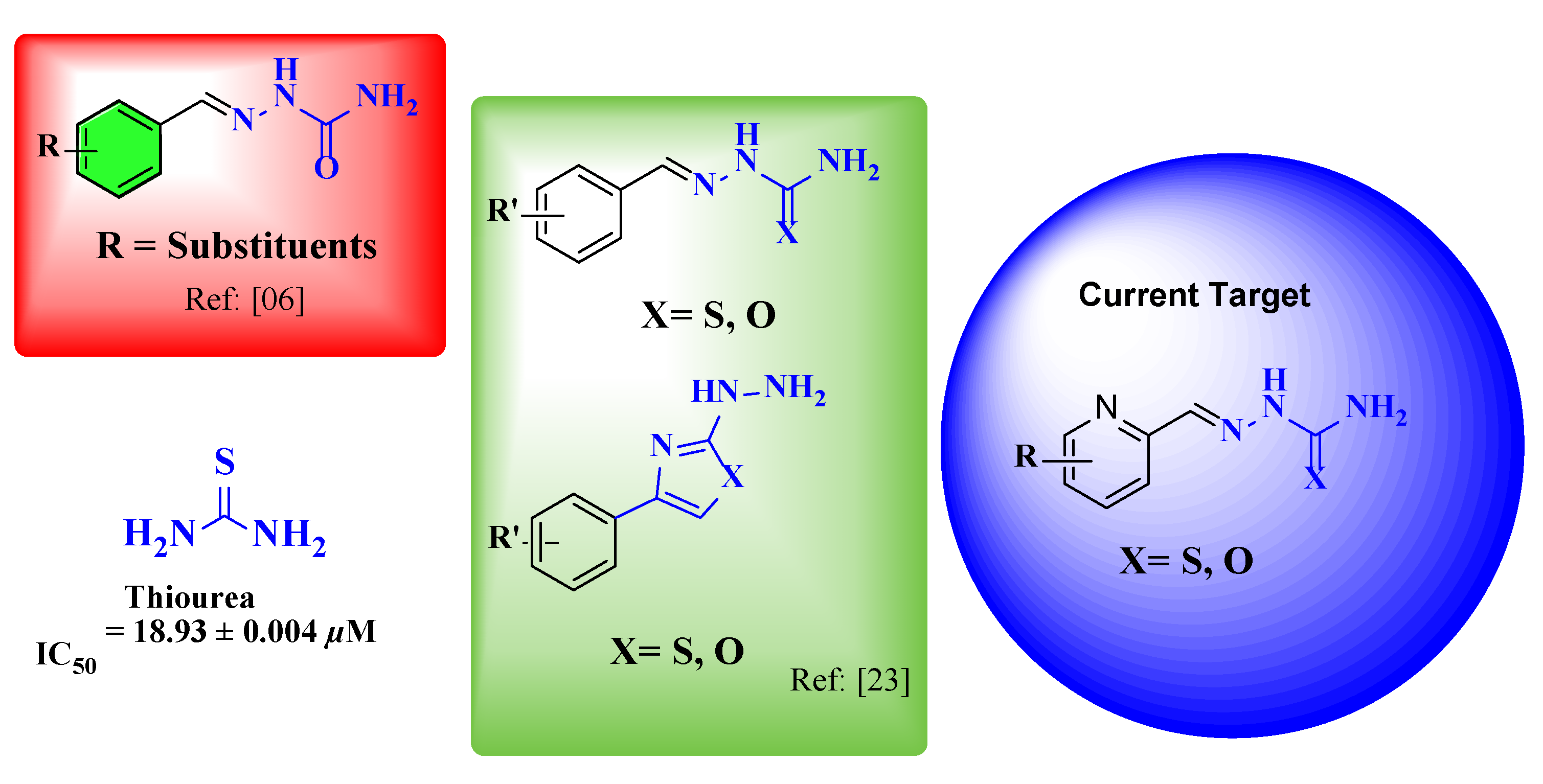
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. The In-Vitro Urease Activity of Pyridine Carboxamide and Carbothioamide and Semicarbazone
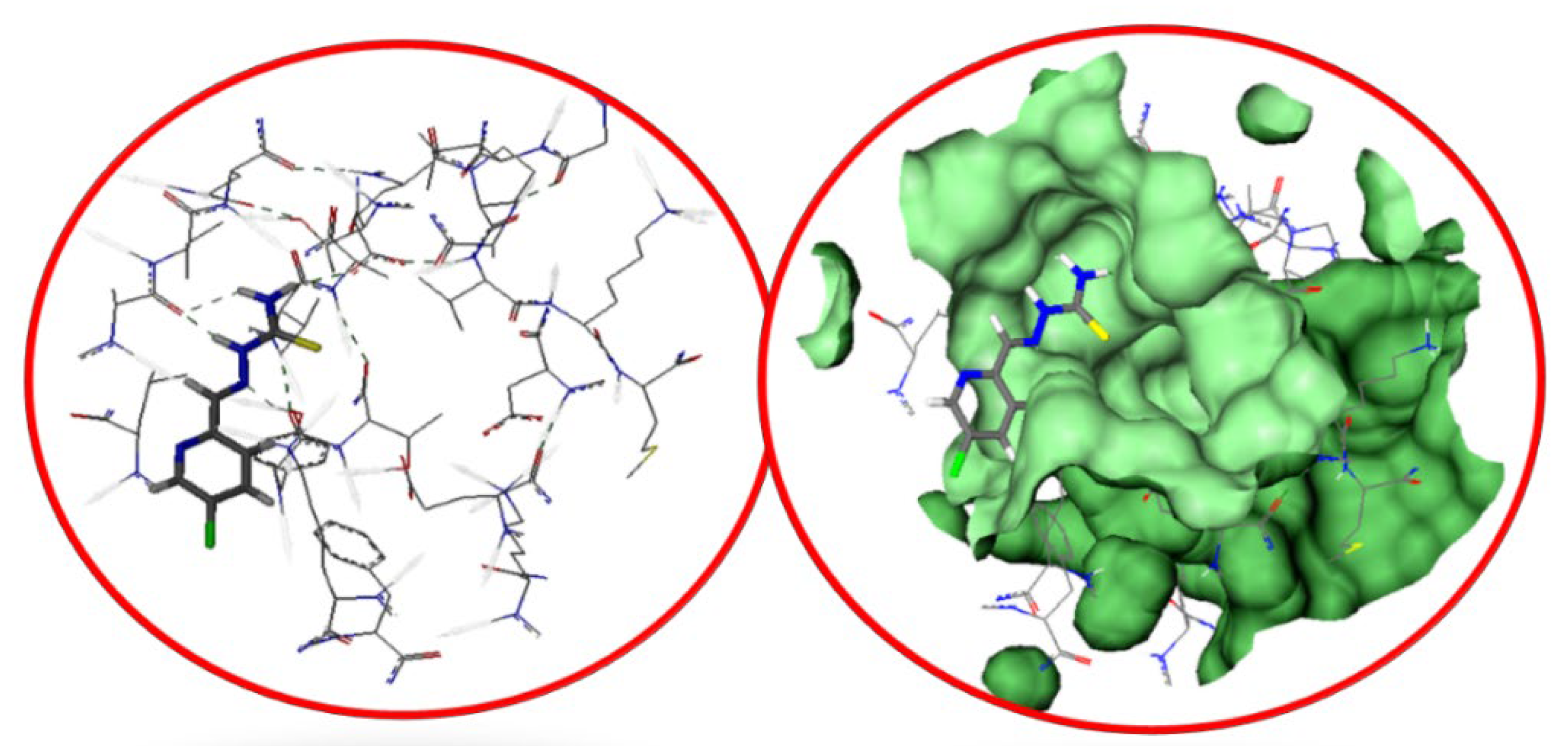
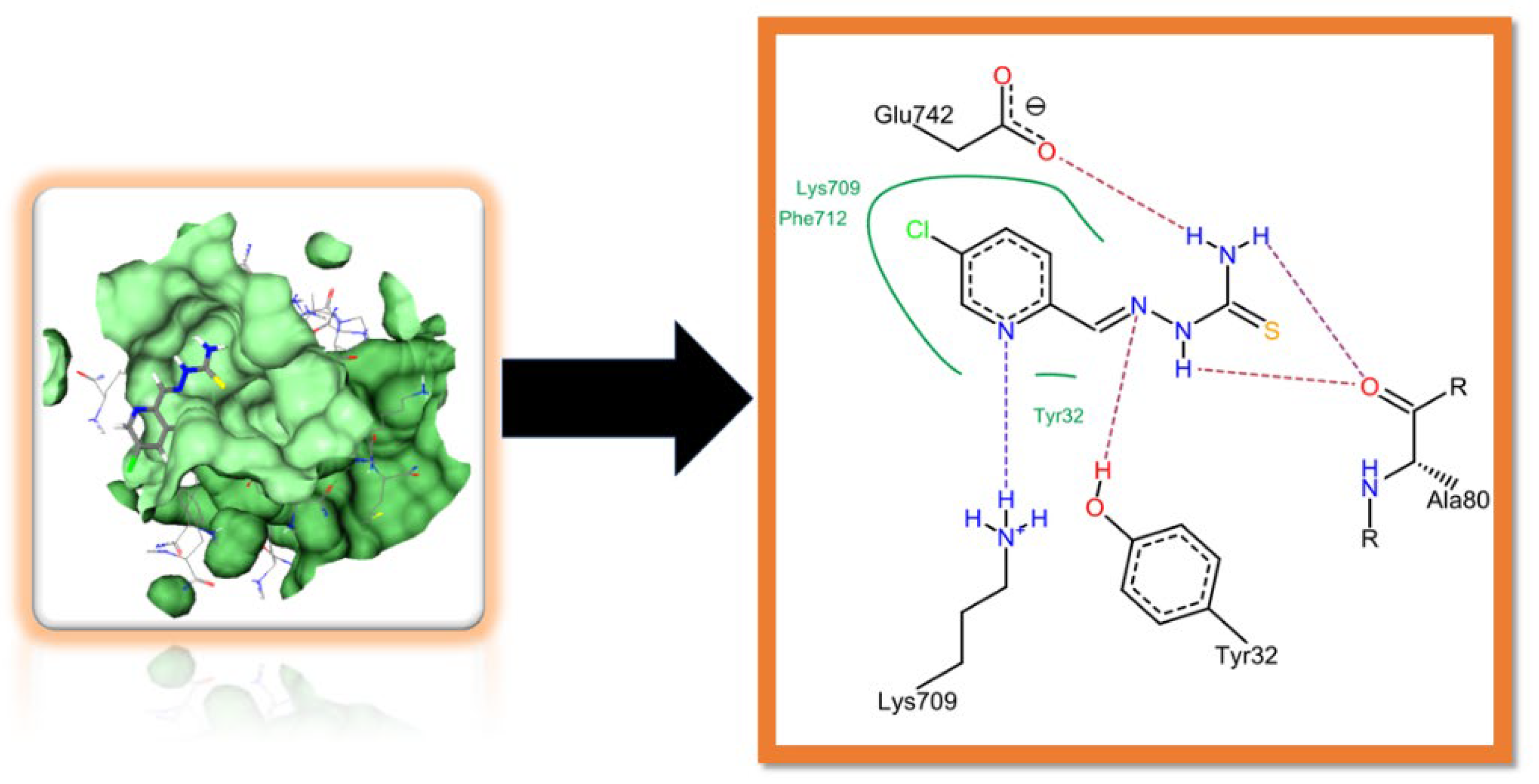
2.3. Molecular Docking Studies
2.4. Enzyme Kinetics Study
2.5. ADME Profile
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Pyridine Carboxamide and Carbothioamide
3.1.2. Characterization Data of Pyridine Carboxamide and Carbothioamide
Pyridine-2-yl-methylene thiosemicarbazide (Rx-1) [45]
6-Methylpyridine-2-yl methylene hydrazine-1-carbothioamide (Rx-2) [46]
6-Bromopyridine-2-yl methylene hydrazine-1-carbothioamide (Rx-3)
6-Methoxypyridine-2-yl methylene thiosemicarbazide (Rx-4)
6-Trifluoromethyl pyridine-2-yl methylene hydrazine-1-carbothioamide (Rx-5)
5-Chloropyridine-2-yl methylene hydrazinecarbothioamide (Rx-6)
2-Pyridine-2-yl-methylene hydrazine carboxamide (Rx-7)
6-Methylpyridine-2-yl methylene hydrazinecarboxamide (Rx-8)
6-Bromopyridine-2-yl methylene hydrazinecarboxamide (Rx-9)
6-Methoxypyridine-2-yl methylene hydrazine carboxamide (Rx-10)
6-Trifluoromethyl pyridine-2-yl methylene hydrazine carboxamide (Rx-11)
5-Chloropyridine-2yl-methylene hydrazine carboxamide (Rx-12)
3.1.3. Urease Enzyme Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, K.C.; Sutar, A.K. Catalytic activities of Schiff base transition metal complexes. Coord. Chem. Rev. 2008, 252, 1420–1450. [Google Scholar] [CrossRef]
- Abuamer, K.M.; Maihub, A.A.; El-Ajaily, M.M.; Etorki, A.M.; Abou-Krisha, M.M.; Almagani, M.A. The role of aromatic Schiff bases in the dyes techniques. Int. J. Org. Chem. 2014, 4, 7–15. [Google Scholar] [CrossRef]
- Rasool, R.; Hasnain, S.; Nishat, N. Metal-based Schiff base polymers: Preparation, spectral, thermal and their in vitro biological investigation. Des. Monomers Polym. 2014, 17, 217–226. [Google Scholar] [CrossRef]
- Mak, J.Y.W.; Xu, W.; Reid, R.C.; Corbett, A.J.; Meehan, B.S.; Wang, H.; Chen, Z.; Rossjohn, J.; McCluskey, J.; Liu, L.; et al. Stabilizing short-lived Schiff base derivatives of 5-aminouracils that activate mucosal-associated invariant T cells. Nat. Commun. 2017, 8, 14599. [Google Scholar] [CrossRef]
- Kaya, S.; Erkan, S.; Karakaş, D. Computational investigation of molecular structures, spectroscopic properties and antitumor-antibacterial activities of some Schiff bases. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 244, 118829. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Atlı, Ö.; Ilgın, S.; Demirel, R.; Özdemir, A.; Kaplancıklı, Z.A. Synthesis and biological evaluation of new naphthalene substituted thiosemicarbazone derivatives as potent antifungal and anticancer agents. Eur. J. Med. Chem. 2016, 108, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Jacob, Í.T.; Gomes, F.O.; de Miranda, M.D.; de Almeida, S.; da Cruz-Filho, I.J.; Peixoto, C.A.; da Silva, T.G.; Moreira, D.R.; de Melo, C.M.; de Oliveira, J.F. Anti-inflammatory activity of novel thiosemicarbazone compounds indole-based as COX inhibitors. Pharmacol. Rep. 2021, 73, 907–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, W.; Shan, Y.; Liu, F.; Xu, X.; Yang, Y.; Zhang, Q.; Zhang, Y.; Kuang, H.; Wang, Z.; et al. Design, synthesis and anticancer activity of novel nopinone-based thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2360–2363. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.F.; da Silva, A.L.; Vendramini-Costa, D.B.; Amorim, C.A.d.; Campos, J.F.; Ribeiro, A.G.; de Moura, R.O.; Neves, J.L.; Ruiz, A.L.T.G.; de Carvalho, J.E. Synthesis of thiophene-thiosemicarbazone derivatives and evaluation of their in vitro and in vivo antitumor activities. Eur. J. Med. Chem. 2015, 104, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Liu, Q.; Wang, S.; Zeng, B.; Du, G.; Zhang, C.; Li, Y. Synthesis, cytotoxicity, and in vivo antitumor activity study of parthenolide semicarbazones and thiosemicarbazones. Bioorg. Med. Chem. 2020, 28, 115557. [Google Scholar] [CrossRef] [PubMed]
- Nazari, S.; Safari, F.; Mamaghani, M.B.; Bazgir, A. Synthesis and evaluation of in vitro cytotoxic effects of triazol/spiroindolinequinazolinedione, triazol/indolin-3-thiosemicarbazone and triazol/thiazol-indolin-2-one conjugates. DARU J. Pharm. Sci. 2020, 28, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Saremi, K.; Rad, S.K.; Khalilzadeh, M.; Hussaini, J.; Majid, N.A. In vivo acute toxicity and anti-gastric evaluation of a novel dichloro Schiff base: Bax and HSP70 alteration. Acta Biochim. Biophys. Sin. 2020, 52, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, N.; Haribabu, J.; Dhanabalan, A.K.; Swaminathan, S.; Sun, S.; Dibwe, D.F.; Bhuvanesh, N.; Awale, S.; Karvembu, R. Thiosemicarbazone (s)-anchored water soluble mono-and bimetallic Cu (II) complexes: Enzyme-like activities, biomolecular interactions, anticancer property and real-time live cytotoxicity. Dalton Trans. 2020, 49, 9411–9424. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-L.; Han, X.; Zhang, G.-N. Synthesis, crystal structures, and urease inhibitory activities of three novel thiocyanato-bridged polynuclear Schiff base cadmium (II) complexes. J. Inorg. Gen. Chem. 2008, 634, 142–146. [Google Scholar] [CrossRef]
- Akhtar, T.; Hameed, S.; Khan, K.M.; Choudhary, M. Syntheses, urease inhibition, and antimicrobial studies of some chiral 3-substituted-4-amino-5-thioxo-1H, 4H-1, 2, 4-triazoles. Med. Chem. 2008, 4, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J. Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. J. Infect. Dis. 1990, 161, 626–633. [Google Scholar] [CrossRef]
- De Oliveira, R.B.; de Souza-Fagundes, E.M.; Soares, R.P.; Andrade, A.A.; Krettli, A.U.; Zani, C.L. Synthesis and antimalarial activity of semicarbazone and thiosemicarbazone derivatives. Eur. J. Med. Chem. 2008, 43, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Matongo, F.; Nwodo, U.U. In vitro assessment of Helicobacter pylori ureases inhibition by honey fractions. Arch. Med. Res. 2014, 45, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Qazi, S.U.; Rahman, S.U.; Awan, A.N.; Al-Rashida, M.; Alharthy, R.D.; Asari, A.; Hameed, A.; Iqbal, J. Semicarbazone derivatives as urease inhibitors: Synthesis, biological evaluation, molecular docking studies and in-silico ADME evaluation. Bioorg. Chem. 2018, 79, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Cabral, J.M.S. Kinetic studies of the urease-catalyzed hydrolysis of urea in a buffer-free system. Appl. Biochem. Biotechnol. 1994, 49, 217–240. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Cabral, J.M. Review properties and applications of urease. Biocatal. Biotransform. 2002, 20, 1–14. [Google Scholar] [CrossRef]
- Salar, U.; Nizamani, A.; Arshad, F.; Khan, K.M.; Fakhri, M.I.; Perveen, S.; Ahmed, N.; Choudhary, M.I. Bis-coumarins; non-cytotoxic selective urease inhibitors and antiglycation agents. Bioorg. Chem. 2019, 91, 103170. [Google Scholar] [CrossRef]
- Kafarski, P.; Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nishida, M.; Ito, K.; Tomita, M. Characterization of silica particles prepared via urease-catalyzed urea hydrolysis and activity of urease in sol–gel silica matrix. Appl. Surf. Sci. 2012, 262, 69–75. [Google Scholar] [CrossRef]
- Burne, R.A.; Chen, Y.-Y.M. Infection, Bacterial ureases in infectious diseases. Microbes Infect. 2000, 2, 533–542. [Google Scholar] [CrossRef]
- Figura, N. Identifiable Helicobacter pylori strains or factors important in the development of duodenal ulcer disease. Helicobacter 1997, 2, 3–12. [Google Scholar] [CrossRef]
- Syrjänen, K.; Eskelinen, M.; Peetsalu, A.; Sillakivi, T.; Sipponen, P.; Härkönen, M.; Paloheimo, L.; Mäki, M.; Tiusanen, T.; Suovaniemi, O.; et al. GastroPanel® biomarker assay: The most comprehensive test for Helicobacter pylori infection and its clinical sequelae. a critical review. Anticancer. Res. 2019, 39, 1091–1104. [Google Scholar] [CrossRef]
- Kataria, R.; Khatkar, A. Lead molecules for targeted urease inhibition: An updated review from 2010–2018. Curr. Protein Pept. Sci. 2019, 20, 1158–1188. [Google Scholar] [CrossRef]
- Mobley, H. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. S1), 57–64. [Google Scholar] [CrossRef] [PubMed]
- Maroney, M.J.; Ciurli, S. Nonredox nickel enzymes. Chem. Rev. 2014, 114, 4206–4228. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Modolo, L.V.; de Souza, A.X.; Horta, L.P.; Araujo, D.P.; de Fátima, A. An overview on the potential of natural products as ureases inhibitors: A review. J. Adv. Res. 2015, 6, 35–44. [Google Scholar] [CrossRef]
- Arshia, A.; Khan, A.; Khan, K.M.; Saad, S.M.; Siddiqui, N.I.; Javaid, S.; Perveen, S.; Choudhary, M.I. Synthesis and urease inhibitory activities of benzophenone semicarbazones/thiosemicarbazones. Med. Chem. Res. 2016, 25, 2666–2679. [Google Scholar] [CrossRef]
- Kosikowska, P.; Berlicki, Ł. Urease inhibitors as potential drugs for gastric and urinary tract infections: A patent review. Expert Opin. Ther. Patents 2011, 21, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, M.T.; Khan, A.; Islam, M.; Halim, S.A.; Khiat, M.; Anwar, M.U.; Hussain, J.; Hameed, A.; Pasha, A.R.; Khan, F.A.; et al. Synthesis, characterization and molecular docking of some novel hydrazonothiazolines as urease inhibitors. Bioorg. Chem. 2020, 94, 103404. [Google Scholar] [CrossRef] [PubMed]
- Qazi, S.U.; Naz, A.; Imran, A.; Iqbal, J. Urease inhibitory kinetics, molecular docking, SAR and ADME studies of imine analogues. New J. Chem. 2022, 46, 3512–3520. [Google Scholar] [CrossRef]
- Hamad, A.; Khan, M.A.; Ahmad, I.; Imran, A.; Khalil, R.; Al-Adhami, T.; Rahman, K.M.; Quratulain; Zahra, N.; Shafiq, Z. Probing sulphamethazine and sulphamethoxazole based Schiff bases as urease inhibitors; synthesis, characterization, molecular docking and ADME evaluation. Bioorg. Chem. 2020, 105, 104336. [Google Scholar] [CrossRef]
- Spassov, V.; Yan, L. Accelrys Software Inc. Discovery Studio Modeling Environment, Release 4.0. Proteins Struct. Funct. Bioinform. 2013, 81, 704–714. [Google Scholar] [CrossRef]
- Waldrop, G.L. A qualitative approach to enzyme inhibition. Biochem. Mol. Biol. Educ. 2009, 37, 11–15. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.; Katsogiannou, A.; Skourtis, D.; Iatrou, H.; Tzeli, D.; Vassiliou, S.; Javornik, U.; Plavec, J.; Mavromoustakos, T. Conformational Properties of New Thiosemicarbazone and Thiocarbohydrazone Derivatives and Their Possible Targets. Molecules 2022, 27, 2537. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Parmar, S.; Kumar, Y. Synthesis, spectroscopic, and antimicrobial studies on bivalent zinc and mercury complexes of 2-formylpyridine thiosemicarbazone. Bioinorg. Chem. Appl. 2009, 2009, 851316. [Google Scholar] [CrossRef]
- Ali, M.A.; Dey, K.K.; Nazimuddin, M.; Smith, F.E.; Butcher, R.J.; Jasinski, J.P.; Jasinski, J.M. The preparation and characterization of some copper (II) complexes of the 6-methyl-2-formylpyridine thiosemicarbazone and the X-ray crystal structure of the chloro(6-methyl-2-formylpyridinethiosemicabazonato) copper (II) complex. Polyhedron 1996, 15, 3331–3339. [Google Scholar] [CrossRef]
- Weatherburn, M.W. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 1967, 39, 971–974. [Google Scholar] [CrossRef]
- Svane, S.; Sigurdarson, J.J.; Finkenwirth, F.; Eitinger, T.; Karring, H. Inhibition of urease activity by different compounds provides insight into the modulation and association of bacterial nickel import and ureolysis. Sci. Rep. 2020, 10, 8503. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No | Compound Name | Structure | IC50 ± SEM (µM) |
|---|---|---|---|
| 1. | RX-1 |  | 3.23 ± 0.015 |
| 2. | RX-2 |  | 6.41 ± 0.023 |
| 3. | RX-3 |  | 3.13 ± 0.034 |
| 4. | RX-4 |  | 4.21 ± 0.022 |
| 5. | RX-5 |  | 4.93 ± 0.012 |
| 6. | RX-6 |  | 1.07 ± 0.043 |
| 7. | RX-7 |  | 2.18 ± 0.058 |
| 8. | RX-8 |  | 3.41 ± 0.011 |
| 9. | RX-9 |  | 14.49 ± 0.067 |
| 10. | RX-10 |  | 5.52 ± 0.072 |
| 11. | RX-11 |  | 5.96 ± 0.005 |
| 12. | RX-12 |  | 4.07 ± 0.003 |
| 13. | * Thiourea | 18.93 ± 0.004 |
| S. No. | Comp. No. | Molecular Weight | LogP | LogS | tPSA | HBD | HBA | Rotatable Bonds | GI Absorption | Oral Bioavailability (VEBER) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Rx-1 | 180.23 | 1.24 | −2.01 | 95.39 | 2 | 2 | 3 | High | Yes |
| 2. | Rx-2 | 194.26 | 1.50 | −1.73 | 95.39 | 2 | 2 | 3 | High | Yes |
| 3. | Rx-3 | 259.13 | 1.63 | −2.53 | 95.39 | 2 | 2 | 3 | High | Yes |
| 4. | Rx-4 | 210.26 | 1.74 | −1.68 | 104.62 | 2 | 3 | 4 | High | Yes |
| 5. | Rx-5 | 248.23 | 1.46 | −2.26 | 95.39 | 2 | 5 | 4 | High | Yes |
| 6. | Rx-6 | 214.68 | 1.49 | −2.59 | 95.39 | 2 | 2 | 3 | High | Yes |
| 7. | Rx-7 | 164.16 | −0.09 | −0.94 | 80.37 | 2 | 3 | 3 | High | Yes |
| 8. | Rx-8 | 178.19 | 0.29 | −1.25 | 80.37 | 2 | 3 | 3 | High | Yes |
| 9. | Rx-9 | 243.06 | 0.33 | −2.05 | 80.37 | 2 | 3 | 3 | High | Yes |
| 10. | Rx-10 | 194.19 | 0.57 | −1.20 | 89.60 | 2 | 4 | 4 | High | Yes |
| 11. | Rx-11 | 232.16 | 0.25 | −1.78 | 80.37 | 2 | 6 | 4 | High | Yes |
| 12. | Rx-12 | 198.61 | 0.17 | −2.11 | 80.37 | 2 | 3 | 3 | High | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naseer, A.; Osra, F.A.; Awan, A.N.; Imran, A.; Hameed, A.; Ali Shah, S.A.; Iqbal, J.; Zakaria, Z.A. Exploring Novel Pyridine Carboxamide Derivatives as Urease Inhibitors: Synthesis, Molecular Docking, Kinetic Studies and ADME Profile. Pharmaceuticals 2022, 15, 1288. https://doi.org/10.3390/ph15101288
Naseer A, Osra FA, Awan AN, Imran A, Hameed A, Ali Shah SA, Iqbal J, Zakaria ZA. Exploring Novel Pyridine Carboxamide Derivatives as Urease Inhibitors: Synthesis, Molecular Docking, Kinetic Studies and ADME Profile. Pharmaceuticals. 2022; 15(10):1288. https://doi.org/10.3390/ph15101288
Chicago/Turabian StyleNaseer, Ayesha, Faisal Abdulrhman Osra, Asia Naz Awan, Aqeel Imran, Abdul Hameed, Syed Adnan Ali Shah, Jamshed Iqbal, and Zainul Amiruddin Zakaria. 2022. "Exploring Novel Pyridine Carboxamide Derivatives as Urease Inhibitors: Synthesis, Molecular Docking, Kinetic Studies and ADME Profile" Pharmaceuticals 15, no. 10: 1288. https://doi.org/10.3390/ph15101288
APA StyleNaseer, A., Osra, F. A., Awan, A. N., Imran, A., Hameed, A., Ali Shah, S. A., Iqbal, J., & Zakaria, Z. A. (2022). Exploring Novel Pyridine Carboxamide Derivatives as Urease Inhibitors: Synthesis, Molecular Docking, Kinetic Studies and ADME Profile. Pharmaceuticals, 15(10), 1288. https://doi.org/10.3390/ph15101288