
In Vitro and In Vivo Evaluation of Oral Controlled Release Formulation of BCS Class I Drug Using Polymer Matrix System

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
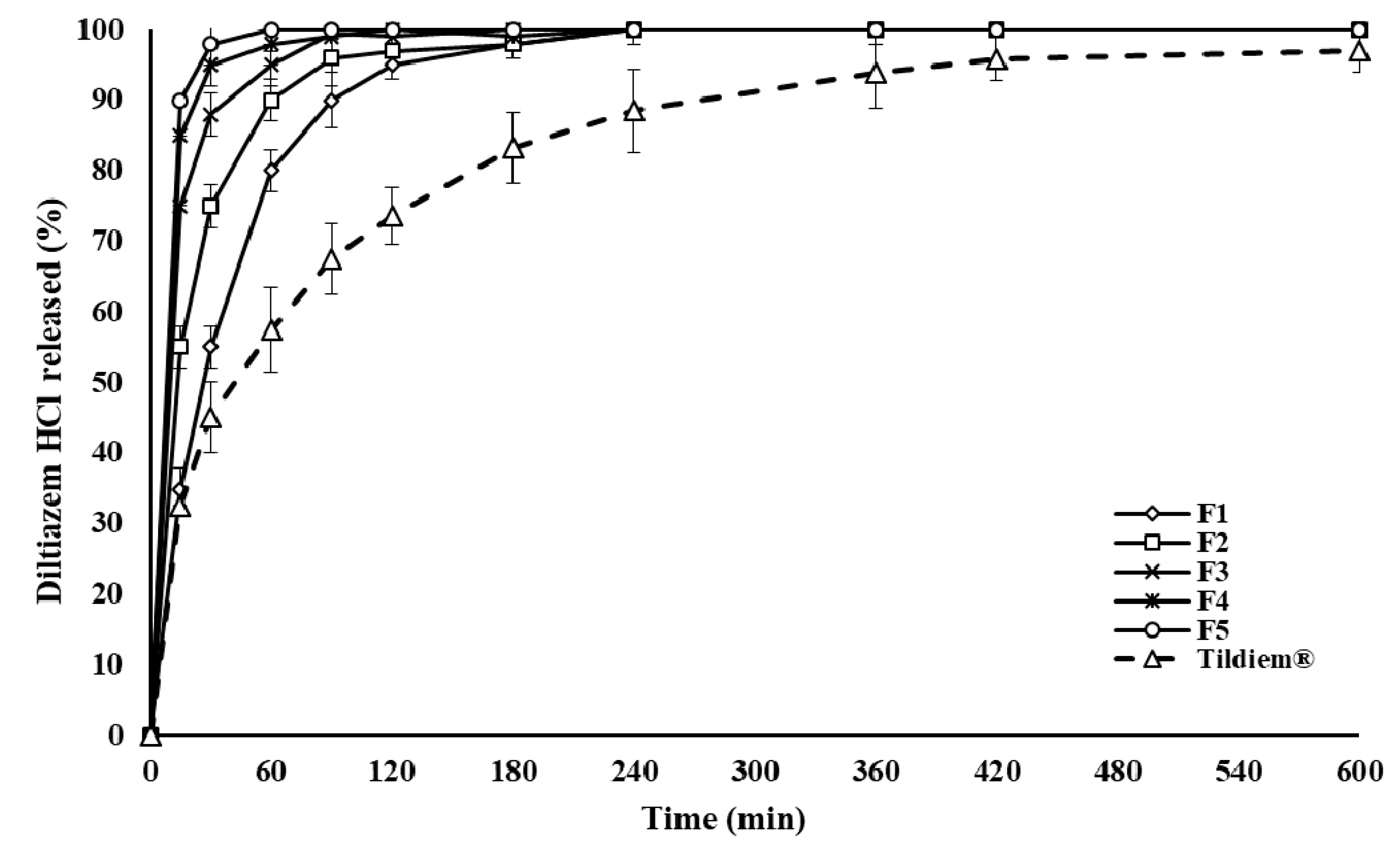
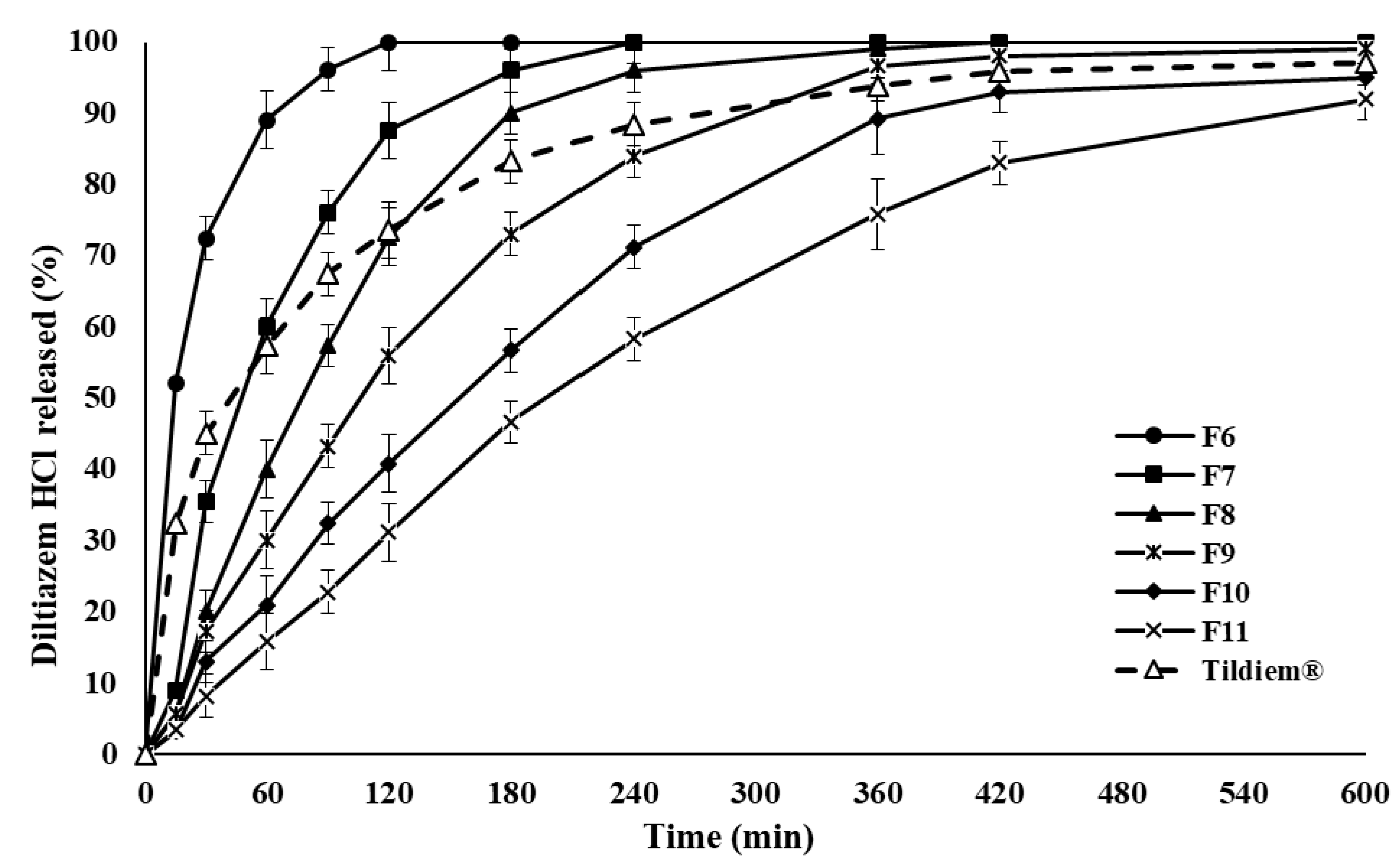
2.1. Drug Release and In Vitro Evaluation
2.2. Mechanism of Drug Release from Polymer Matrix
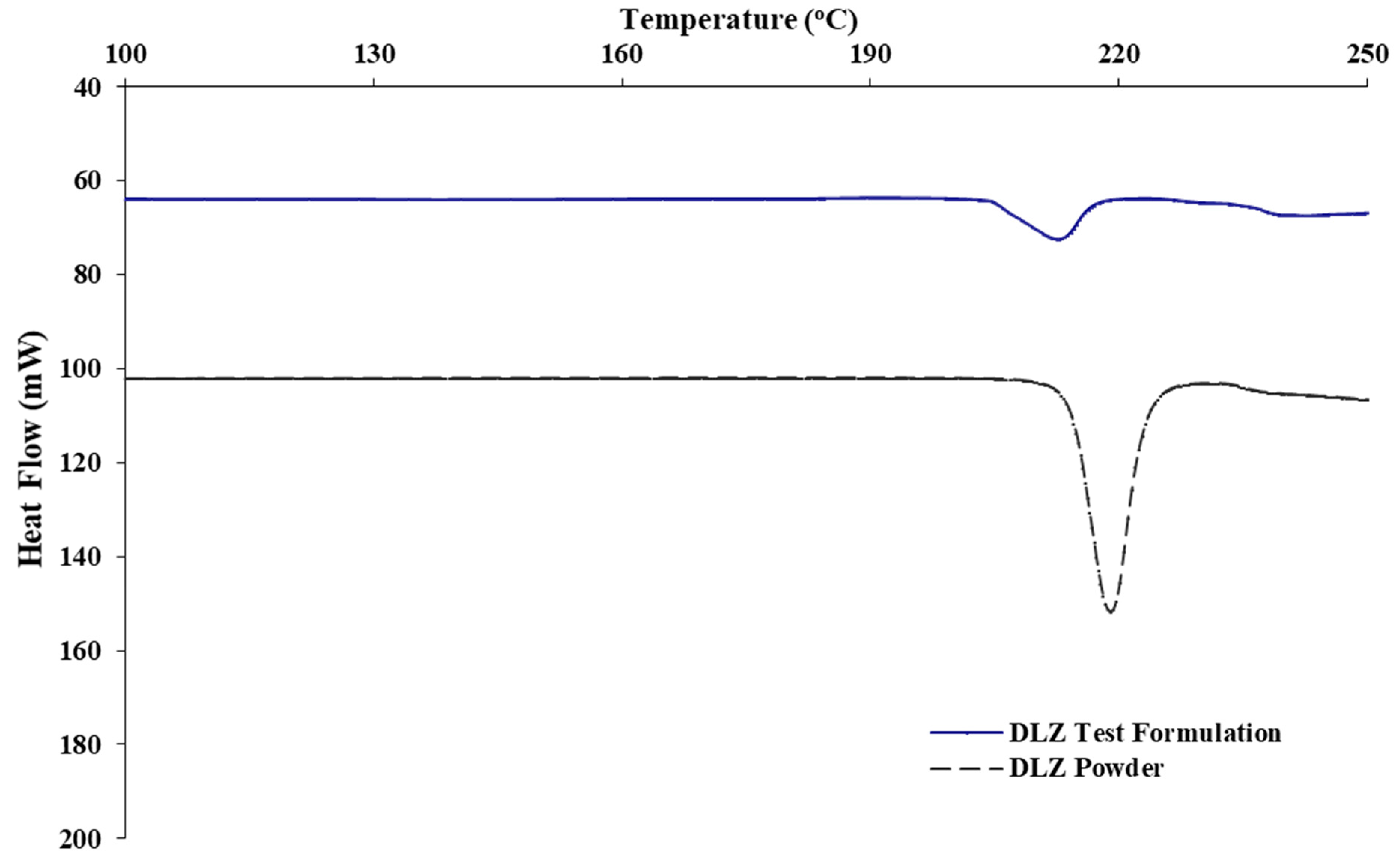
2.3. Thermal Profiles of DLZ in P-188 Polymer Matrix
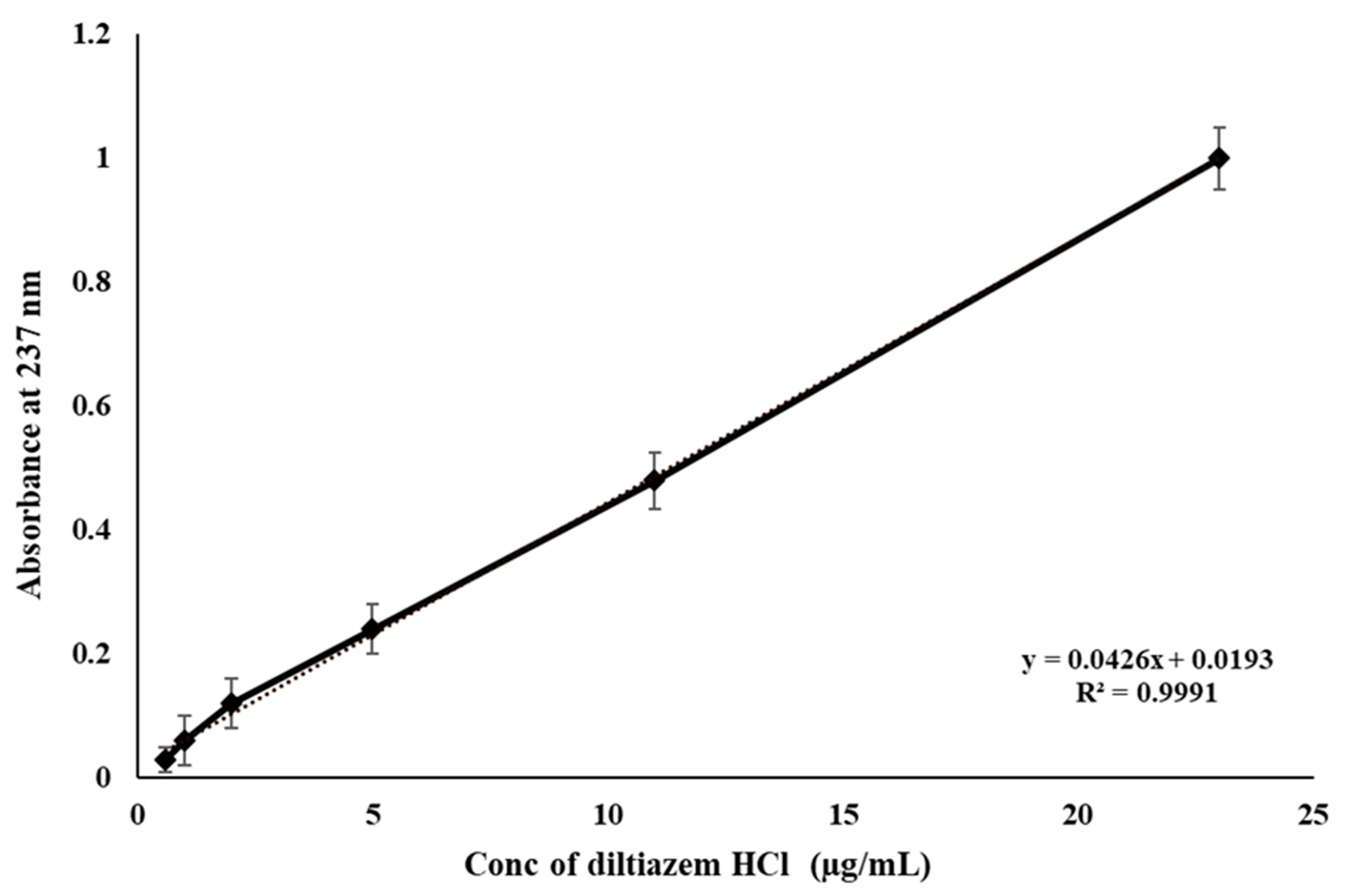
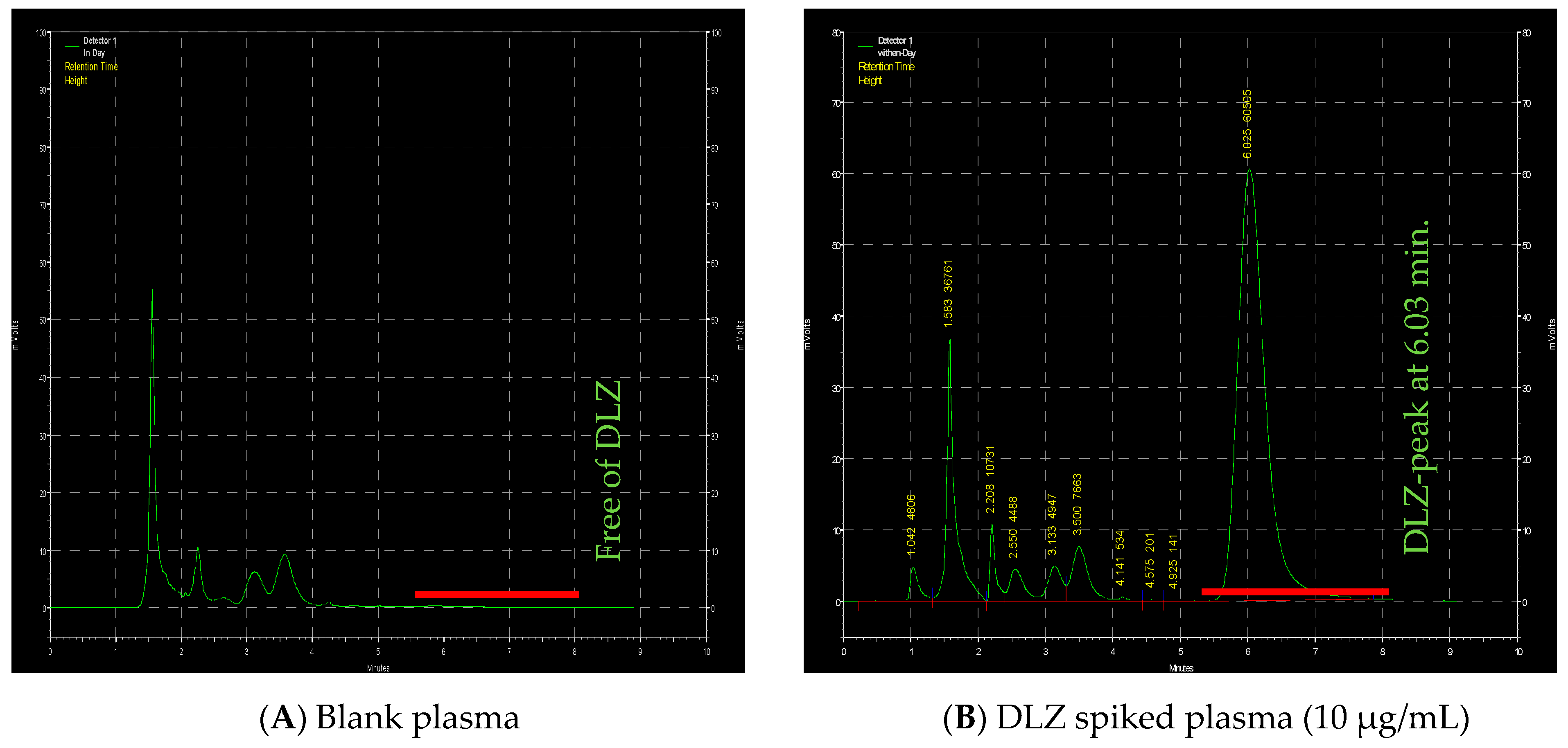
2.4. HPLC
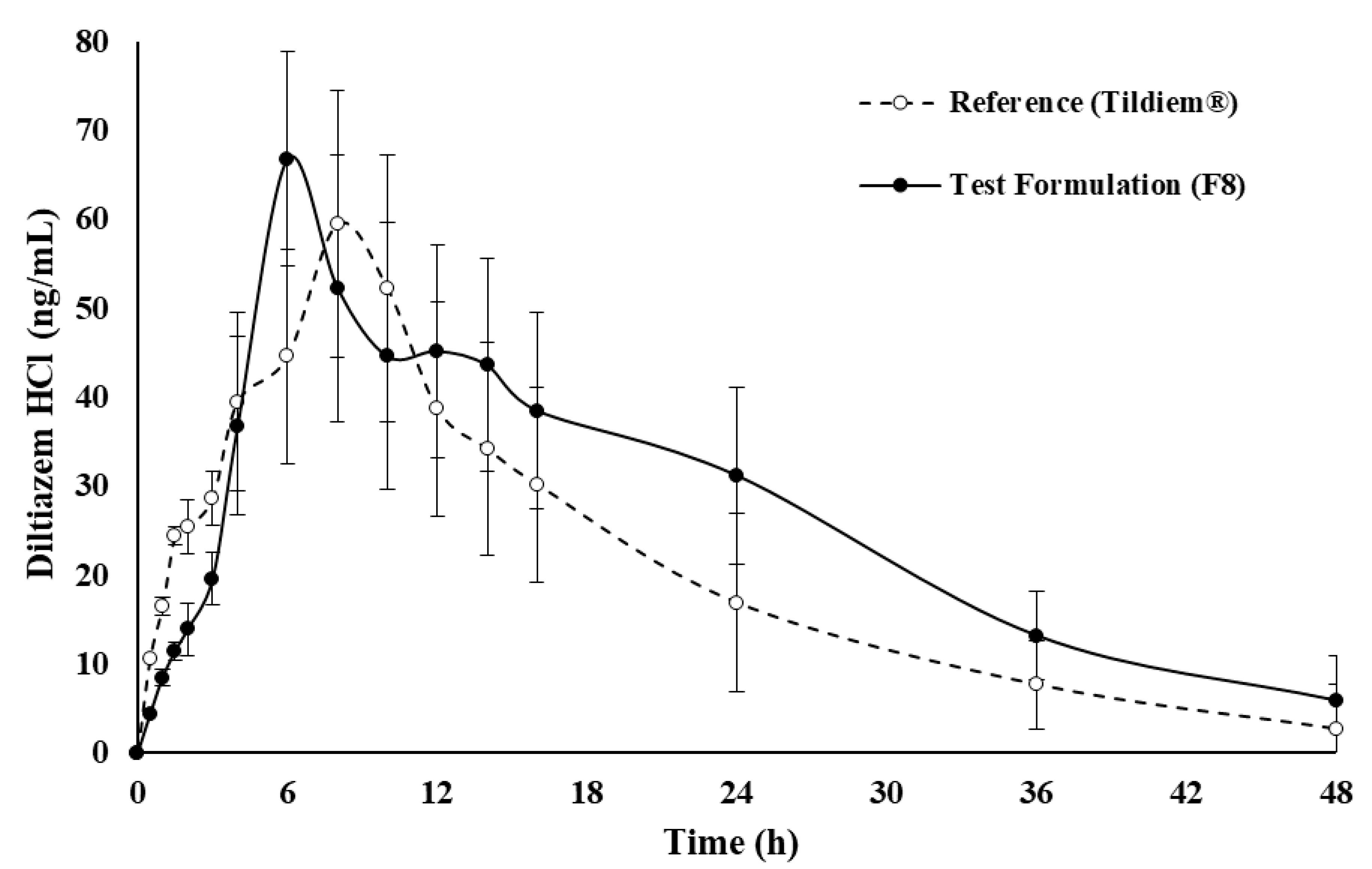
2.5. In Vivo Assessment of Drug
3. Discussion
4. Material and Methods
4.1. Chemicals
4.2. Preparation of Controlled Release Polymer Matrix Containing DLZ
4.3. Drug Content Dermination
4.4. Dissolution Studies
4.5. Thermal Analysis
4.6. In Vivo Animal Study
4.7. HPLC Method for In Vivo Assessments and PK Parameters Determination
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parriman, M.; Campolo, A.; Waller, A.P.; Lacombe, V.A. Adverse Metabolic Effects of Diltiazem Treatment during Diabetic Cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2018, 24, 193–203. [Google Scholar] [CrossRef]
- Claas, S.; Glasser, S.P. Long-acting diltiazem HCL for the chronotherapeutic treatment of hypertension and chronic stable angina pectoris. Expert Opin. Pharmacother. 2005, 6, 765–776. [Google Scholar] [CrossRef]
- Shahi, S.R.; Gulecha, B.; Shivanikar, S.S.; Shinde, S.B.; Zadbuke, N.S. Design and development of controlled porosity osmotic tablet of diltiazem hydrochloride. J. Adv. Pharm. Technol. Res. 2012, 3, 229–236. [Google Scholar] [CrossRef]
- Bojanić, V.; Bojanić, Z.; Najman, S.; Savić, T.; Jakovljević, V.; Najman, S.; Jančić, S. Diltiazem prevention of toxic effects of monosodium glutamate on ovaries in rats. Gen. Physiol. Biophys. 2009, 28, 149–154. [Google Scholar]
- Mircioiu Anuta, V.; Nicolescu, A.; Fotaki, N.; Mircioiu, C.; Mircioiu, I.; Mircioiu, I. In Vitro–In Vivo Correlations Based on In Vitro Dissolution of Parent Drug Diltiazem and Pharmacokinetics of its Metabolite. Pharmaceutics 2019, 11, 344. [Google Scholar] [CrossRef]
- Giri, T.K.; Choudhary, C.; Alexander, A.; Uddin, A.; Badwaik, H.; Tripathy, M.; Tripathi, D. Sustained Release of Diltiazem Hydrochloride from Cross-linked Biodegradable IPN Hydrogel Beads of Pectin and Modified Xanthan Gum. Indian J. Pharm. Sci. 2013, 75, 619–627. [Google Scholar]
- Markl, D.; Zeitler, J.A. A Review of Disintegration Mechanisms and Measurement Techniques. Pharm. Res. 2017, 34, 890–917. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Jung, H.; Li, X. Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J. 2015, 17, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Laracuente, M.-L.; Yu, M.H.; McHugh, K.J. Zero-order drug delivery: State of the art and future prospects. J. Control. Release 2020, 327, 834–856. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Bannerjee, S.; Bhati, L.; Pandey, S.; Pandey, P.; Sriwastawa, B. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Vidyadhara, S.; Balakrishna, T.; Prasad, M.B.; Sasidhar, R.L.C.; Trilochani, P. Development and evaluation of diltiazem hydro-chloride controlled-release pellets by fluid bed coating process. J. Adv. Pharm. Technol. Res. 2013, 4, 101–107. [Google Scholar] [CrossRef]
- Park, K. Controlled drug delivery systems: Past forward and future back. J. Control. Release 2014, 190, 3–8. [Google Scholar] [CrossRef]
- Liu, L.; Shen, T.; Yang, Y.; Gao, B.; Li, Y.C.; Xie, J.; Tang, Y.; Zhang, S.; Wang, Z.; Chen, J. Bio-based Large Tablet Controlled-Release Urea: Syn-thesis, Characterization, and Controlled-Released Mechanisms. J. Agric. Food Chem. 2018, 66, 11265–11272. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ma, Y.; Zhu, J. Dry powder coated osmotic drug delivery system. Eur. J. Pharm. Sci. 2018, 111, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Dergunov, S.A.; Durbin, J.; Pattanaik, S.; Pinkhassik, E. pH-Mediated Catch and Release of Charged Molecules with Porous Hollow Nanocapsules. J. Am. Chem. Soc. 2014, 136, 2212–2215. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, C.G.; Bloemen, V.; Mignon, A. Synthetic, Natural, and Semisynthetic Polymer Carriers for Controlled Nitric Oxide Release in Dermal Applications: A Review. Polymers 2021, 13, 760. [Google Scholar] [CrossRef]
- Al Hanbali, O.A.; Khan, H.M.S.; Sarfraz, M.; Arafat, M.; Ijaz, S.; Hameed, A. Transdermal patches: Design and current approaches to painless drug delivery. Acta Pharm. 2019, 69, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Arafat, M. Approaches to Achieve an Oral Controlled Releases Drug Delivery System Using Polymers: A Recent Review. Int. J. Pharma. Pharm. Sci. 2015, 7, 16–21. [Google Scholar]
- Aranaz, I.; Paños, I.; Peniche, C.; Heras, Á.; Acosta, N. Chitosan Spray-Dried Microparticles for Controlled Delivery of Venlafaxine Hydrochloride. Molecules 2017, 22, 1980. [Google Scholar] [CrossRef]
- Lemieux, M.; Gosselin, P.; Mateescu, M.A. Carboxymethyl high amylose starch as excipient for controlled drug release: Mechanistic study and the influence of degree of substitution. Int. J. Pharm. 2009, 382, 172–182. [Google Scholar] [CrossRef]
- Shi, C.; Tao, F.; Cui, Y. Cellulose-based film modified by succinic anhydride for the controlled release of domperidone. J. Biomater. Sci. Polym. Ed. 2018, 29, 1233–1249. [Google Scholar] [CrossRef]
- Martínez, A.D.B.; Galán, I.C.R.; Bellver, M.V.M. Application of a Biodegradable Polyesteramide Derived from L-Alanine as Novel Excipient for Controlled Release Matrix Tablets. AAPS PharmSciTech 2017, 18, 3286–3295. [Google Scholar] [CrossRef]
- Peng, D.; Huang, K.; Liu, Y.; Liu, S. Preparation of novel polymeric microspheres for controlled release of finasteride. Int. J. Pharm. 2007, 342, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Bostanudin, M.F.; Arafat, M.; Sarfraz, M.; Górecki, D.C.; Barbu, E. Butylglyceryl Pectin Nanoparticles: Synthesis, Formulation and Characterization. Polymers 2019, 11, 789. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for Drug Delivery Systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Benyerbah, N.; Ispas-Szabo, P.; Sakeer, K.; Chapdelaine, D.; Mateescu, M.A. Ampholytic and Polyelectrolytic Starch as Matrices for Controlled Drug Delivery. Pharmaceutics 2019, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Khandai, M.; Sharma, A.; Patra, C.; Patro, V.; Sen, K.K. Effects of drug solubility on the release kinetics of water soluble and insoluble drugs from HPMC based matrix formulations. Acta Pharm. 2009, 59, 313–323. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Chen, K.; Wen, H.; Ouyang, D.; Li, X.; Gao, Y.; Pan, W.; Yang, X. Design and Evaluation of Hydrophilic Matrix System Containing Polyethylene Oxides for the Zero-Order Controlled Delivery of Water-Insoluble Drugs. AAPS PharmSciTech 2016, 18, 82–92. [Google Scholar] [CrossRef]
- Bacaita, E.S.; Agop, M. A multiscale mechanism of drug release from polymeric matrices: Confirmation through a nonlinear theoretical model. Phys. Chem. Chem. Phys. 2016, 18, 21809–21816. [Google Scholar] [CrossRef]
- Arafat, M.; Sarfraz, M.; AbuRuz, S. Development and In Vitro Evaluation of Controlled Release Viagra® Containing Poloxamer-188 Using Gastroplus™ PBPK Modeling Software for In Vivo Predictions and Pharmacokinetic Assessments. Pharmaceuticals 2021, 14, 479. [Google Scholar] [CrossRef]
- Tehsin, K.; Muhammad, S.; Mosab, A.; Momir, M.; Nisar, U.R. Rahman Cross-linked guar gum and sodium borate based microspheres as co-lon-targeted anticancer drug delivery systems for 5-fluorouracil. Pak. J. Pharm. Sci. 2017, 30, 2329–2336. [Google Scholar]
- Al-Hanbali, O.A.; Hamed, R.; Arafat, M.; Bakkour, Y.; Al-Matubsi, H.; Mansour, R.; Al-Bataineh, Y.; Aldhoun, M.; Sarfraz, M.; Dardas, A.K.Y. Formulation and evaluation of diclofenac con-trolled release matrix tablets made of HPMC and Poloxamer 188 polymer: An assessment on mechanism of drug release. Pak. J. Pharm. Sci. 2018, 31, 345–351. [Google Scholar]
- Bodratti, A.M.; Alexandridis, P. Formulation of Poloxamers for Drug Delivery. J. Funct. Biomater. 2018, 9, 11. [Google Scholar] [CrossRef]
- Russo, E.; Villa, C. Poloxamer Hydrogels for Biomedical Applications. Pharmaceutics 2019, 11, 671. [Google Scholar] [CrossRef]
- Phillips, D.M.; Haut, R.C. The use of a non-ionic surfactant (P188) to save chondrocytes from necrosis following impact loading of chondral explants. J. Orthop. Res. 2004, 22, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Larrañeta, E.; Isasi, J.R. Phase Behavior of Reverse Poloxamers and Poloxamines in Water. Langmuir 2013, 29, 1045–1053. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C. Poloxamine-based nanomaterials for drug delivery. Front. Biosci. 2010, E2, 424–440. [Google Scholar] [CrossRef]
- Szafraniec, J.; Antosik, A.; Knapik-Kowalczuk, J.; Chmiel, K.; Kurek, M.; Gawlak, K.; Odrobińska, J.; Paluch, M.; Jachowicz, R. The Self-Assembly Phenomenon of Poloxamers and Its Effect on the Dissolution of a Poorly Soluble Drug from Solid Dispersions Obtained by Solvent Methods. Pharmaceutics 2019, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Moloughney, J.G. Poloxamer 188 (P188) as a Membrane Resealing Reagent in Biomedical Applications. Recent Patents Biotechnol. 2012, 6, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Jin, H.; Cheng, Y.; Cheng, Z.; Xu, Y. The Controlled Release and Anti-Inflammatory Activity of a Tetramethylpyrazine-Loaded Thermosensitive Poloxamer Hydrogel. Pharm. Res. 2019, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Tabor, Z.; Tarasiuk, J.; Strach, B.; Pociecha, K.; Wyska, E.; Wronski, S.; Łyszczarz, E.; Jachowicz, R. The impact of polymers on 3D microstructure and controlled release of sildenafil citrate from hydrophilic matrices. Eur. J. Pharm. Sci. 2018, 119, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, M.M.; Alwattar, J.K.; Elmaradny, H.A.; Alwattar, J.K. Optimization, physicochemical characterization and in vivo assessment of spray dried emulsion: A step toward bioavailability augmentation and gastric toxicity minimization. Int. J. Pharm. 2015, 496, 766–779. [Google Scholar] [CrossRef]
- Ivanova, N.A.; Trapani, A.; Di Franco, C.; Mandracchia, D.; Trapani, G.; Franchini, C.; Corbo, F.; Tripodo, G.; Kolev, I.N.; Stoyanov, G.S.; et al. In vitro and ex vivo studies on diltiazem hydrochloride-loaded microsponges in rectal gels for chronic anal fissures treatment. Int. J. Pharm. 2018, 557, 53–65. [Google Scholar] [CrossRef]
- Elgaied-Lamouchi, D.; Descamps, N.; Lefèvre, P.; Mackin-Mohamour, A.R.; Neut, C.; Siepmann, F.; Siepmann, J.; Muschert, S. Robustness of Controlled Release Tablets Based on a Cross-linked Pregelatinized Potato Starch Matrix. AAPS Pharm. Sci. Tech. 2020, 2, 148. [Google Scholar] [CrossRef]
- Arafat, M. Simple HPLC validated method for the determination of diltiazem hydrochloride in human plasma. Int. J. Pharma. Pharm. Sci. 2014, 6, 213–216. [Google Scholar]
- Prabakaran, D. Effect of hydrophilic polymers on the release of diltiazem hydrochloride from elementary osmotic pumps. Int. J. Pharm. 2003, 259, 173–179. [Google Scholar] [CrossRef]
- Giuliano, E.; Paolino, D.; Fresta, M.; Cosco, D. Drug-Loaded Biocompatible Nanocarriers Embedded in Poloxamer 407 Hydrogels as Therapeutic Formulations. Medicines 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Sriamornsak, P.; Thirawong, N.; Weerapol, Y.; Nunthanid, J.; Sungthongjeen, S. Swelling and erosion of pectin matrix tablets and their impact on drug release behavior. Eur. J. Pharm. Biopharm. 2007, 67, 211–219. [Google Scholar] [CrossRef]
- Adibkia, K.; Ghanbarzadeh, S.; Mohammadi, G.; Khiavi, H.Z.; Sabzevari, A.; Barzegar-Jalali, M. Drug Release Kinetic Analysis and Prediction of Release Data via Polymer Molecular Weight in Sustained Release Diltiazem Matrices. Drug Res. 2013, 64, 118–123. [Google Scholar] [CrossRef]
- Aryani, N.L.D.; Siswodihardjo, S.; Soeratri, W.; Sari, N.F.I. Development, characterization, molecular docking, and in vivo skin penetration of coenzyme Q10 nanostructured lipid carriers using tristearin and stearyl alcohol for dermal delivery. J. Basic Clin. Physiol. Pharmacol. 2021, 32, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, L.; Sun, Y.; Liu, X.; Liu, W.; Du, Y.; Li, L.; Sun, J. Preparation and evaluation of sustained-release diltiazem hydrochloride pellets. Asia J. Pharm. Sci. 2013, 8, 244–251. [Google Scholar] [CrossRef][Green Version]
- Choi, J.S.; Han, H.K. Enhanced oral exposure of diltiazem by the concomitant use of naringin in rats. Int. J. Pharm. 2005, 305, 122–128. [Google Scholar] [CrossRef]
- Tian, J.-L.; Zhao, Y.-Z.; Jin, Z.; Lu, C.-T.; Tang, Q.-Q.; Xiang, Q.; Sun, C.-Z.; Zhang, L.; Xu, Y.-Y.; Gao, H.-S.; et al. Synthesis and characterization of Poloxamer 188-grafted heparin copolymer. Drug Dev. Ind. Pharm. 2010, 36, 832–838. [Google Scholar] [CrossRef]
- Bostanudin, M.F.; Salam, A.; Mahmood, A.; Arafat, M.; Kaharudin, A.N.; Sahudin, S.; Lazim, A.M.; Azfaralariff, A. Formulation and In-Vitro Characterisation of Cross-Linked Amphiphilic Guar Gum Nanocarriers for Percutaneous Delivery of Arbutin. J. Pharm. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Arafat, M.; Kirchhoefer, C.; Mikov, M. Mixed Micelles Loaded with Bile Salt: An Approach to Enhance Intestinal Transport of the BCS Class III Drug Cefotaxime in Rats. Eur. J. Drug Metab. Pharmacokinet. 2016, 42, 635–645. [Google Scholar] [CrossRef]
- Arafat, M.; Fahelelbom, K.M.; Sarfraz, M.K.; Bostanudin, M.F.; Sharif, Q.-U.; Esmaeil, A.; AL Hanbali, O.; Aburuz, S. Comparison between branded and generic furosemide 40 mg tablets using thermal gravimetric analysis and Fourier transform infrared spectroscopy. J. Pharm. Bioallied Sci. 2020, 12, 489–498. [Google Scholar] [CrossRef]
- Arafat, M.; Kirchhoefer, C.; Mikov, M.; Sarfraz, M.; Löbenberg, R. Nanosized Liposomes Containing Bile Salt: A Vesicular Nanocarrier for Enhancing Oral Bioavailability of BCS Class III Drug. J. Pharm. Pharm. Sci. 2017, 20, 305–318. [Google Scholar] [CrossRef]
- Khalid, N.; Sarfraz, M.; Arafat, M.; Akhtar, M.; Löbenberg, R.; Rehman, N.U. Nano-sized Droplets of Self-Emulsifying System for Enhancing Oral Bioavailability of Chemotherapeutic Agent VP-16 in Rats: A Nano Lipid Carrier for BCS Class IV Drugs. J. Pharm. Pharm. Sci. 2018, 21, 398–408. [Google Scholar] [CrossRef]
- Arafat, M.; Ahmed, Z.; Mikov, M. Determination of nifedipine in rat plasma using HPLC-UV detector: A simple method for pharmacokinetics and oral bioavailability studies. Int. J. Pharma. Pharm. Sci. 2016, 8, 98–102. [Google Scholar]
- Arafat, M.; Golocorbin-kon, S.; Mikov, M. The measurement of cefotaxime sodium in rat plasma after oral administration: A sensitive HPLC-UV method. Int. J. Pharma. Pharm. Sci. 2015, 7, 343–346. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | DLZ (%) | P-188t (%) | Total Weight (mg) |
|---|---|---|---|
| F1 | 10 | 90 | 600 |
| F2 | 20 | 80 | 600 |
| F3 | 30 | 70 | 600 |
| F4 | 40 | 60 | 600 |
| F5 | 50 | 50 | 600 |
| Formulations | DLZ (%) | P-188 (%) | HPMC (%) | STA (%) | Total Weight (mg) |
|---|---|---|---|---|---|
| F6 | 20 | 30 | 50 | 0 | 300 |
| F7 | 20 | 30 | 47.5 | 2.5 | 300 |
| F8 | 20 | 30 | 45 | 5 | 300 |
| F9 | 20 | 30 | 42.5 | 7.5 | 300 |
| F10 | 20 | 30 | 40 | 10 | 300 |
| F11 | 20 | 30 | 35 | 15 | 300 |
| Model Name | r2 of F7 | r2 of F8 | r2 of F9 | r2 of F10 |
|---|---|---|---|---|
| Zero order model | 0.9984 | 0.9816 | 0.9843 | 0.9857 |
| First order model | 0.7943 | 0.6778 | 0.6291 | 0.5769 |
| Hixson-Crowell model | 0.9751 | 0.9845 | 0.9877 | 0.9971 |
| Higuchi model | 0.9711 | 0.9874 | 0.9745 | 0.9686 |
| Korsmeyer-Peppas model | 0.4313 | 0.4409 | 0.3990 | 0.3829 |
| Concentration | Recovery | Intra-Day | Inter-Day | |||
|---|---|---|---|---|---|---|
| (µg/mL) | Accuracy | Precision | Accuracy | Precision | Accuracy | Precision |
| (%) | (C.V. %) | (%) | (C.V. %) | (%) | (C.V. %) | |
| 0.25 | 105.1 | 3.4 | 91.8 | 5.2 | 101.8 | 3.8 |
| 0.5 | 104.7 | 5.7 | 106.7 | 3.3 | 98.8 | 7.5 |
| 1 | 99.1 | 3.3 | 95.1 | 6.2 | 102.4 | 5.0 |
| 2 | 98.4 | 7.5 | 92.5 | 5.0 | 94.1 | 4.4 |
| 5 | 103.8 | 3.6 | 103.8 | 3.1 | 103.4 | 5.3 |
| 10 | 101.3 | 3.1 | 99.3 | 4.3 | 99.0 | 4.7 |
| 20 | 104.2 | 2.4 | 102.5 | 0.4 | 101.3 | 1.7 |
| PK Parameters | Commercial Product (Tildiem®) | Test Formulation (F8) |
|---|---|---|
| Cmax (ng/mL) | 59.50 ± 11.7 | 66.81 ± 8.33 |
| Tmax (h) | 8.00 ± 3.27 | 6.00 ± 2.42 |
| MRT0–∞ (h) | 50.5 ± 2.53 | 52.6 ± 3.71 |
| AUC0–∞ (ng·h/mL) | 1109.5 ± 352.7 | 1397.7 ± 352.9 |
| AUC0–t (ng·h/mL) | 1065.1 ± 454.2 | 1300.7 ± 292.4 |
| ke (h−1) | 0.074 ± 0.03 | 0.061 ± 0.04 |
| t½ (h) | 9.30 ± 4.25 | 11.4 ± 3.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arafat, M.; Sarfraz, M.; Bostanudin, M.F.; Esmaeil, A.; Salam, A.; AbuRuz, S. In Vitro and In Vivo Evaluation of Oral Controlled Release Formulation of BCS Class I Drug Using Polymer Matrix System. Pharmaceuticals 2021, 14, 929. https://doi.org/10.3390/ph14090929
Arafat M, Sarfraz M, Bostanudin MF, Esmaeil A, Salam A, AbuRuz S. In Vitro and In Vivo Evaluation of Oral Controlled Release Formulation of BCS Class I Drug Using Polymer Matrix System. Pharmaceuticals. 2021; 14(9):929. https://doi.org/10.3390/ph14090929
Chicago/Turabian StyleArafat, Mosab, Muhammad Sarfraz, Mohammad F. Bostanudin, Anna Esmaeil, Aisha Salam, and Salahdein AbuRuz. 2021. "In Vitro and In Vivo Evaluation of Oral Controlled Release Formulation of BCS Class I Drug Using Polymer Matrix System" Pharmaceuticals 14, no. 9: 929. https://doi.org/10.3390/ph14090929
APA StyleArafat, M., Sarfraz, M., Bostanudin, M. F., Esmaeil, A., Salam, A., & AbuRuz, S. (2021). In Vitro and In Vivo Evaluation of Oral Controlled Release Formulation of BCS Class I Drug Using Polymer Matrix System. Pharmaceuticals, 14(9), 929. https://doi.org/10.3390/ph14090929