
Flavanonol Glycosides from the Stems of Myrsine seguinii and Their Neuroprotective Activities

 , , and
, , and
Abstract
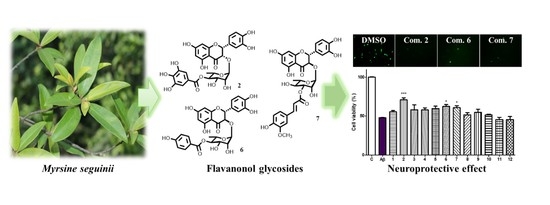
1. Introduction
2. Results
2.1. Authentication of M. seguinii H. Lév
2.2. Isolation of Compounds from M. seguinii Using Molecular Networking
2.3. Structural Determination of Compounds from M. seguinii
2.3.1. (2R,3R)-4″-O-Galloylastilbin (1)
2.3.2. (2R,3S)-4″-O-Galloylisoastilbin (2)
2.3.3. (2R,3R)-4″-O-(3‴,4‴-Dihydroxybenzoyl)astilbin (3)
2.3.4. (2R,3R)-4″-O-Vanilloylastilbin (4)
2.3.5. (2R,3S)-4″-O-Vanilloylisoastilbin (5)
2.3.6. (2R,3R)-4″-O-(4‴-Hydroxybenzoyl)astilbin (6)
2.3.7. (2R,3R)-3″-O-E-Feruloylastilbin (7)
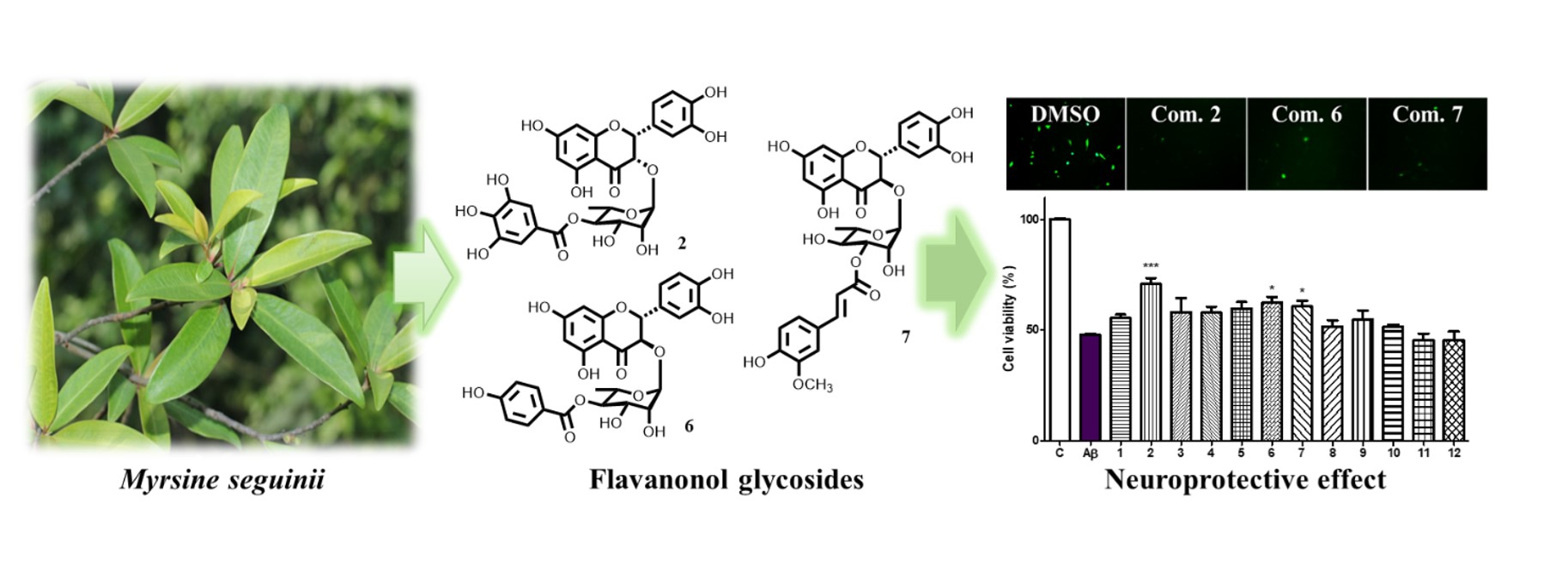
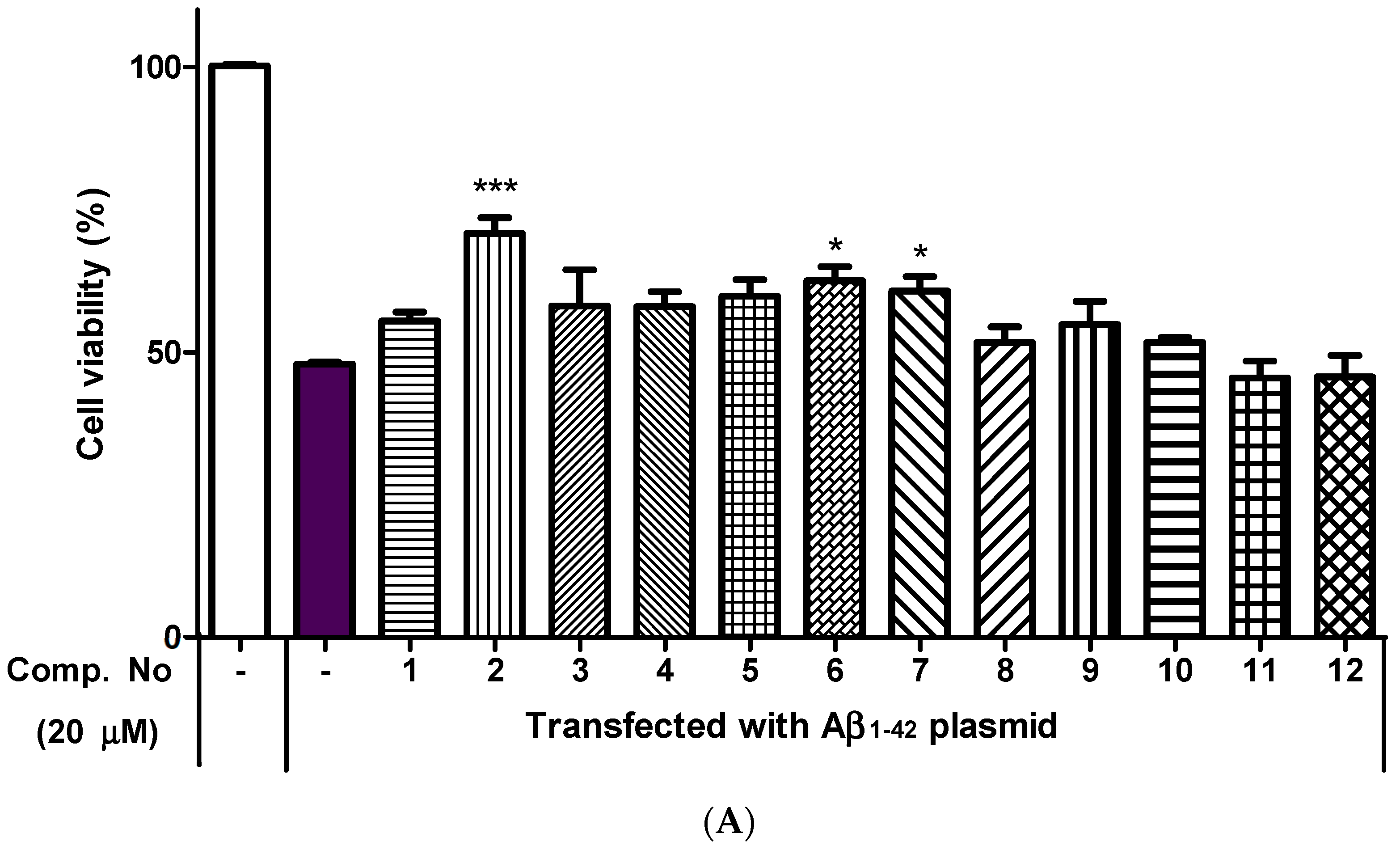
2.4. Neuroprotective Effects of Isolated Compounds (1–12)
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Morphology and DNA Sequencing Analysis of M. seguinii
4.4. LC-MS/MS Analysis and Molecular Networking
4.5. Extraction and Isolation of Flavonoids from M. seguinii
4.6. Spectroscopic and Physical Characteristic of Compounds
4.7. Cell Culture and Cell Viability Assay
4.8. Cytotoxicity Assay of Aβ1–42-Transfected HT22 Cells
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s & Dement. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International; Patterson, C. World Alzheimer Repots 2018; The State of the Art of Dementia Research: New Frontiers; Available online: http://www.alzint.org/resource/world-alzheimer-report-2018/ (accessed on 21 September 2018).
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Barril, X.; Orozco, M.; Luque, F.J. Towards improved acetylcholinesterase inhibitors: A structural and computational approach. Mini Rev. Med. Chem. 2001, 1, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 41, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Chopra, K.; Misra, S.; Kuhad, A. Current perspectives on pharmacotherapy of Alzheimer’s disease. Expert Opin. Pharmacother. 2011, 12, 335–350. [Google Scholar] [CrossRef]
- Scott, L.J.; Goa, K.L. Galantamine. Drugs 2000, 60, 1095–1122. [Google Scholar] [CrossRef] [PubMed]
- Stáhl, B.; Anderberg, A.A. Myrsinaceae. In The Families and Genera of Vascular Plants Flowering Plants. Dicotyledons Celastrales, Oxalidales, Rosales, Cornales, Ericales; Kubitzki, K., Ed.; Springer: Berlin, Germany, 2004; Volume 6, pp. 266–281. [Google Scholar]
- Oga, N.; Sakura, N. Forest vegetation of Chiba Prefecture: Vegetation catalogue of Chiba Prefecture. In Biological Society of Chiba Prefecture (de) Flora and Vegetation of Chiba Prefecture; Inoue Book Co.: Tokyo, Japan, 1975; 387p. (In Japanese) [Google Scholar]
- Makabe, H.; Miyazaki, S.; Kamo, T.; Hirota, M. Myrsinoic acid E, an anti-inflammatory compound from Myrsine seguinii. Biosci. Biotechnol. Biochem. 2003, 67, 2038–2041. [Google Scholar] [CrossRef][Green Version]
- Bloor, S.J.; Qi, L. Cytotoxic saponins from New Zealand Myrsine species. J. Nat. Prod. 1994, 57, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Githiori, J.B.; Höglund, J.; Waller, P.J.; Baker, R.L. Anthelmintic activity of preparations derived from Myrsine Africana and Rapanea melanophloeos against the nematode parasite, Haemonchus contortus, of sheep. J. Ethnopharmacol. 2002, 80, 187–191. [Google Scholar] [CrossRef]
- Hirota, M.; Miyazaki, S.; Minakuchi, T.; Takagi, T.; Shibata, H. Myrsinoic acids B, C, and F, anti-inflammatory compounds from Myrsine seguinii. Biosci. Biotechnol. Biochem. 2002, 66, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.N.; Otsuka, H.; Ide, T.; Hirata, E.; Takushi, A.; Takeda, Y. Three flavonol glycosides from leaves of Myrsine seguinii. Phytochemisty 1997, 46, 943–946. [Google Scholar] [CrossRef]
- Ito, S.; Narise, A.; Shimura, S. Identification of a methioninase inhibitor, myrsinoic acid B, from Myrsine seguinii Lév., and its inhibitory activities. Biosci. Biotechnol. Biochem. 2008, 72, 2411–2414. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsunami, K.; Otsuka, H.; Takeda, Y. Myrseguinosides A-E, five new glycosides from the fruits of Myrsine seguinii. Chem. Pharm. Bull. 2011, 59, 1274–1280. [Google Scholar] [CrossRef]
- Metcalfe, M.J.; Figueiredo-Pereira, M.E. Relationship between tau pathology and neuroinflammation in Alzheimer’s disease. Mt Sinai J. Med. 2010, 77, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.M.; Lee, Y.R.; Lee, B.W.; Zhang, M.; Ryu, B.; Nghiem, D.T.; Pham, H.T.; Oh, W.K. Phenolic constituents of the roots of Rhamnoneuron balansae with senolytic activity. J. Nat. Prod. 2020, 83, 3661–3670. [Google Scholar] [CrossRef]
- Tourdjman, M.; Srihawong, R.; Soy, T.K.; Touch, S.; Hul, S.; Janssens, B.; Galliot-Guilley, M.; Vong, S. Plant poisoning outbreak in the western area of Cambodia, 2005. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 949–951. [Google Scholar] [CrossRef]
- Thongkhao, K.; Tungphatthong, C.; Phadungcharoen, T.; Sukrong, S. The use of plant DNA barcoding coupled with HRM analysis to differentiate edible vegetables from poisonous plants for food safety. Food Control 2020, 109, 106896. [Google Scholar] [CrossRef]
- Jie, C.; Pipoly, J.J. Myrsinacea through Loganiaceae. In Flora of China; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 1996; Volume 15, pp. 1–38. [Google Scholar]
- Huang, H.; Cheng, Z.; Shi, H.; Xin, W.; Wang, T.T.Y.; Yu, L. Isolation and characterization of two flavonoids, engeletin and astilbin, from the leaves of Engelhardia roxburghiana and their potential anti-inflammatory properties. J. Agric. Food Chem. 2011, 59, 4562–4569. [Google Scholar] [CrossRef] [PubMed]
- Kuspradini, H.; Mitsunaga, T.; Ohashi, H. Antimicrobial activity against Streptococcus sobrinus and glucosyltransferase inhibitory activity of taxifolin and some flavanonol rhamnosides from kempas (Koompassia malaccensis) extracts. J. Wood Sci. 2009, 55, 308–313. [Google Scholar] [CrossRef]
- Slade, D.; Ferreira, D.; Marais, J.P.J. Circular dichroism, a powerful tool for the assessment of absolute configuration of flavonoids. Phytochemistry 2005, 66, 2177–2215. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, X.; Zhang, P.; Li, Z.L.; Wang, Y. Antiinflammatory constituents from the roots of Smilax bockii warb. Arch. Pharm. Res. 2005, 28, 395–399. [Google Scholar] [CrossRef]
- Gratani, L. Plant phenotypic plasticity in response to environmental factors. Adv. Bot. 2014, 4, 1–17. [Google Scholar] [CrossRef]
- Yves, C. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621S–629S. [Google Scholar] [CrossRef]
- Xin, W.; Huang, H.; Yu, L.; Shi, H.; Sheng, Y.; Wang, T.T.Y.; Yu, L. Three new flavanonol glycosides from leaves of Engelhardia roxburghiana, and their anti-inflammation, antiproliferative and antioxidant properties. Food Chem. 2012, 132, 788–798. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, R.; Shi, Y.; Zhang, X.; Tian, C.; Xia, D. Antioxidant and anti-inflammatory activities of six flavonoids from Smilax glabra Roxb. Molecules 2020, 25, 5295. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.M.; Ha, T.K.Q.; Doan, T.P.; Dhodary, B.; An, J.P.; Lee, B.W.; Yang, J.L.; Oh, W.K. Neuroprotective effects of triterpenoids from Camellia japonica against amyloid β-induced neuronal damage. J. Nat. Prod. 2020, 83, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- An, J.P.; Ha, T.K.Q.; Kim, H.W.; Ryu, B.; Kim, J.; Park, J.; Lee, C.H.; Oh, W.K. Eudesmane glycosides from Ambrosia artemisiifolia (Common Ragweed) as potential neuroprotective agents. J. Nat. Prod. 2019, 82, 702–713. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 b | 5 a | 6 b | 7 c |
|---|---|---|---|---|---|---|---|
| 2 | 5.08, d (11.1) | 5.48, d (2.0) | 5.10, d (11.2) | 5.09, d (11.0) | 5.47, d (2.1) | 5.09, d (11.1) | 5.12, d (11.0) |
| 3 | 4.64, d (11.1) | 4.16, d (2.2) | 4.63, d (11.1) | 4.63, d (11.0) | 4.16, d (2.2) | 4.63, d (11.1) | 4.61, d (11.0) |
| 4 | |||||||
| 5 | |||||||
| 6 | 5.94, d (1.8) | 6.00, d (1.9) | 5.93, d (1.7) | 5.92, d (1.7) | 6.00, d (2.1) | 5.94, d (2.0) | 5.93, d (2.2) |
| 7 | |||||||
| 8 | 5.91, d (1.9) | 5.93, d (1.9) | 5.91, d (1.7) | 5.90, d (1.6) | 5.94, d (2.1) | 5.92, d (1.9) | 5.91, d (2.1) |
| 9 | |||||||
| 10 | |||||||
| 1′ | |||||||
| 2′ | 6.87, dd (8.1, 1.7) | 6.85, dd (8.3, 1.8) | 6.99, d (1.6) | 6.98, d (1.3) | 6.87, br s | 6.99, d (1.8) | 6.98, d (2.0) |
| 3′ | 6.83, d (8.1) | 6.89, d (8.1) | |||||
| 4′ | |||||||
| 5′ | 6.84, overlap | 6.82, d (8.1) | 6.87, br s | 6.85, overlap | 6.80, d (8.0) | ||
| 6′ | 6.99, d (1.5) | 7.08, d (1.6) | 6.87, d (8.1, 1.7) | 6.87, overlap | 7.03, br s | 6.87, dd (8.1, 1.8) | 6.87, dd (8.1, 2.0) |
| 1″ | 4.06, s | 5.02, br s | 4.07, br s | 4.08, s | 5.02, d (1.2) | 4.08, br s | 4.03, d (1.3) |
| 2″ | 3.59, br s | 3.72, m | 3.60, m | 3.59, m | 3.74, dd (3.3, 1.5) | 3.60, m | 3.76, dd (3.1, 1.8) |
| 3″ | 3.93, dd (9.8, 1.3) | 3.68, dd (9.7, 3.3) | 3.94, dd (9.8, 3.2) | 3.95, dd (9.8, 3.1) | 3.70, dd (9.8, 3.4) | 3.94, dd (9.8, 3.2) | 5.08, dd (10.0, 3.2) |
| 4″ | 5.04, t (9.9) | 4.85, m | 5.04, t (9.8) | 5.07, t (10.0) | 4.95, t (9.9) | 5.07, t (9.9) | 3.59, m |
| 5″ | 4.59, d (9.9, 6.3) | 2.43, dq (12.4, 6.1) | 4.58, d (9.9, 6.3) | 4.56, dq (12.4, 6.1) | 2.43, dd (9.9, 6.2) | 4.58, dd (9.9, 6.2) | 4.45, dd (9.7, 6.2) |
| 6″ | 1.09, d (6.2) | 0.81, d (6.2) | 1.10, d (6.3) | 1.09, d (6.2) | 0.82, d (6.3) | 1.09, d (6.2) | 1.24, d (6.2) |
| 1‴ | |||||||
| 2‴ | 7.10, s | 7.11, s | 7.48, br s | 7.57, d (1.5) | 7.55, d (1.9) | 7.92, d (8.7) | 7.19, d (1.8) |
| 3‴ | 6.85, overlap | ||||||
| 4‴ | |||||||
| 5‴ | 6.84, overlap | 6.87, overlap | 6.96, d (8.4) | 6.85, overlap | 6.81, d (8.0) | ||
| 6‴ | 7.10, s | 7.11, s | 7.46, dd (8.1, 2.0) | 7.60, dd (8.3, 1.7) | 7.66, dd (8.3, 2.0) | 7.92, d (8.7) | 7.08, dd (8.2, 1.8) |
| 7‴ | 7.68, d (15.9) | ||||||
| 8‴ | 6.41, d (15.9) | ||||||
| 9‴ | |||||||
| OCH3 | 3.90, s | 3.96, s | 3.90, s |
| No. | 1 a | 2 a | 3 a | 4 b | 5 a | 6 b | 7 c |
|---|---|---|---|---|---|---|---|
| 2 | 84.0 | 82.0 | 84.0 | 84.0 | 82.0 | 84.0 | 83.9 |
| 3 | 78.6 | 74.7 | 78.7 | 78.9 | 74.7 | 78.8 | 78.9 |
| 4 | 196.1 | 193.6 | 196.1 | 196.1 | 193.8 | 196.1 | 195.8 |
| 5 | 165.6 | 164.6 | 165.6 | 164.2 | 164.2 | 165.6 | 165.6 |
| 6 | 97.5 | 96.4 | 97.5 | 97.5 | 97.5 | 97.5 | 97.5 |
| 7 | 168.9 | 169.5 | 168.9 | 168.8 | 169.2 | 168.8 | 168.8 |
| 8 | 96.4 | 97.6 | 96.3 | 96.3 | 96.3 | 96.3 | 96.3 |
| 9 | 164.2 | 166.4 | 164.2 | 165.6 | 166.3 | 164.2 | 164.2 |
| 10 | 102.4 | 101.6 | 102.5 | 102.5 | 101.6 | 102.5 | 102.5 |
| 1′ | 129.2 | 129.2 | 129.2 | 129.2 | 128.8 | 129.2 | 129.2 |
| 2′ | 120.5 | 118.9 | 115.5 | 115.5 | 118.9 | 115.6 | 115.4 |
| 3′ | 116.3 | 116.2 | 146.1 | 146.6 | 146.6 | 146.6 | 146.6 |
| 4′ | 147.5 | 146.6 | 147.5 | 147.5 | 146.6 | 147.5 | 147.4 |
| 5′ | 146.6 | 146.3 | 116.3 | 116.3 | 116.1 | 116.4 | 116.3 |
| 6′ | 115.5 | 115.2 | 120.5 | 120.5 | 114.9 | 120.6 | 120.5 |
| 1″ | 102.0 | 99.2 | 102.1 | 102.2 | 99.4 | 102.2 | 102.1 |
| 2″ | 71.9 | 72.1 | 71.9 | 71.9 | 72.2 | 71.9 | 69.7 |
| 3″ | 70.4 | 70.3 | 70.4 | 70.3 | 70.2 | 70.4 | 75.4 |
| 4″ | 75.5 | 74.9 | 75.5 | 75.7 | 74.9 | 75.5 | 71.3 |
| 5″ | 68.5 | 67.9 | 68.4 | 68.6 | 68.0 | 68.6 | 70.7 |
| 6″ | 17.7 | 17.7 | 17.8 | 17.8 | 17.7 | 17.8 | 17.9 |
| 1‴ | 121.5 | 121.4 | 122.6 | 122.6 | 122.4 | 122.3 | 127.8 |
| 2‴ | 110.2 | 110.6 | 117.6 | 113.7 | 113.8 | 132.9 | 111.7 |
| 3‴ | 146.5 | 146.3 | 146.6 | 148.7 | 148.7 | 116.2 | 149.3 |
| 4‴ | 139.8 | 139.8 | 151.8 | 152.9 | 152.9 | 163.6 | 150.6 |
| 5‴ | 146.5 | 146.3 | 115.9 | 115.9 | 116.2 | 116.2 | 116.5 |
| 6‴ | 110.2 | 110.6 | 123.8 | 125.3 | 125.9 | 132.9 | 124.1 |
| 7‴ | 168.2 | 168.1 | 168.1 | 167.9 | 168.0 | 167.9 | 146.9 |
| 8‴ | 115.8 | ||||||
| 9‴ | 168.9 | ||||||
| OCH3 | 56.5 | 56.7 | 56.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-J.; Park, E.-J.; Lee, B.-W.; Cho, H.-M.; Pham, T.-L.-G.; Hoang, Q.-H.; Pan, C.-H.; Oh, W.-K. Flavanonol Glycosides from the Stems of Myrsine seguinii and Their Neuroprotective Activities. Pharmaceuticals 2021, 14, 911. https://doi.org/10.3390/ph14090911
Lee H-J, Park E-J, Lee B-W, Cho H-M, Pham T-L-G, Hoang Q-H, Pan C-H, Oh W-K. Flavanonol Glycosides from the Stems of Myrsine seguinii and Their Neuroprotective Activities. Pharmaceuticals. 2021; 14(9):911. https://doi.org/10.3390/ph14090911
Chicago/Turabian StyleLee, Hee-Ju, Eun-Jin Park, Ba-Wool Lee, Hyo-Moon Cho, Thi-Linh-Giang Pham, Quynh-Hoa Hoang, Cheol-Ho Pan, and Won-Keun Oh. 2021. "Flavanonol Glycosides from the Stems of Myrsine seguinii and Their Neuroprotective Activities" Pharmaceuticals 14, no. 9: 911. https://doi.org/10.3390/ph14090911
APA StyleLee, H.-J., Park, E.-J., Lee, B.-W., Cho, H.-M., Pham, T.-L.-G., Hoang, Q.-H., Pan, C.-H., & Oh, W.-K. (2021). Flavanonol Glycosides from the Stems of Myrsine seguinii and Their Neuroprotective Activities. Pharmaceuticals, 14(9), 911. https://doi.org/10.3390/ph14090911