
GPR55 and GPR119 Receptors Contribute to the Processing of Neuropathic Pain in Rats

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
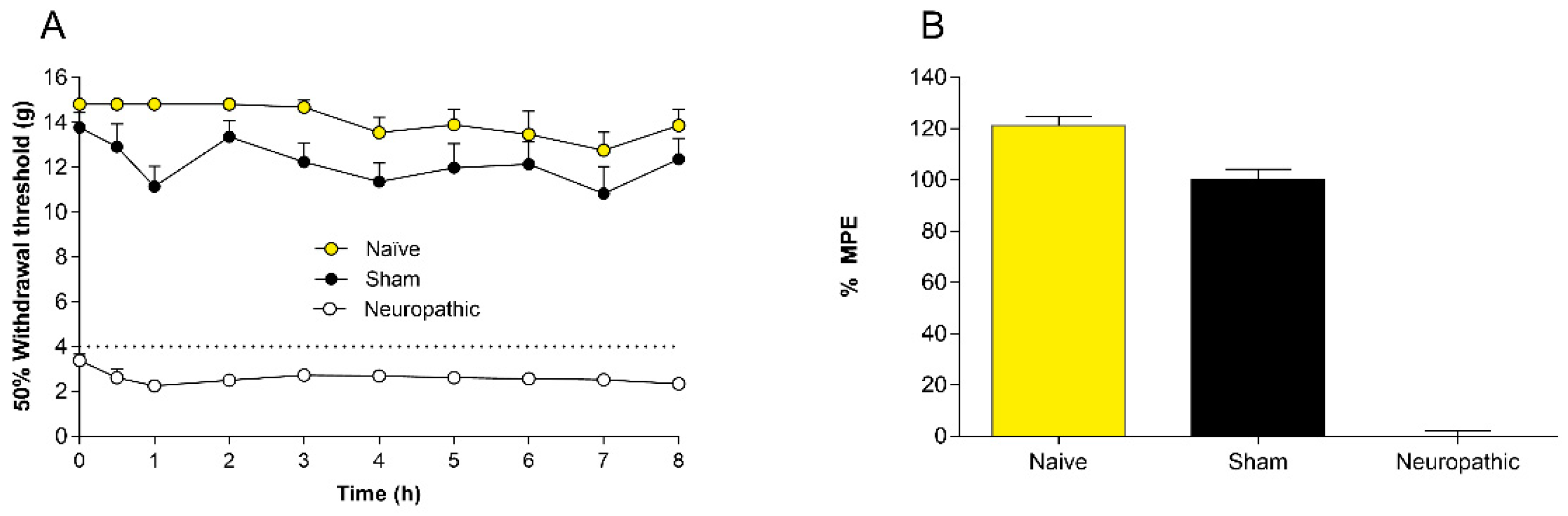
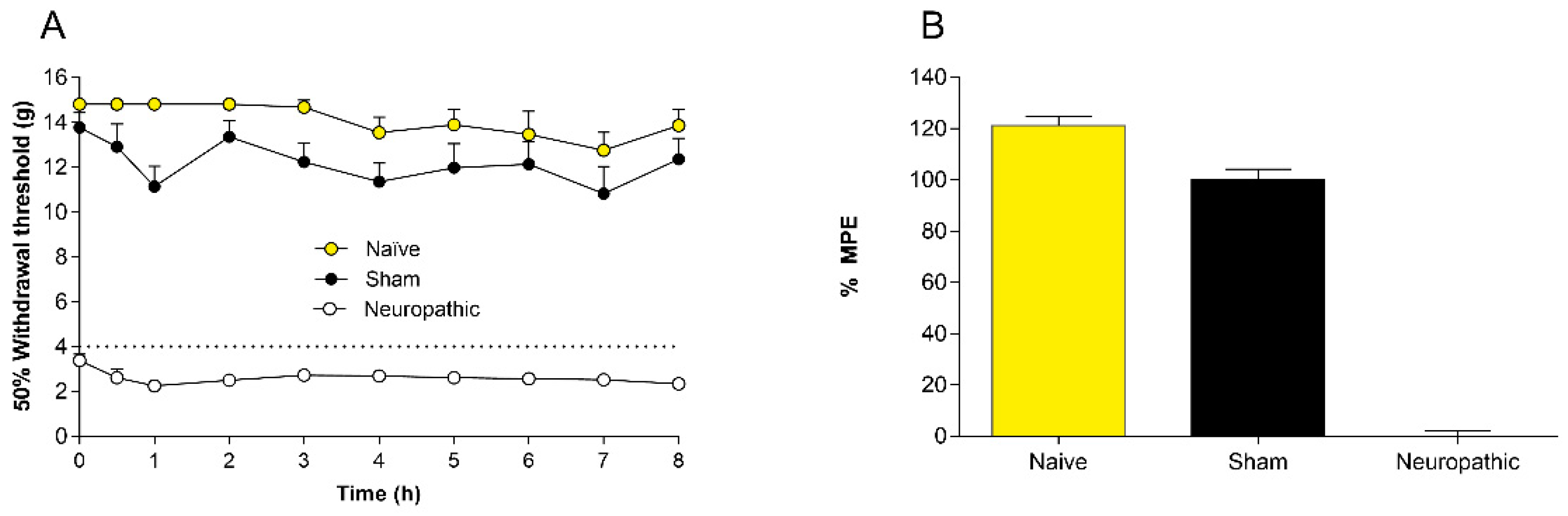
2.1. Controls
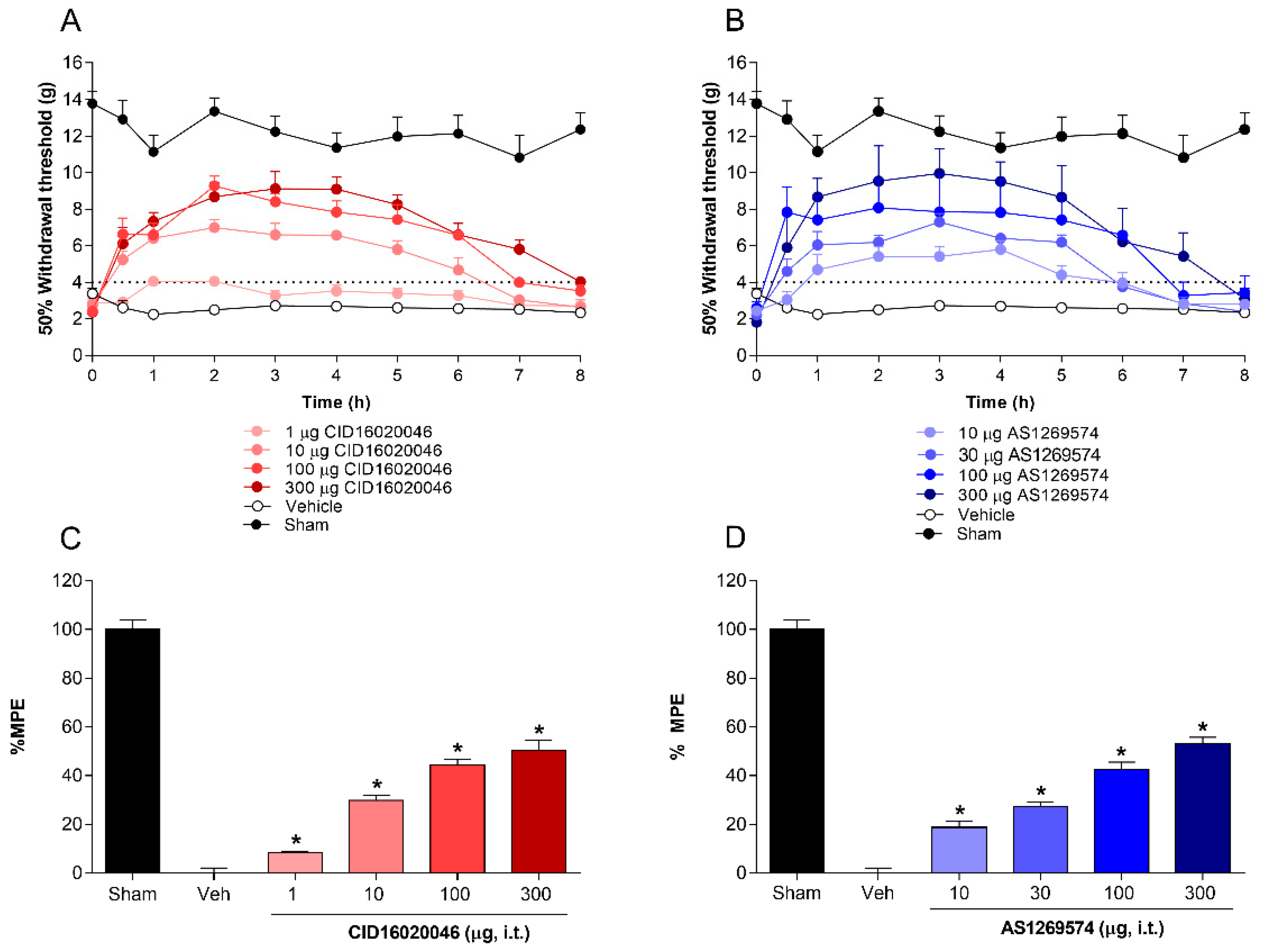
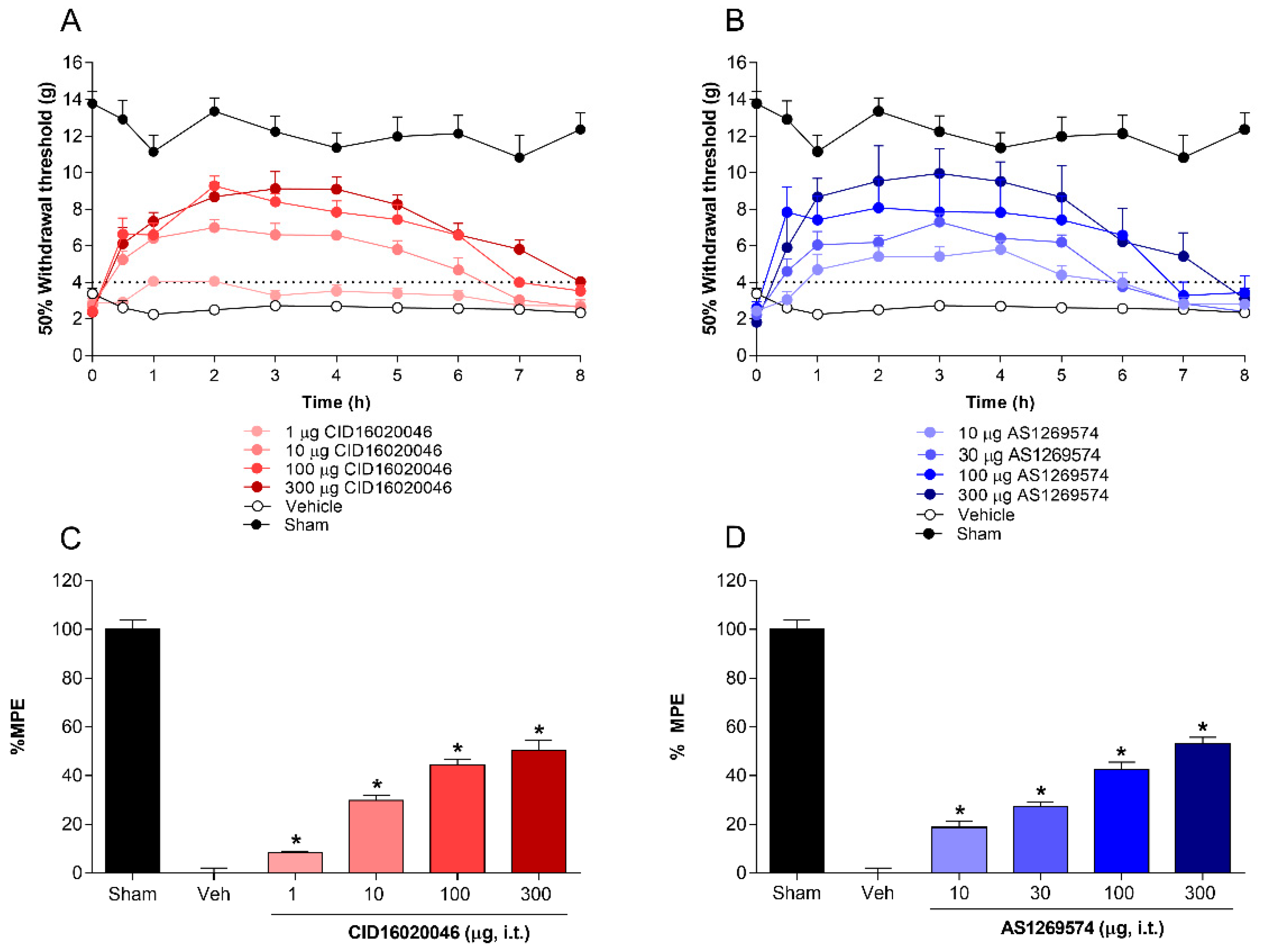
2.2. Antiallodynic Effect of Selective Antagonist GPR55 (CID16020046) and Selective Agonist GPR119 (AS1269574) in L5-L6 Spinal Nerve-Ligated Rats
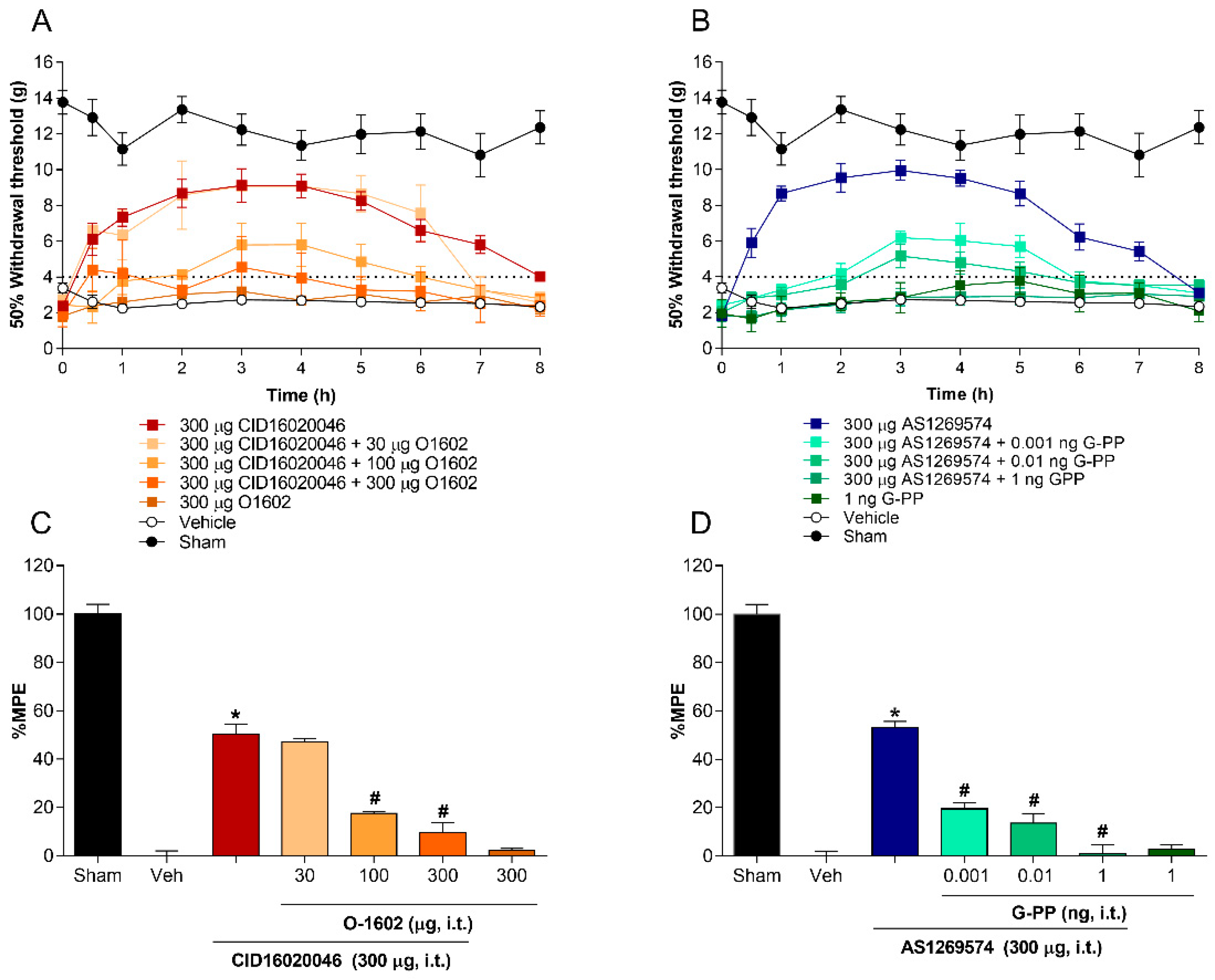
2.3. Co-Administration of Agonist GPR55 (O-1602) or G-Protein Antagonist Peptide Reverses the Antiallodynic Effect Induced by Antagonist GPR55 (CID16020046) and Agonist GPR119 (AS1269574), Respectively, in L5-L6 Spinal Nerve-Ligated Rats
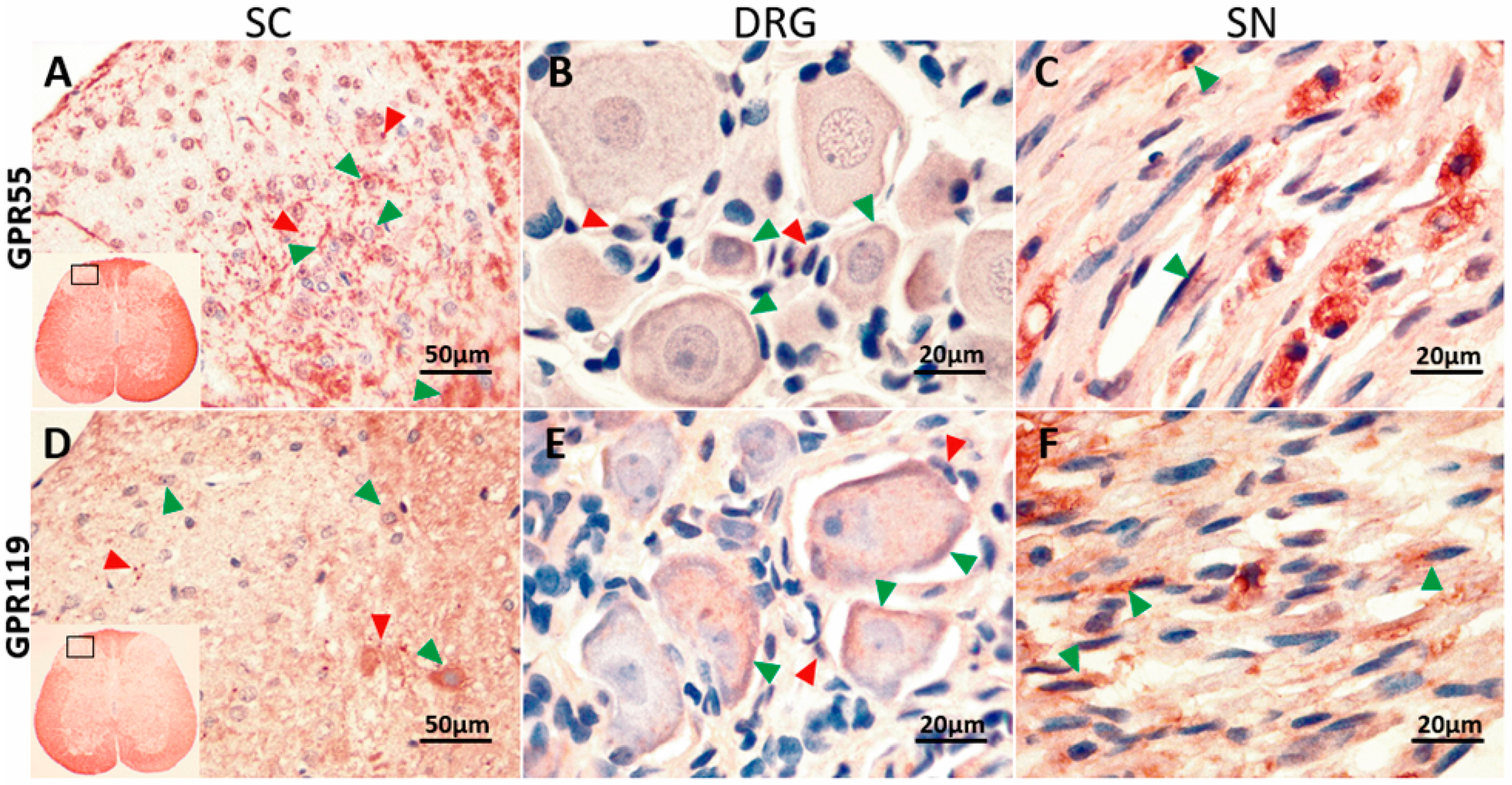
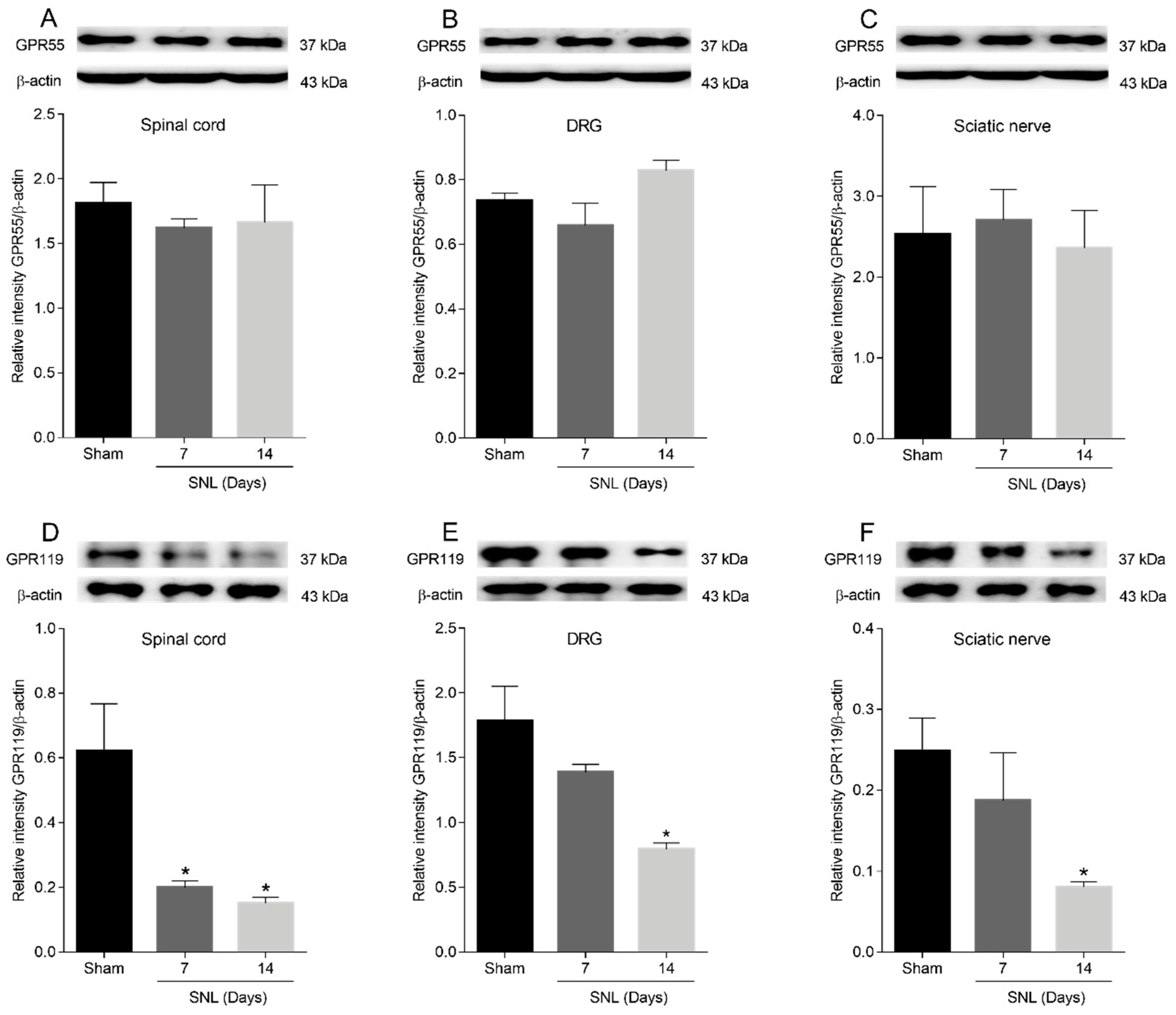
2.4. GPR55 or GPR119 Localization in the Spinal Cord, Dorsal Root Ganglia and Sciatic Nerve
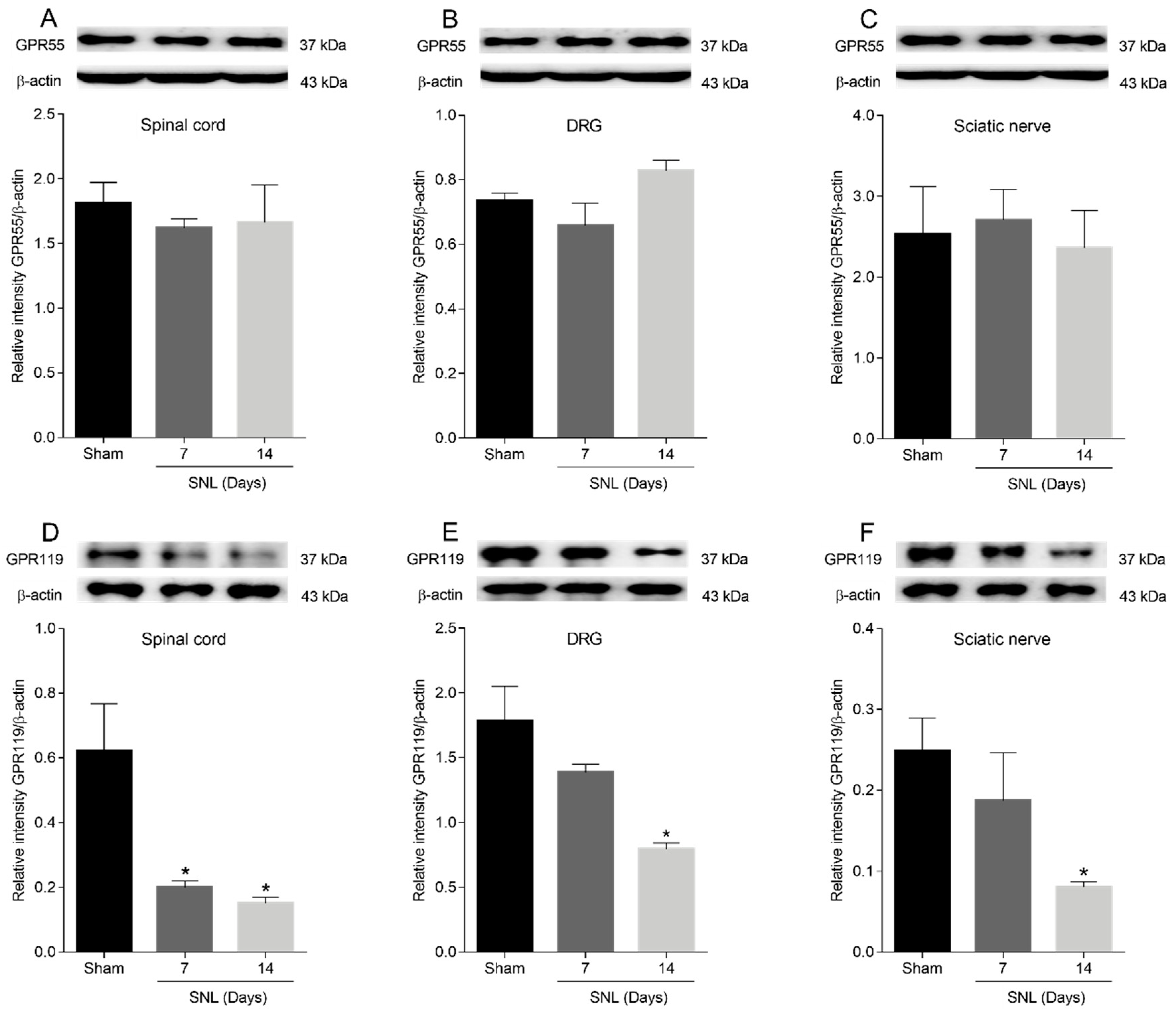
2.5. GPR55 or GPR119 Modulation in the Lumbar Ipsilateral Spinal Cord and Left Sciatic Nerve
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs
4.2.1. Induction of Spinal Nerve Ligation and Measurement of Tactile Allodynia
4.2.2. Intrathecal Administration
4.3. Immunohistochemistry
4.4. Western Blot Analysis
4.5. Experimental Design
4.6. Data Analysis and Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Issue
References
- van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The neuropathic pain: An overview of the current treatment and future therapeutic approaches. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419838383. [Google Scholar] [CrossRef] [Green Version]
- Nourbakhsh, F.; Atabaki, R.; Roohbakhsh, A. The role of orphan G protein-coupled receptors in the modulation of pain: A review. Life Sci. 2018, 212, 59–69. [Google Scholar] [CrossRef]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Im, D.-S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. Int. J. Mol. Sci. 2021, 22, 1034. [Google Scholar] [CrossRef]
- Tudurí, E.; López, M.; Diéguez, C.; Nadal, A.; Nogueiras, R. GPR55 and the regulation of glucose homeostasis. Int. J. Biochem. Cell Biol. 2017, 88, 204–207. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Gangadharan, V.; Selvaraj, D.; Kurejova, M.; Njoo, C.; Gritsch, S.; Škoricová, D.; Horstmann, H.; Offermanns, S.; Brown, A.J.; Kuner, T.; et al. A novel biological role for the phospholipid lysophosphatidylinositol in nociceptive sensitization via activation of diverse G-protein signalling pathways in sensory nerves in vivo. Pain 2013, 154, 2801–2812. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. J. Cereb. Blood Flow Metab. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Deliu, E.; Sperow, M.; Console-Bram, L.; Carter, R.L.; Tilley, D.G.; Kalamarides, D.J.; Kirby, L.G.; Brailoiu, G.C.; Brailoiu, E.; Benamar, K.; et al. The Lysophosphatidylinositol Receptor GPR55 Modulates Pain Perception in the Periaqueductal Gray. Mol. Pharmacol. 2015, 88, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Malek, N.; Kostrzewa, M.; Makuch, W.; Pajak, A.; Kucharczyk, M.; Piscitelli, F.; Przewlocka, B.; Di Marzo, V.; Starowicz, K. The multiplicity of spinal AA-5-HT anti-nociceptive action in a rat model of neuropathic pain. Pharmacol. Res. 2016, 111, 251–263. [Google Scholar] [CrossRef]
- Chiocchetti, R.; Galiazzo, G.; Tagliavia, C.; Stanzani, A.; Giancola, F.; Menchetti, M.; Militerno, G.; Bernardini, C.; Forni, M.; Mandrioli, L. Cellular Distribution of Canonical and Putative Cannabinoid Receptors in Canine Cervical Dorsal Root Ganglia. Front. Vet. Sci. 2019, 6, 313. [Google Scholar] [CrossRef]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 2008, 139, 225–236. [Google Scholar] [CrossRef]
- Armin, S.; Muenster, S.; Abood, M.; Benamar, K. GPR55 in the brain and chronic neuropathic pain. Behav. Brain Res. 2021, 406, 113248. [Google Scholar] [CrossRef]
- Carey, L.M.; Gutierrez, T.; Deng, L.; Lee, W.-H.; Mackie, K.; Hohmann, A.G. Inflammatory and Neuropathic Nociception is Preserved in GPR55 Knockout Mice. Sci. Rep. 2017, 7, 944. [Google Scholar] [CrossRef] [Green Version]
- Bonini, J.A.; Borowsky, B.E.; Adham, N.; Boyle, N.; Thompson, T.O. DNA Encoding SNORF25 Receptor. U.S. Patent 6,221,660-B1, 24 April 2001. [Google Scholar]
- Chu, Z.L.; Jones, R.M.; He, H.; Carroll, C.; Gutierrez, V.; Lucman, A.; Moloney, M.; Gao, H.; Mondala, H.; Bagnol, D.; et al. A role for beta-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology 2007, 148, 2601–2609. [Google Scholar] [CrossRef]
- Bonini, J.A.; Borowsky, B.E.; Adham, N.; Boyle, N.; Thompson, T.O. Methods of Identifying Compounds that Bind to SNORF25 Receptors. U.S. Patent 6,468,756-B1, 22 October 2002. [Google Scholar]
- Console-Bram, L.; Brailoiu, E.; Brailoiu, G.C.; Sharir, H.; Abood, M.E. Activation of GPR18 by cannabinoid compounds: A tale of biased agonism. Br. J. Pharmacol. 2014, 171, 3908–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabisiak, A.; Fabisiak, N.; Mokrowiecka, A.; Malecka-Panas, E.; Jacenik, D.; Kordek, R.; Zielińska, M.; Kieć-Kononowicz, K.; Fichna, J. Novel selective agonist of GPR18, PSB-KK-1415 exerts potent anti-inflammatory and anti-nociceptive activities in animal models of intestinal inflammation and inflammatory pain. Neurogastroenterol. Motil. 2021, 33, e14003. [Google Scholar] [CrossRef]
- Kargl, J.; Brown, A.J.; Andersen, L.; Dorn, G.; Schicho, R.; Waldhoer, M.; Heinemann, A. A Selective Antagonist Reveals a Potential Role of G Protein–Coupled Receptor 55 in Platelet and Endothelial Cell Function. J. Pharmacol. Exp. Ther. 2013, 346, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Henstidge, C.M.; Balenga, N.A.B.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-α-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Lanuti, M.; De Bardi, M.; Battistini, L.; Maccarrone, M. The differential characterization of GPR55 receptor in human peripheral blood reveals a distinctive expression in monocytes and NK cells and a proinflammatory role in these innate cells. Int. Immunol. 2015, 27, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Okine, B.N.; Mc Laughlin, G.; Gaspar, J.C.; Harhen, B.; Roche, M.; Finn, D.P. Antinociceptive Effects of the GPR55 Antagonist CID16020046 Injected into the Rat Anterior Cingulate Cortex. Neuroscience 2020, 443, 19–29. [Google Scholar] [CrossRef]
- Djouhri, L.; Lawson, S.N. Abeta-fiber nociceptive primary afferent neurons: A review of incidence and properties in relation to other afferent A-fiber neurons in mammals. Brain Res. Rev. 2004, 46, 131–145. [Google Scholar] [CrossRef]
- Wei, Z.; Fei, Y.; Su, W.; Chen, G. Emerging Role of Schwann Cells in Neuropathic Pain: Receptors, Glial Mediators and Myelination. Front. Cell. Neurosci. 2019, 13, 116. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-Y.; Sun, Y.-N.; Wang, F.-T.; Li, Q.; Su, L.; Zhao, Z.-F.; Meng, X.-L.; Zhao, H.; Wu, X.; Sun, Q.; et al. Activation of satellite glial cells in lumbar dorsal root ganglia contributes to neuropathic pain after spinal nerve ligation. Brain Res. 2012, 1427, 65–77. [Google Scholar] [CrossRef]
- Polidoro, G.; Galiazzo, G.; Giancola, F.; Papadimitriou, S.; Kouki, M.; Sabattini, S.; Rigillo, A.; Chiocchetti, R. Expression of cannabinoid and cannabinoid-related receptors in the oral mucosa of healthy cats and cats with chronic gingivostomatitis. J. Feline Med. Surg. 2020, 23, 679–691. [Google Scholar] [CrossRef]
- Dunn, S.L.; Wilkinson, J.M.; Crawford, A.; Bunning, R.A.; Le Maitre, C.L. Expression of Cannabinoid Receptors in Human Osteoarthritic Cartilage: Implications for Future Therapies. Cannabis Cannabinoid Res. 2016, 1, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Suardíaz, M.; Estivill-Torrús, G.; Goicoechea, C.; Bilbao, A.; de Fonseca, F.R. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain 2007, 133, 99–110. [Google Scholar] [CrossRef]
- Wang, X.; Miyares, R.L.; Ahern, G.P. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J. Physiol. 2005, 564, 541–547. [Google Scholar] [CrossRef]
- Mukai, H.; Munekata, E.; Higashijima, T. G protein antagonists. A novel hydrophobic peptide competes with receptor for G protein binding. J. Biol. Chem. 1992, 267, 16237–16243. [Google Scholar] [CrossRef]
- McKillop, A.M.; Moran, B.M.; Abdel-Wahab, Y.H.A.; Gormley, N.M.; Flatt, P. Metabolic effects of orally administered small-molecule agonists of GPR55 and GPR119 in multiple low-dose streptozotocin-induced diabetic and incretin-receptor-knockout mice. Diabetologia 2016, 59, 2674–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivisto, A.; Jalava, N.; Bratty, R.; Pertovaara, A. TRPA1 Antagonists for Pain Relief. Pharmaceuticals 2018, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Hassing, H.A.; Fares, S.; Larsen, O.; Pad, H.; Hauge, M.; Jones, R.M.; Schwartz, T.W.; Hansen, H.S.; Rosenkilde, M.M. Biased signaling of lipids and allosteric actions of synthetic molecules for GPR119. Biochem. Pharmacol. 2016, 119, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; O’Neill, K.; Lan, H.; Pang, L.; Shan, L.X.; Hawes, B.E.; Hedrick, J.A. Endogenous and synthetic agonists of GPR119 differ in signalling pathways and their effects on insulin secretion in MIN6c4 insulinoma cells. Br. J. Pharmacol. 2008, 155, 1056–1065. [Google Scholar] [CrossRef] [Green Version]
- Millan, M.J. The induction of pain: An integrative review. Prog. Neurobiol. 1999, 57, 1–164. [Google Scholar] [CrossRef]
- Kim, H.-J.; Yoon, H.-J.; Park, J.-W.; Che, X.; Jin, X.; Choi, J.-Y. G protein-coupled receptor 119 is involved in RANKL-induced osteoclast differentiation and fusion. J. Cell. Physiol. 2019, 234, 11490–11499. [Google Scholar] [CrossRef]
- Grill, M.; Högenauer, C.; Blesl, A.; Haybaeck, J.; Golob-Schwarzl, N.; Ferreirós, N.; Thomas, D.; Gurke, R.; Trötzmüller, M.; Köfeler, H.C.; et al. Members of the endocannabinoid system are distinctly regulated in inflammatory bowel disease and colorectal cancer. Sci. Rep. 2019, 9, 2358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y.; Li, J.; Xie, X. Discovery and characterization of novel small molecule agonists of G protein-coupled receptor 119. Acta Pharmacol. Sin. 2014, 35, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Ohishi, T.; Matsui, T.; Shibasaki, M. Identification of a novel GPR119 agonist, AS1269574, with in vitro and in vivo glucose-stimulated insulin secretion. Biochem. Biophys. Res. Commun. 2010, 400, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Chung, J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Xu, J.J.; Walla, B.C.; Diaz, M.F.; Fuller, G.N.; Gutstein, H.B. Intermittent Lumbar Puncture in Rats: A Novel Method for the Experimental Study of Opioid Tolerance. Anesth. Analg. 2006, 103, 714–720. [Google Scholar] [CrossRef]
- Notter, T.; Panzanelli, P.; Pfister, S.; Mircsof, D.; Fritschy, J.-M. A protocol for concurrent high-quality immunohistochemical and biochemical analyses in adult mouse central nervous system. Eur. J. Neurosci. 2013, 39, 165–175. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz-Álvarez, L.E.; Zúñiga-Romero, Ángel; Huerta-Cruz, J.C.; Flores-Murrieta, F.J.; Reyes-García, J.G.; Araiza-Saldaña, C.I.; Rocha-González, H.I. Antiallodynic interaction and motor performance of the pregabalin/thioctic acid and pregabalin/α-tocopherol combinations in neonatal streptozotocin-induced diabetic rats. Drug Dev. Res. 2018, 79, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zúñiga-Romero, Á.; Rivera-Plata, Q.; Arrieta, J.; Flores-Murrieta, F.J.; Rodríguez-Silverio, J.; Reyes-García, J.G.; Huerta-Cruz, J.C.; Ramírez-Martínez, G.; Rocha-González, H.I. GPR55 and GPR119 Receptors Contribute to the Processing of Neuropathic Pain in Rats. Pharmaceuticals 2022, 15, 67. https://doi.org/10.3390/ph15010067
Zúñiga-Romero Á, Rivera-Plata Q, Arrieta J, Flores-Murrieta FJ, Rodríguez-Silverio J, Reyes-García JG, Huerta-Cruz JC, Ramírez-Martínez G, Rocha-González HI. GPR55 and GPR119 Receptors Contribute to the Processing of Neuropathic Pain in Rats. Pharmaceuticals. 2022; 15(1):67. https://doi.org/10.3390/ph15010067
Chicago/Turabian StyleZúñiga-Romero, Ángel, Quetzali Rivera-Plata, Jesús Arrieta, Francisco Javier Flores-Murrieta, Juan Rodríguez-Silverio, Juan Gerardo Reyes-García, Juan Carlos Huerta-Cruz, Gustavo Ramírez-Martínez, and Héctor Isaac Rocha-González. 2022. "GPR55 and GPR119 Receptors Contribute to the Processing of Neuropathic Pain in Rats" Pharmaceuticals 15, no. 1: 67. https://doi.org/10.3390/ph15010067
APA StyleZúñiga-Romero, Á., Rivera-Plata, Q., Arrieta, J., Flores-Murrieta, F. J., Rodríguez-Silverio, J., Reyes-García, J. G., Huerta-Cruz, J. C., Ramírez-Martínez, G., & Rocha-González, H. I. (2022). GPR55 and GPR119 Receptors Contribute to the Processing of Neuropathic Pain in Rats. Pharmaceuticals, 15(1), 67. https://doi.org/10.3390/ph15010067