
In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation





Abstract
:1. Introduction
2. Results and Discussion
2.1. Validation of Docking Method by Re-Docking and Cross-Docking
Analysis of Virtual Screening Accuracy
2.2. Selection of Hits after Consensus Approach
2.3. Pharmacokinetic Analysis
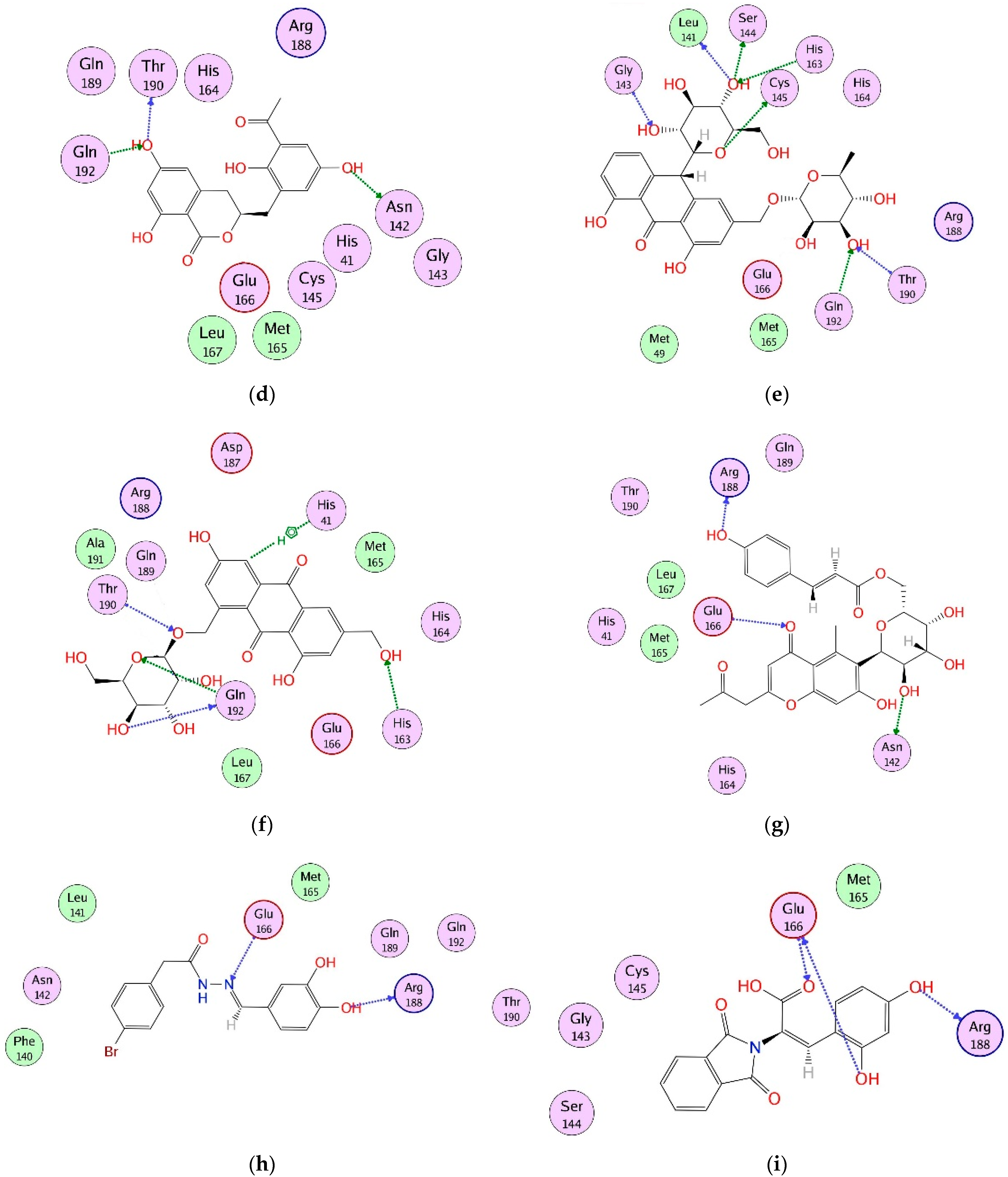
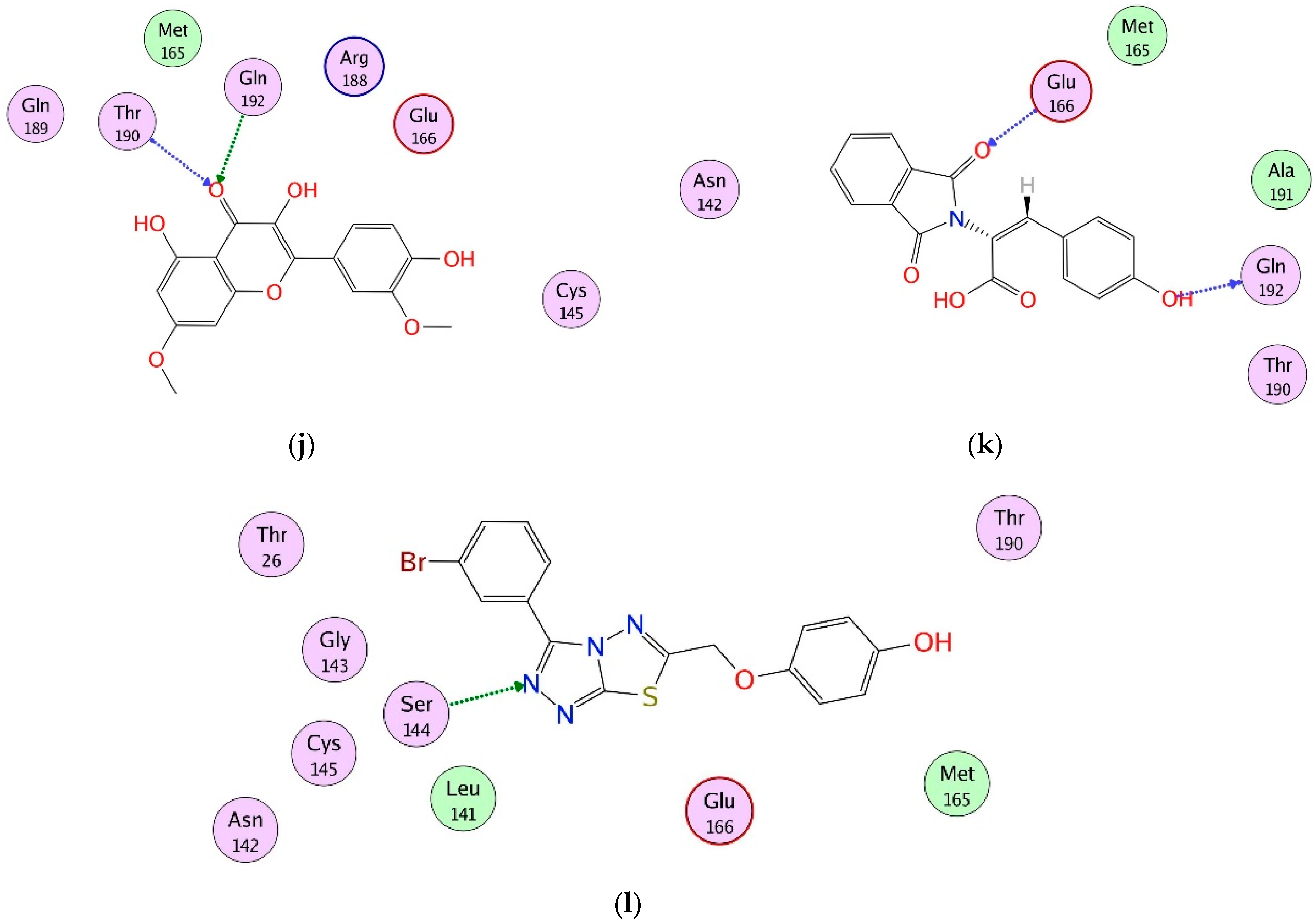
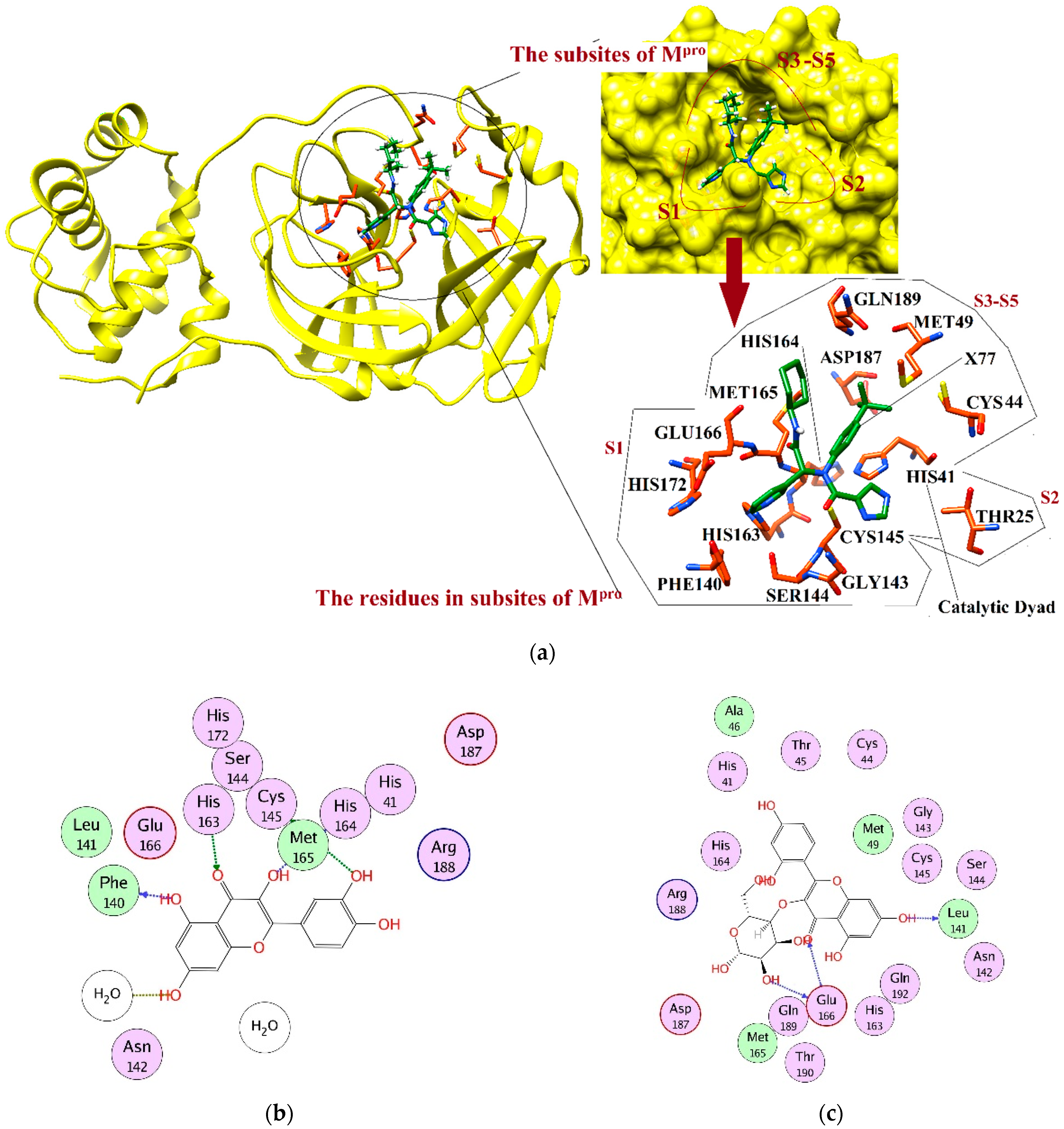
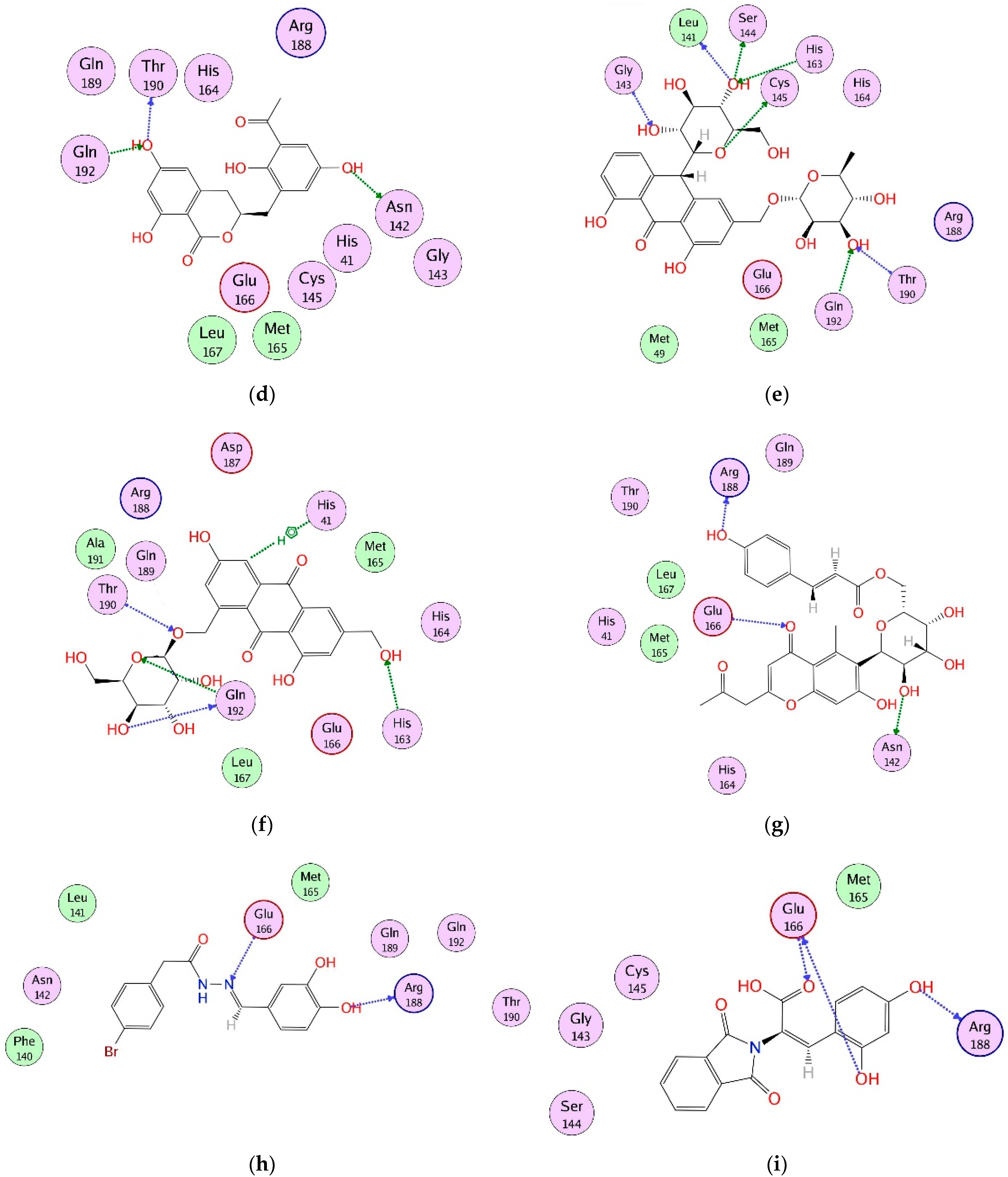
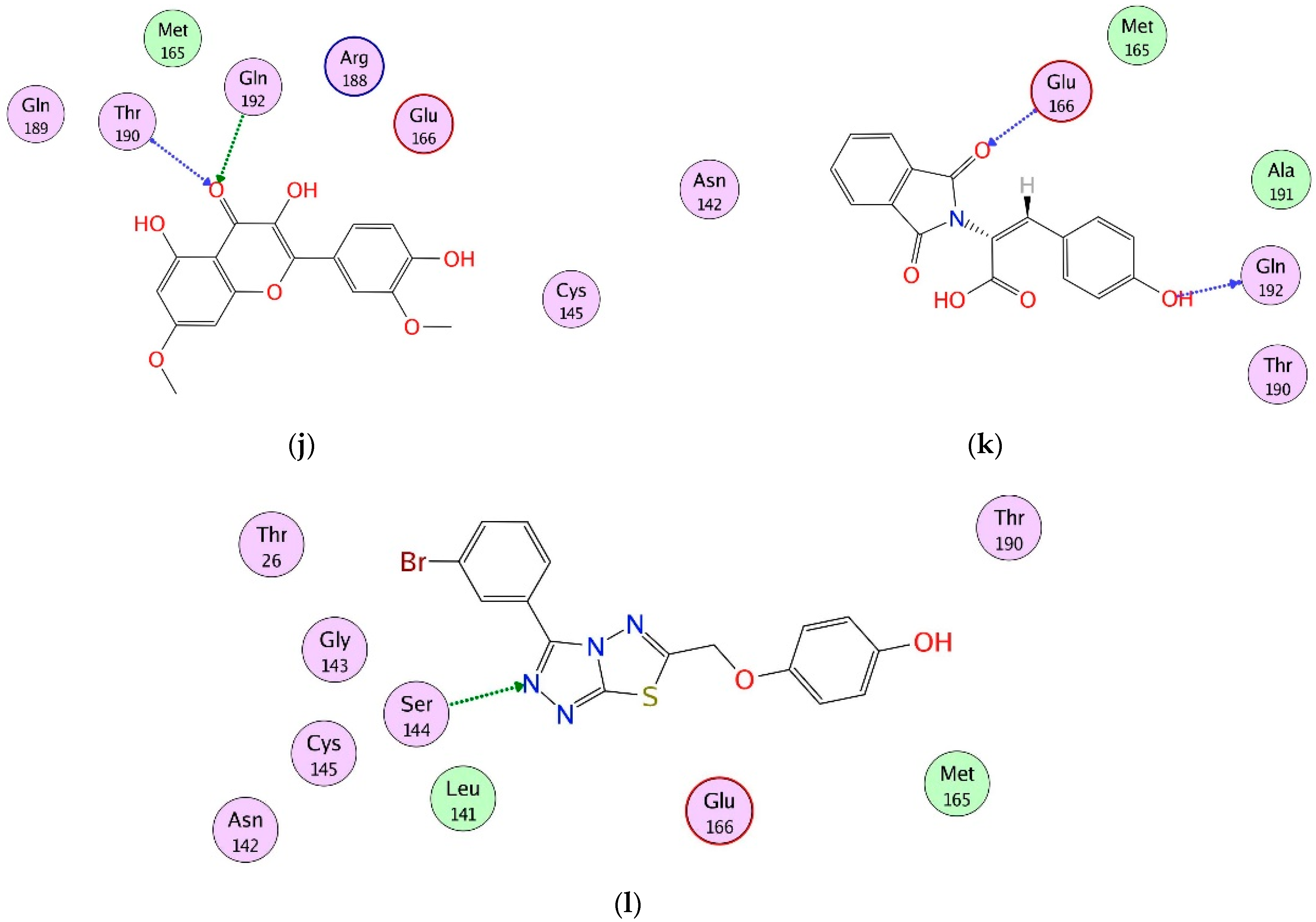
2.4. Interaction Analysis
2.5. Molecular Dynamic Simulation
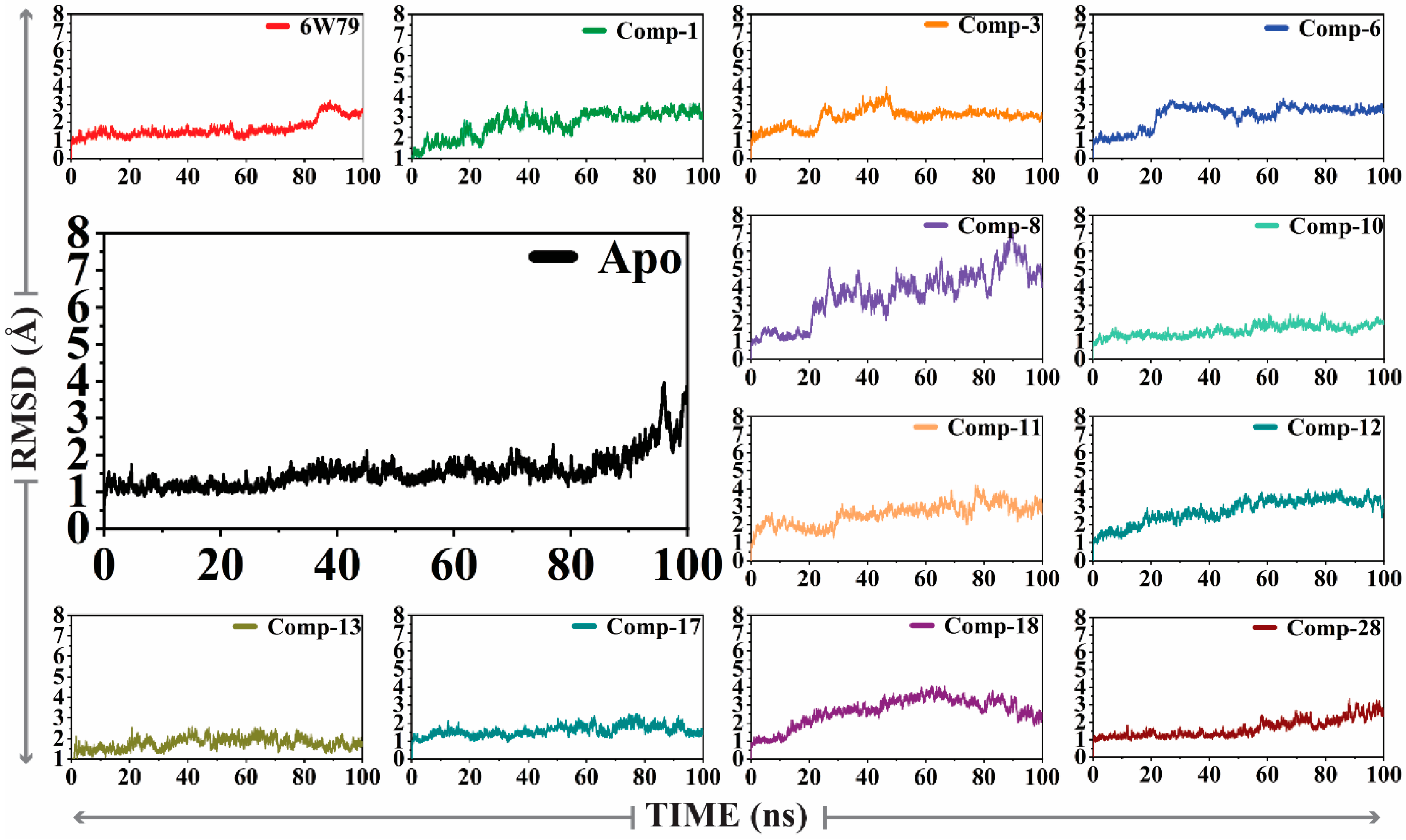
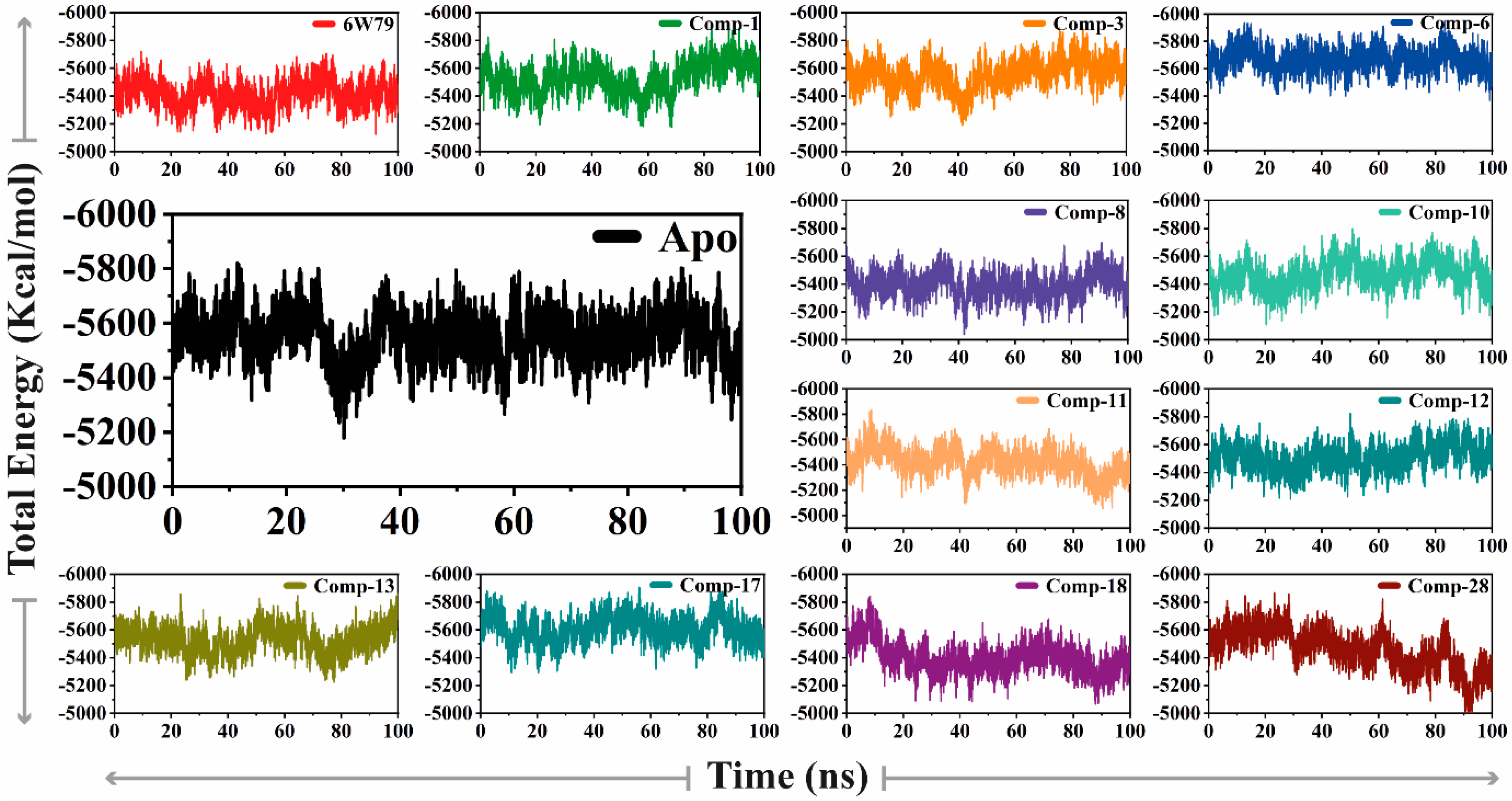
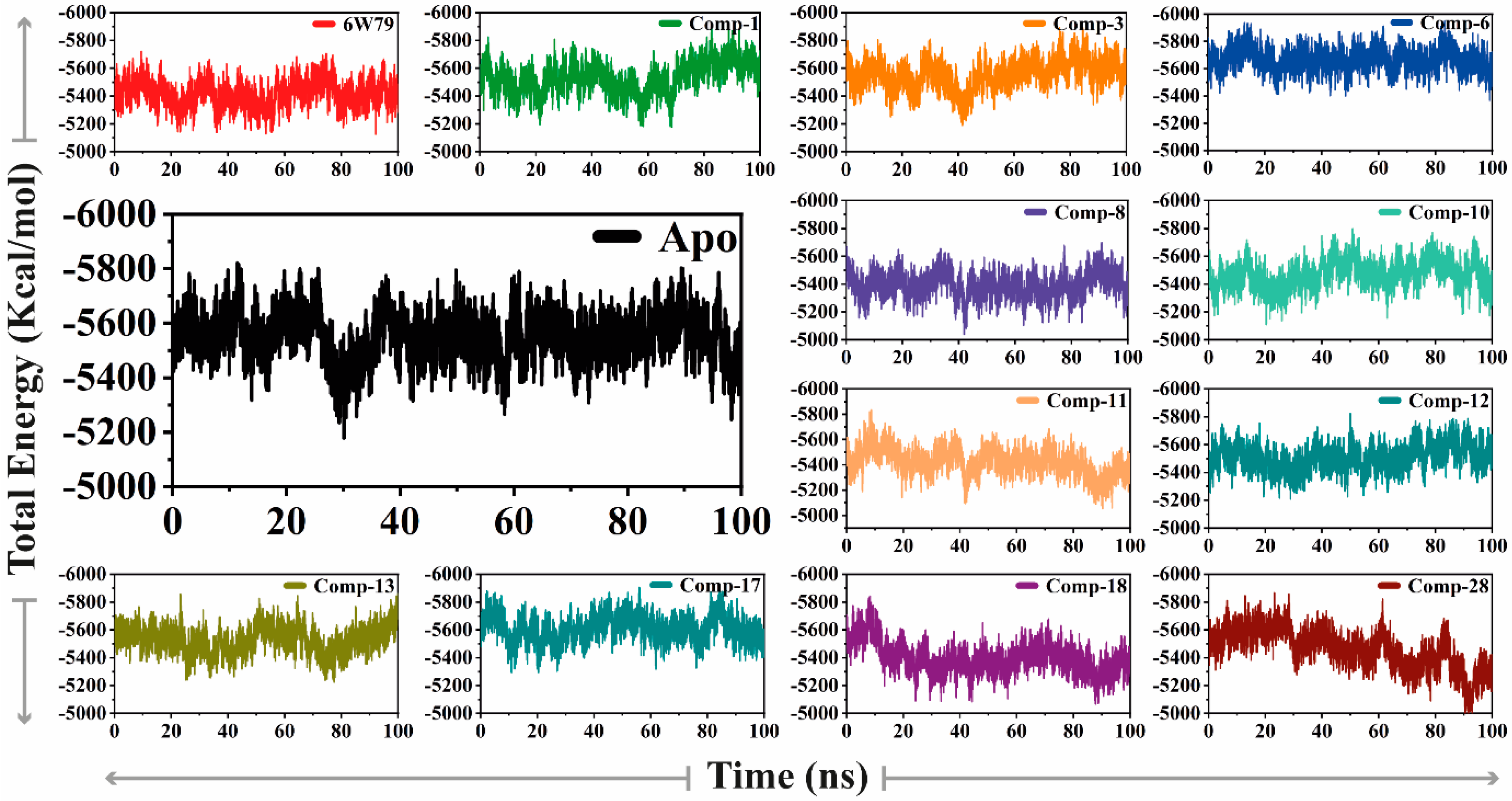
2.5.1. Convergence of Mpro Free and Inhibited States
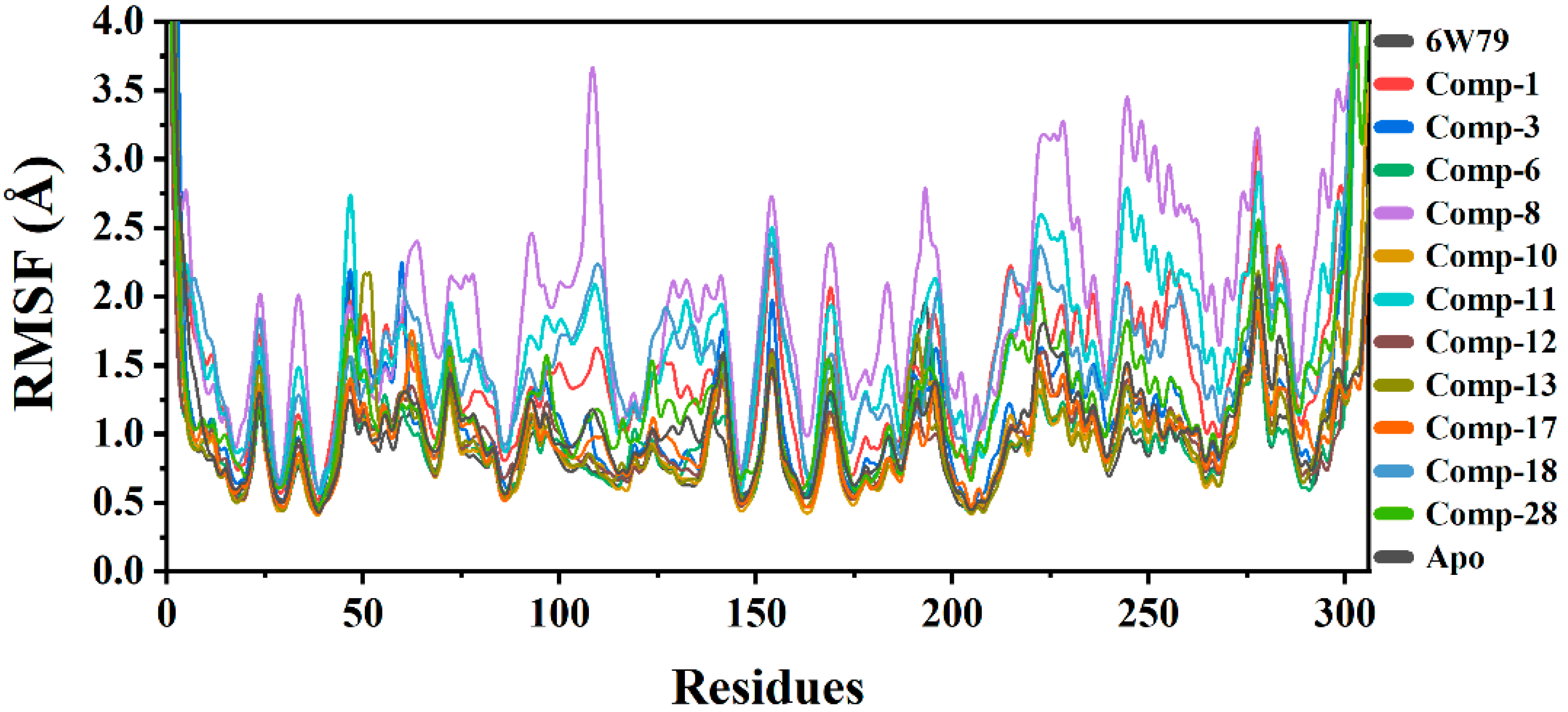
2.5.2. Root Mean Square Fluctuation (RMSF)
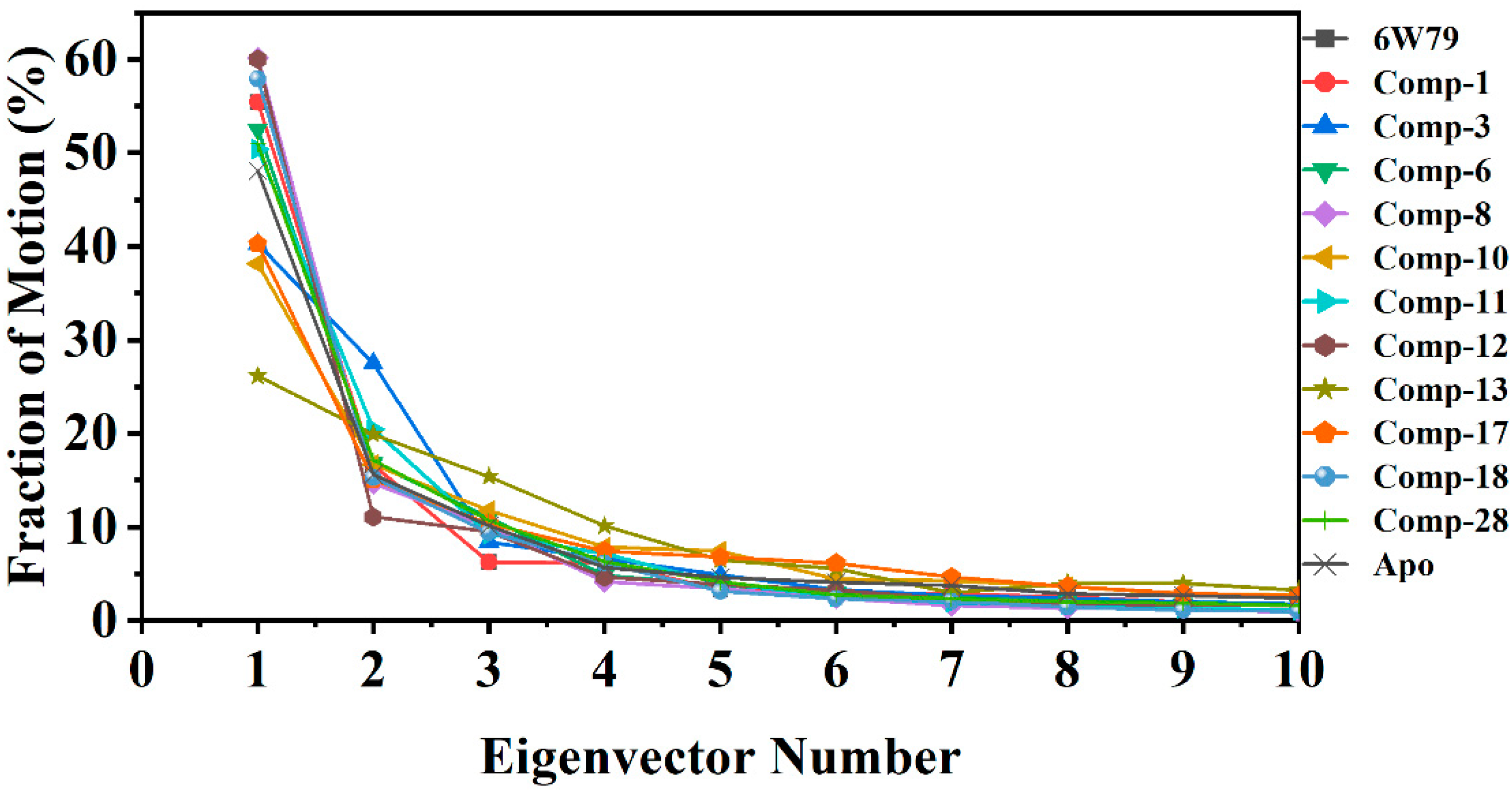
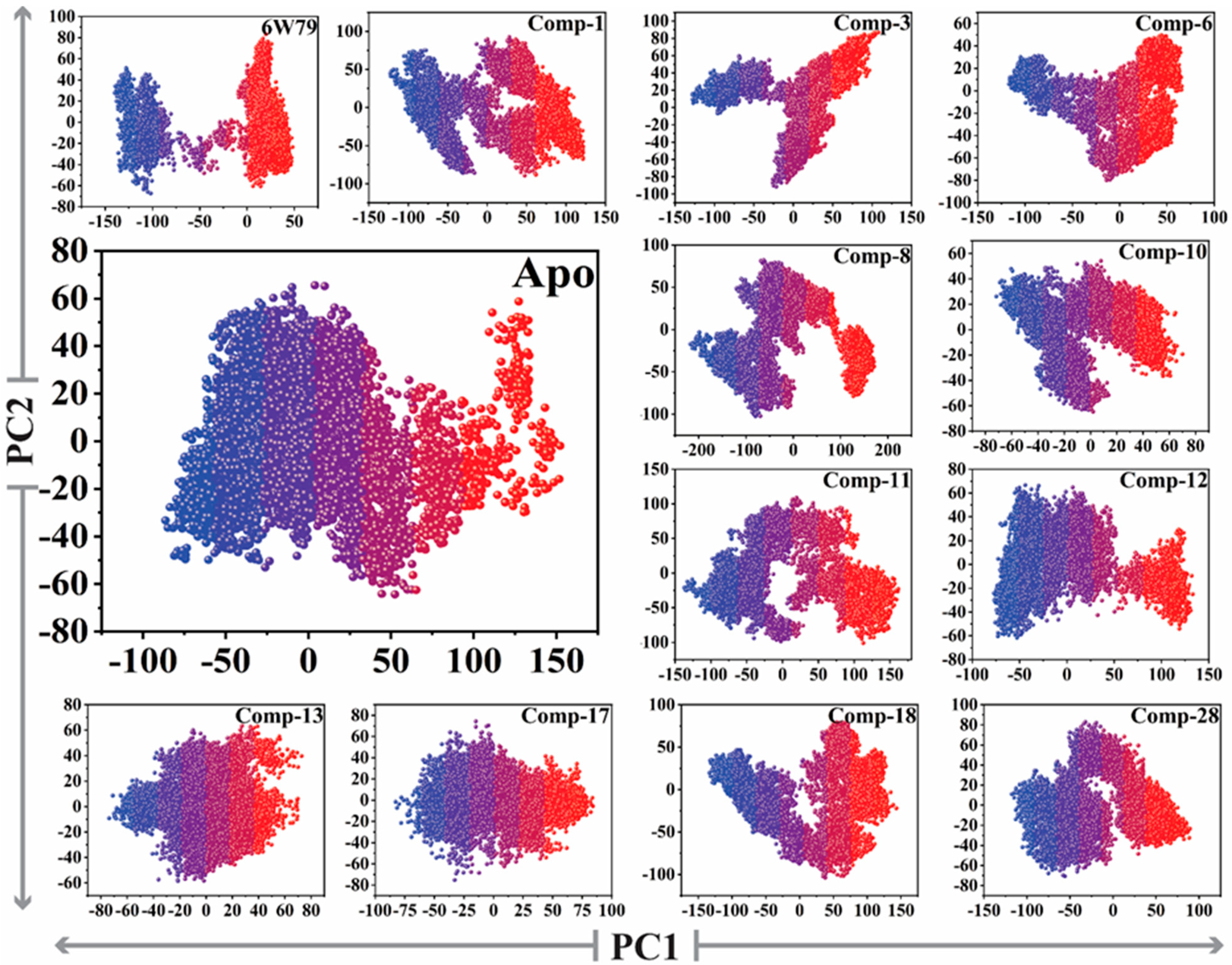
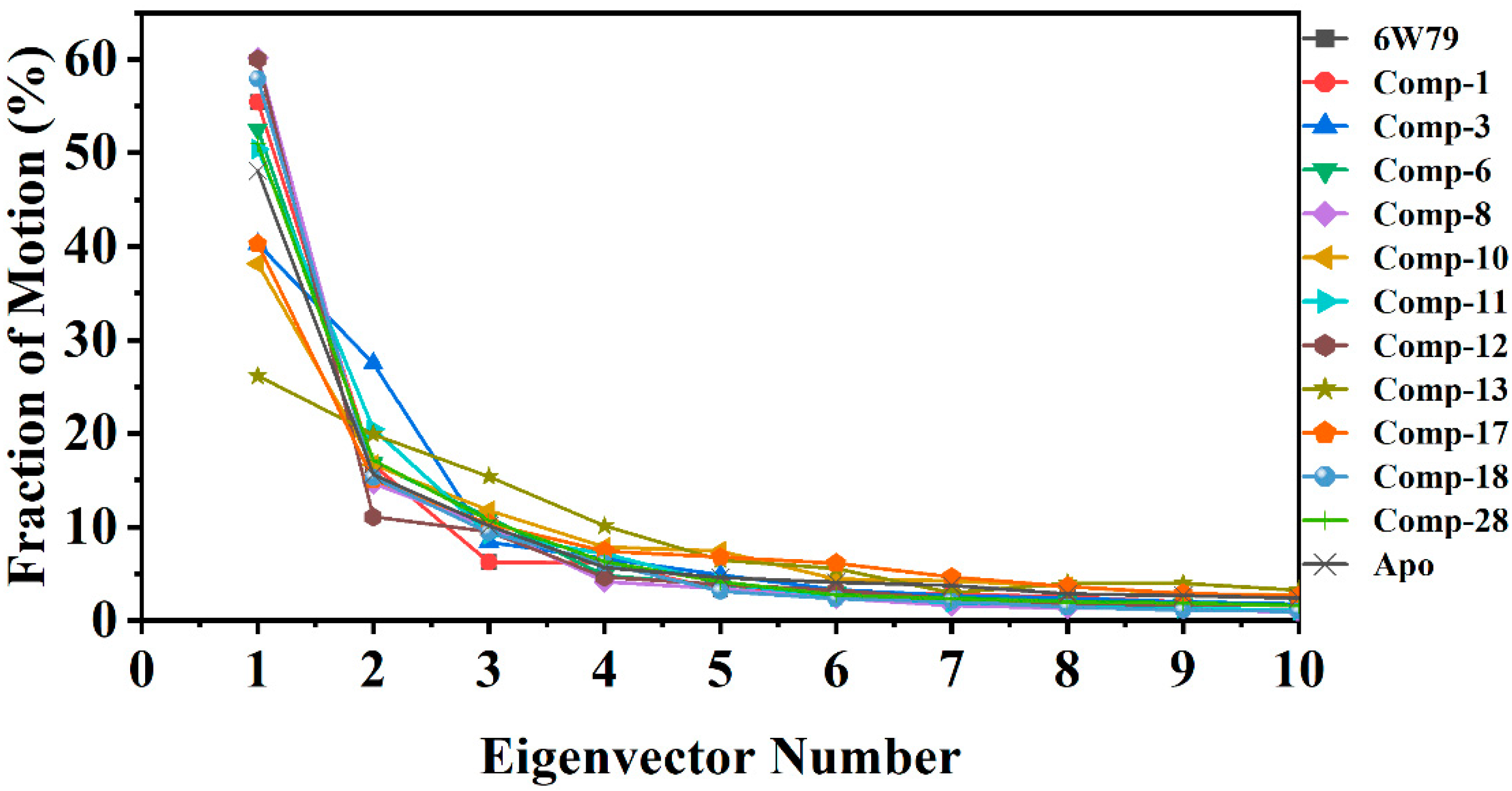
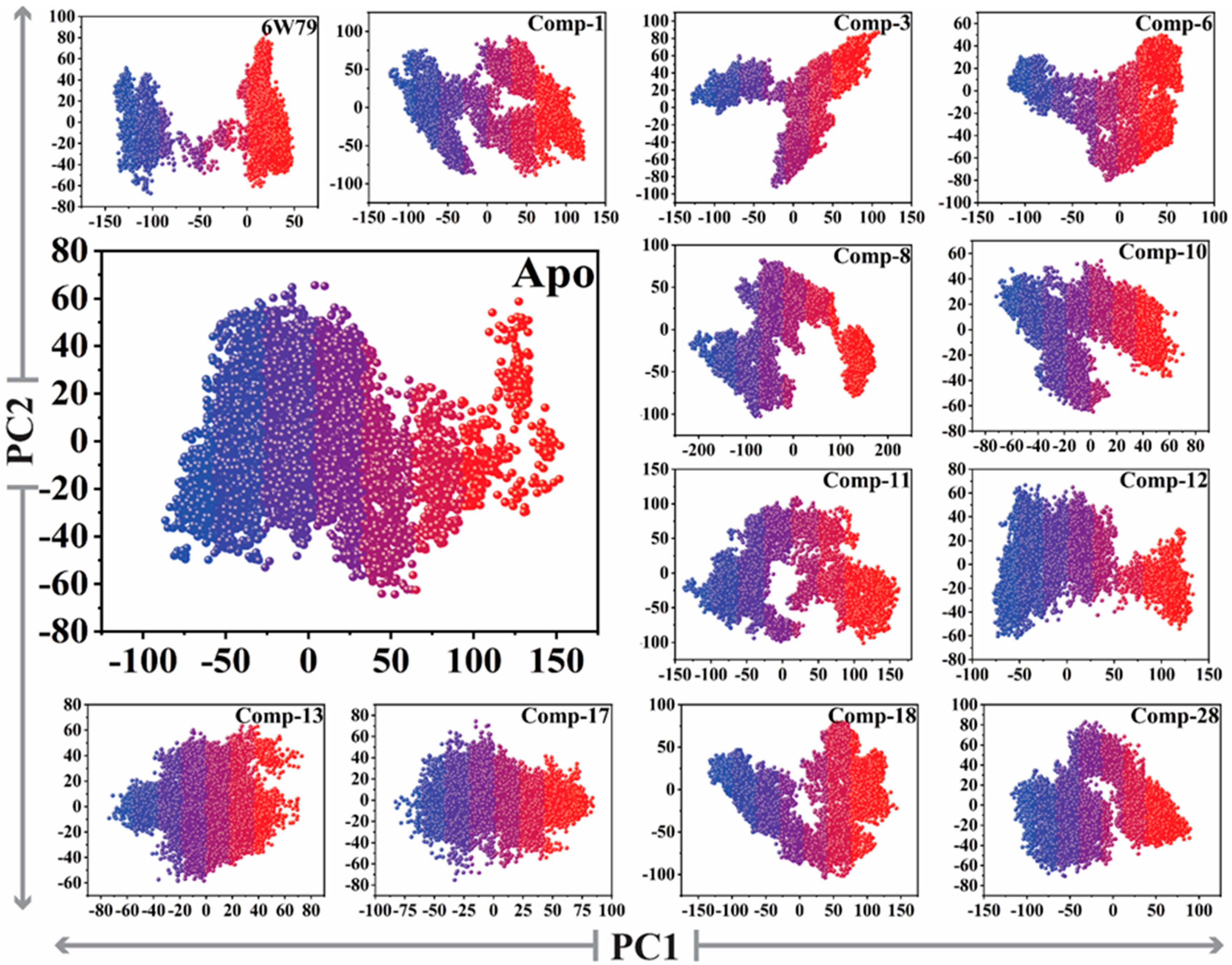
2.5.3. Protein Motions and Trajectories Clustering
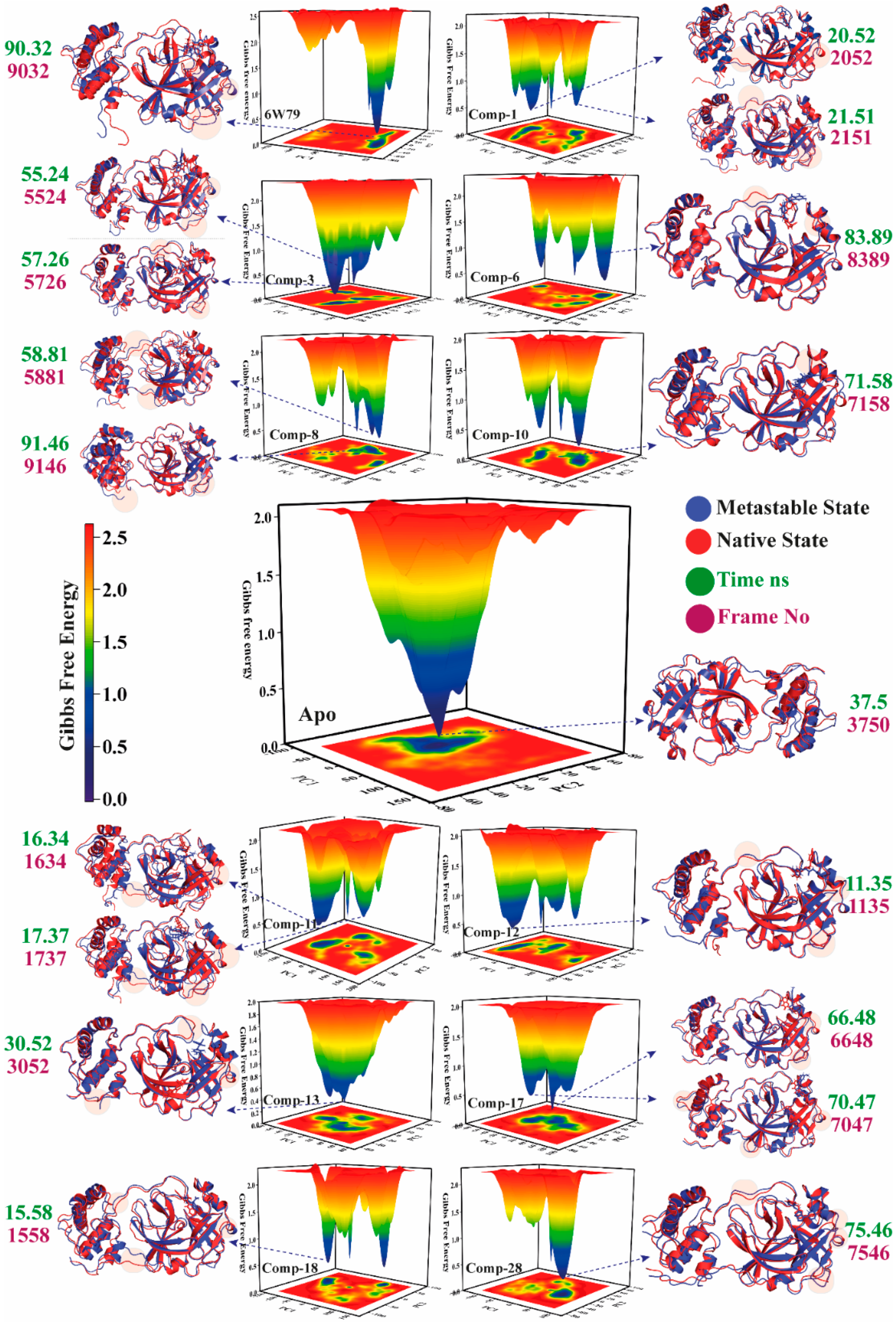
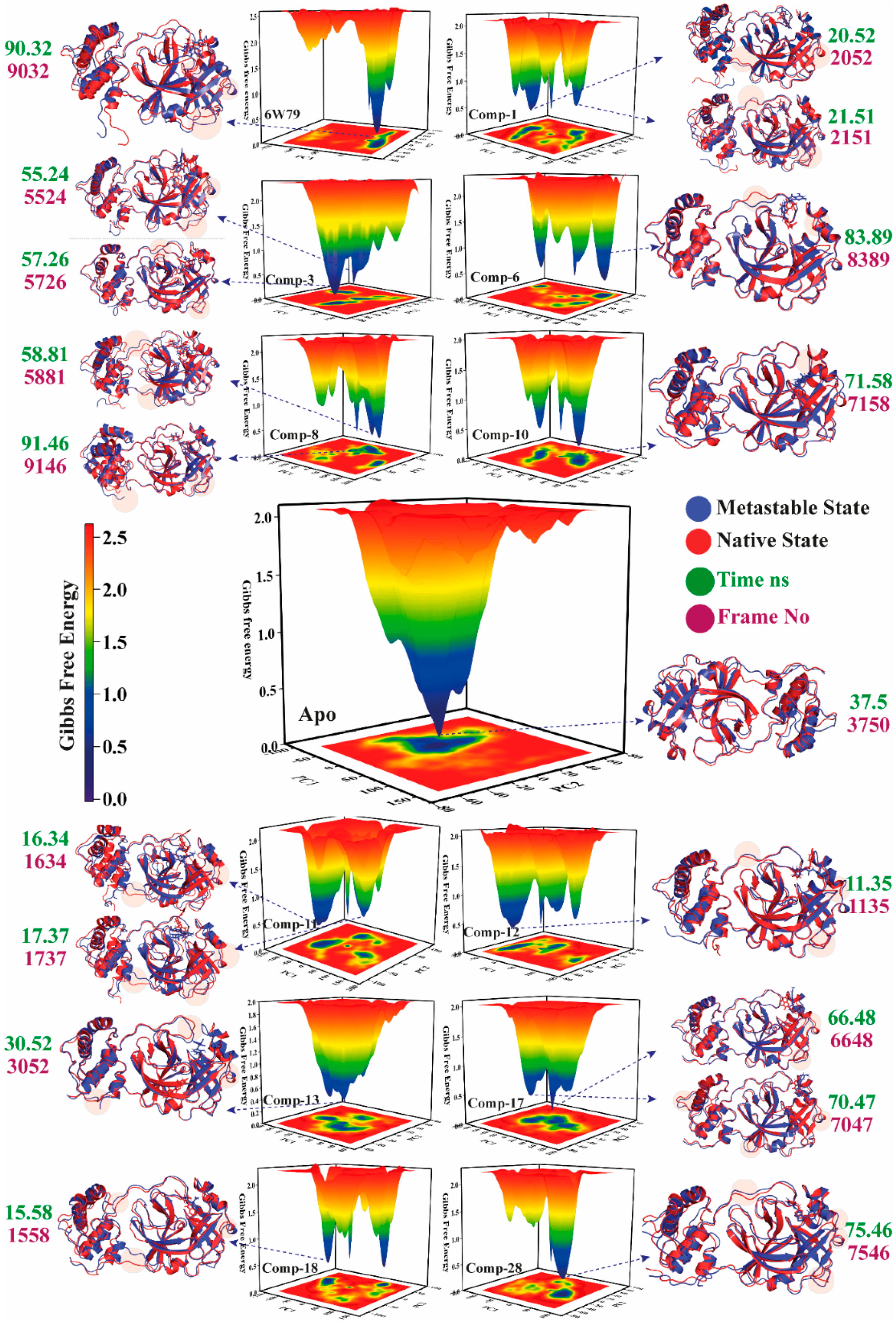
2.5.4. Metastable to Native State Transition Pathway
2.5.5. Dynamic Cross-Correlated Map Analysis
2.5.6. Binding Free Energy Calculations
3. Materials and Methods
3.1. Preparation of Protein’s Structures for Docking
Preparation of Compound Database for Docking
3.2. Structure-Based Screening by Molecular Docking
Analysis Measures and Conformational Sampling after Virtual Screening
3.3. Prediction of Pharmacokinetic Properties
3.4. Molecular Dynamic Simulation
3.4.1. Post-Dynamic Evaluation
3.4.2. MD Trajectories Unsupervised Clustering and Free Energy Landscape
3.4.3. Dynamic Cross-Correlation Analysis (DCC)
3.4.4. MM/GBSA Free Energy Calculation
3.4.5. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 30 June 2021).
- Monitoring Knowledge, Risk Perceptions, Preventive Behaviours, and Public Trust in the Current Coronavirus Outbreak in Georgia Analytical Report of the First, Second and Third Wave Studies, WHO Report. May 2020, pp. 1–61. Available online: www.unicef.org/georgia/media/4736/file/COVID-19-Study-Analytical-Report-1-st-2nd-and-3rd-waves-Eng.pdf (accessed on 15 May 2021).
- Taboada, M.; González, M.; Alvarez, A.; Eiras, M.; Costa, J.; Álvarez, J.; Seoane-Pillado, T. First, second and third wave of COVID-19. What have we changed in the ICU management of these patients? J. Infect. 2021, 82, e14–e15. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [Green Version]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Inf. 2021, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Asghari, A.; Naseri, M.; Safari, H.; Saboory, E.; Parsamanesh, N. The novel insight of SARS-CoV-2 molecular biology and pathogenesis and therapeutic options. DNA Cell Biol. 2020, 39, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, function, and evolution of Coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Artika, I.M.; Dewantari, A.K.; Wiyatno, A. Molecular biology of coronaviruses: Current knowledge. Heliyon 2020, 6, e04743. [Google Scholar] [CrossRef]
- Alsobaie, S. Understanding the molecular biology of SARS-CoV2 and the COVID-19 pandemic: A Review. Infect. Drug Res. 2021, 14, 2259–2268. [Google Scholar] [CrossRef]
- Ali, M.J.; Hanif, M.; Haider, M.A.; Ahmed, M.U.; Sundas, F.N.U.; Hirani, A.; Khan, I.A.; Anis, K.; Karim, A.H. Treatment options for COVID-19: A review. Front. Med. 2020, 7, 480. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19—Final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Chloroquine and hydroxychloroquine in covid-19: Use of these drugs is premature and potentially harmful. BMJ 2020, 369, m1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R. Chloroquine and hydroxychloroquine for COVID-19: Perspectives on their failure in repurposing. J. Clin. Pharm. Ther. 2021, 46, 17–27. [Google Scholar] [CrossRef]
- Samaee, H.; Mohsenzadegan, M.; Ala, S.; Maroufi, S.S.; Moradimaj, P. Tocilizumab for treatment patients with COVID-19: Recommended medication for novel disease. Int. Immunopharmacol. 2020, 89, 107018. [Google Scholar] [CrossRef]
- Rosas, I.O.; Bräu, N.; Waters, M.; Go, R.C.; Hunter, B.D.; Bhagani, S.; Skiest, D.; Aziz, M.S.; Cooper, N.; Douglas, I.S.; et al. Tocilizumab inh patients with severe Covid-19 pneumonia. N. Engl. J. Med. 2021, 384, 1503–1516. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of Lopinavir–Ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, L. 15 Drugs Being Tested to Treat COVID-19 and How They Would Work. Nat. Med. 2020. Available online: www.nature.com/articles/d41591-020-00019-9 (accessed on 20 May 2021).
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic treatments for Coronavirus disease 2019 (COVID-19), A review. JAMA 2020, 323, 1824–1836. [Google Scholar] [PubMed]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. Anti-Coronavirus Vaccines: Past Investigations on SARS-CoV-1 and MERS-CoV, the Approved Vaccines from BioNTech/Pfizer, Moderna, Oxford/AstraZeneca and others under Development Against SARSCoV-2 Infection. Curr. Med. Chem. 2021, 28. [Google Scholar] [CrossRef] [PubMed]
- Roviello, V.; Musumeci, D.; Mokhir, A.; Roviello, G.N. Evidence of Protein Binding by a Nucleopeptide Based on a Thyminedecorated L-Diaminopropanoic Acid through CD and In Silico Studies. Curr. Med. Chem. 2021, 28. [Google Scholar] [CrossRef]
- Vicidomini, C.; Roviello, V.; Roviello, G.N. In Silico Investigation on the Interaction of Chiral Phytochemicals from Opuntia ficus-indica with SARS-CoV-2 Mpro. Symmetry 2021, 13, 1041. [Google Scholar] [CrossRef]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Wang, X.; Jochmans, D.; Neyts, J.; Młynarski, W.; et al. SARS-CoV-2 M pro inhibitors and activity based probes for patient-sample imaging. Nat. Chem. Biol. 2021, 17, 222–228. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-Ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Paasche, A.; Zipper, A.; Schäfer, S.; Ziebuhr, J.; Schirmeister, T.; Engels, B. Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochemistry 2014, 53, 5930–5946. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Maple, H.J.; Clayden, N.; Baron, A.; Stacey, C.; Felix, R. Developing degraders: Principles and perspectives on design and chemical space. MedChemComm 2019, 10, 1755–1764. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Egan, W.J.; Walters, W.P.; Murcko, M.A. Guiding molecules towards drug-likeness. Curr. Opin. Drug Disc. Dev. 2002, 5, 540–549. [Google Scholar]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Singh, P.K. Analyzing FDA-approved drugs for compliance of pharmacokinetic principles: Should there be a critical screening parameter in drug designing protocols? Exp. Opin. Drug Metab. Toxicol. 2021, 17, 351–354. [Google Scholar] [CrossRef]
- Mesecar, A. A taxonomically driven approach to development of potent, broad-spectrum inhibitors of coronavirus main protease including SARS-CoV-2 (COVID-19). Unpublished work. 2020. [Google Scholar]
- Molecular Operating Environment Version 2014.09; Chemical Computing Group: Montreal, QC, Canada, 2014.
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L., III; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Available online: https://www.unizwa.edu.om/index.php?contentid=1038 (accessed on 5 January 2021).
- Edelsbrunner, H. Weighted Alpha Shapes; Report UIUCDCS-R-92-1760; Department of Computer Science, University of Illinois, Urbana Champagne: Champaign, IL, USA, 1992. [Google Scholar]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, R.; Powell, M.J. A rapidly convergent descent method for minimization. Comput. J. 1963, 6, 163–168. [Google Scholar] [CrossRef]
- Shewchuk, J.R. An Introduction to the Conjugate Gradient Method without the Agonizing Pain; Department of Computer Science, Carnegie-Mellon University: Pittsburgh, PA, USA, 1994. [Google Scholar]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D., III; Darden, T.A.; Duke, R.E. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Sengupta, A.; Li, Z.; Song, L.F.; Li, P.; Merz, K.M., Jr. Parameterization of Monovalent Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Inf. Model. 2021, 61, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building water models: A different approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Allaire, G.; Dapogny, C.; Frey, P. A mesh evolution algorithm based on the level set method for geometry and topology optimization. Struct. Multidiscip. Optim. 2013, 48, 711–715. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef] [Green Version]
- Hunenberger, P.H. Thermostat algorithms for molecular dynamics simulations. Adv. Polym. Sci. 2005, 173, 105–149. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Altona, C.T.; Sundaralingam, M. Conformational analysis of the sugar ring in nucleosides and nucleotides. New description using the concept of pseudo rotation. J. Am. Chem. Soc. 1972, 94, 8205–8212. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E.J. UCSF Chimera- A visualization system for exploratory research and analysis. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC.: New York, NY, USA; Available online: https://pymol.org/ (accessed on 15 March 2021).
- Origin (Pro), OriginLab Corporation, Northampton, Massachusetts, United States of America. 2021. Available online: www.originlab.com (accessed on 15 March 2021).
- Williams, T.; Kelley, C. Gnuplot 5.4, An Interactive Plotting Program, User Manual. 2020. Available online: www.gnuplot.info (accessed on 15 March 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex Name | X Coordinates | Y Coordinates | Frame No | Time ns |
|---|---|---|---|---|
| Apo–Mpro | 1.859 | −0.699 | 3750 | 37.5 |
| Mpro–X77 | 29.197 | −20.599 | 9032 | 90.32 |
| Mpro–1 | −73.840 | 3.321 | 2052 | 20.52 |
| −71.395 | 1.503 | 2151 | 21.51 | |
| Mpro–3 | 1.713 | −50.468 | 5524 | 55.24 |
| 6.430 | −46.903 | 5726 | 57.26 | |
| Mpro–6 | 34.503 | 34.145 | 8389 | 83.89 |
| Mpro–8 | 7.810 | 45.191 | 5881 | 58.81 |
| 136.017 | −19.440 | 9146 | 91.46 | |
| Mpro–10 | 28.891 | 3.061 | 7158 | 71.58 |
| Mpro–11 | −89.371 | −31.716 | 1634 | 16.34 |
| −86.385 | −25.418 | 1737 | 17.37 | |
| Mpro–12 | −54.007 | −18.018 | 1135 | 11.35 |
| Mpro–13 | −29.833 | 3.606 | 3052 | 30.52 |
| Mpro–17 | 27.298 | −4.897 | 6648 | 66.48 |
| 33.972 | −6.394 | 7047 | 70.47 | |
| Mpro–18 | −91.981 | 0.876 | 1558 | 15.58 |
| Mpro–28 | 37.636 | −1.614 | 7546 | 75.46 |
| Complex Name | Kcal/mol | ||||
|---|---|---|---|---|---|
| ∆VDW | ∆EEL | ∆EGB | ∆SASA | ∆G TOTAL | |
| Mpro–X77 | −46.7396 | −7.2011 | 22.3537 | −5.6612 | −37.2483 |
| Mpro–1 | −27.9473 | −11.8236 | 20.6925 | −3.7064 | −22.7848 |
| Mpro–3 | −36.3657 | −28.7266 | 43.2732 | −5.0843 | −26.9034 |
| Mpro–6 | −34.5680 | −15.4295 | 31.3375 | −4.2467 | −22.9067 |
| Mpro–8 | −41.0277 | −15.6216 | 36.141 | −6.0766 | −26.5848 |
| Mpro–10 | −10.8292 | −5.7753 | 11.5464 | −1.4387 | −6.4968 |
| Mpro–11 | −39.9829 | −15.1839 | 25.9731 | −4.4549 | −33.6485 |
| Mpro–12 | −25.5687 | −13.1445 | 22.4730 | −3.8878 | −20.1279 |
| Mpro–13 | −15.3599 | −5.7322 | 13.8263 | −2.135 | −9.4012 |
| Mpro–17 | −25.1406 | −18.6810 | 25.3125 | −3.5204 | −22.0295 |
| Mpro–18 | −33.2958 | −9.3268 | 22.2269 | −4.3144 | −24.7101 |
| Mpro–28 | −41.1238 | −7.7362 | 19.6643 | −4.4767 | −33.6723 |
| S. No. | System Composition (Complexes) | Temperature (K) | Force Fields | Water Model | Time (ns) |
|---|---|---|---|---|---|
| 1 | Full length apo–Mpro | 300 | FF19SB | Octahedral OPC | 100 |
| 2 | Mpro–X77 (6W79) | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 3 | Mpro–1 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 4 | Mpro–3 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 5 | Mpro–6 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 6 | Mpro–8 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 7 | Mpro–10 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 8 | Mpro–11 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 9 | Mpro–12 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 10 | Mpro–13 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 11 | Mpro–17 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 12 | Mpro–18 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
| 13 | Mpro–28 | 300 | FF19SB+Gaff2 | Octahedral OPC | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halim, S.A.; Waqas, M.; Khan, A.; Al-Harrasi, A. In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation. Pharmaceuticals 2021, 14, 896. https://doi.org/10.3390/ph14090896
Halim SA, Waqas M, Khan A, Al-Harrasi A. In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation. Pharmaceuticals. 2021; 14(9):896. https://doi.org/10.3390/ph14090896
Chicago/Turabian StyleHalim, Sobia Ahsan, Muhammad Waqas, Ajmal Khan, and Ahmed Al-Harrasi. 2021. "In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation" Pharmaceuticals 14, no. 9: 896. https://doi.org/10.3390/ph14090896
APA StyleHalim, S. A., Waqas, M., Khan, A., & Al-Harrasi, A. (2021). In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation. Pharmaceuticals, 14(9), 896. https://doi.org/10.3390/ph14090896