
Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds

 ,
,  , ,
, ,  ,
,  ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
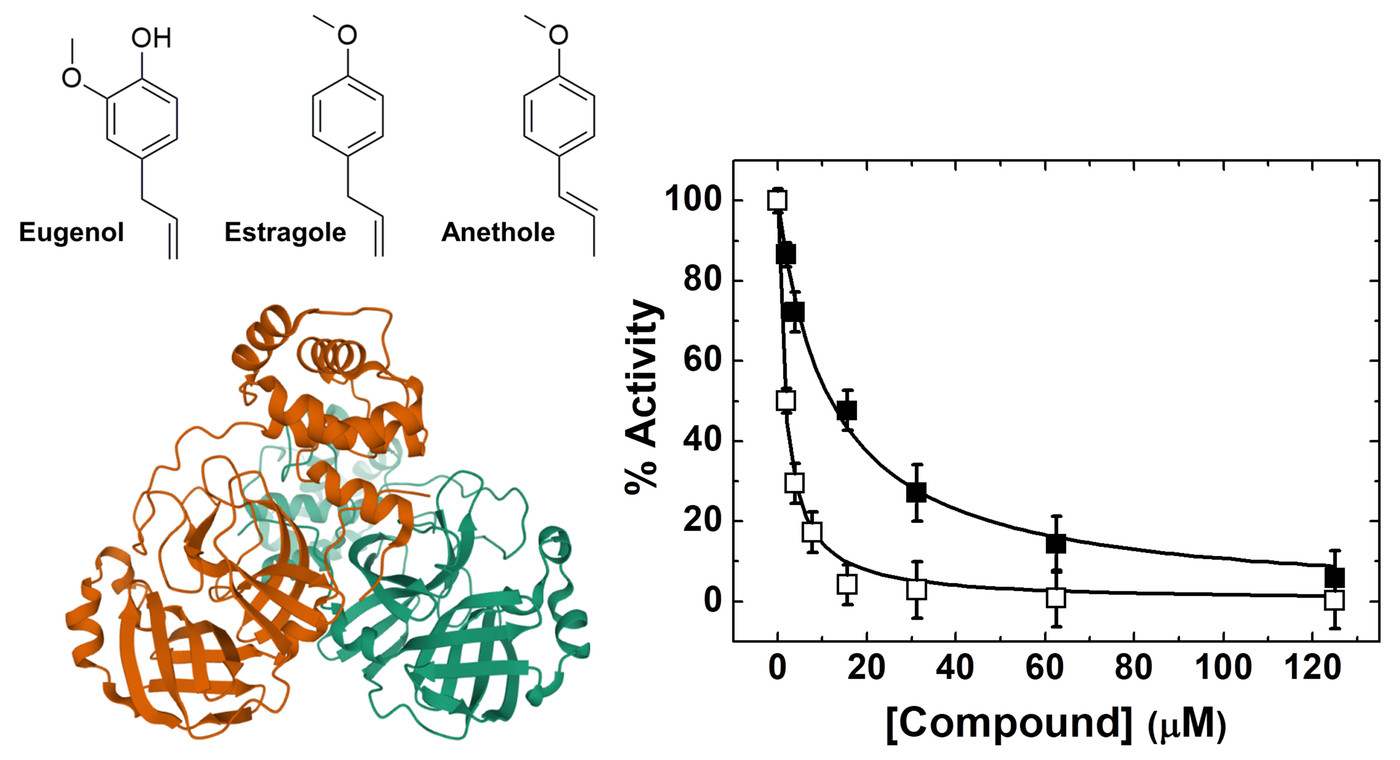
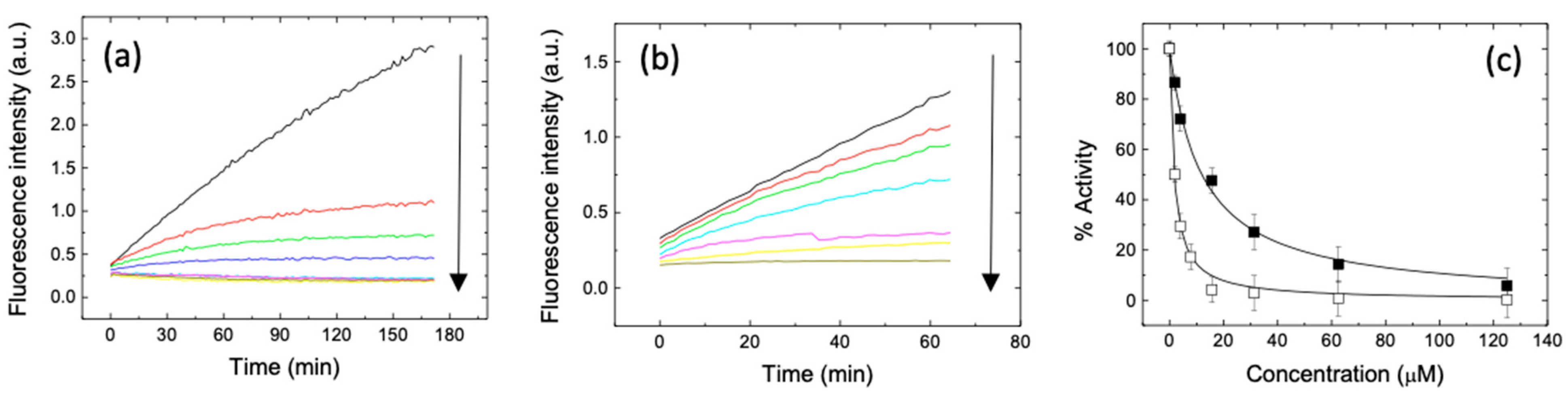
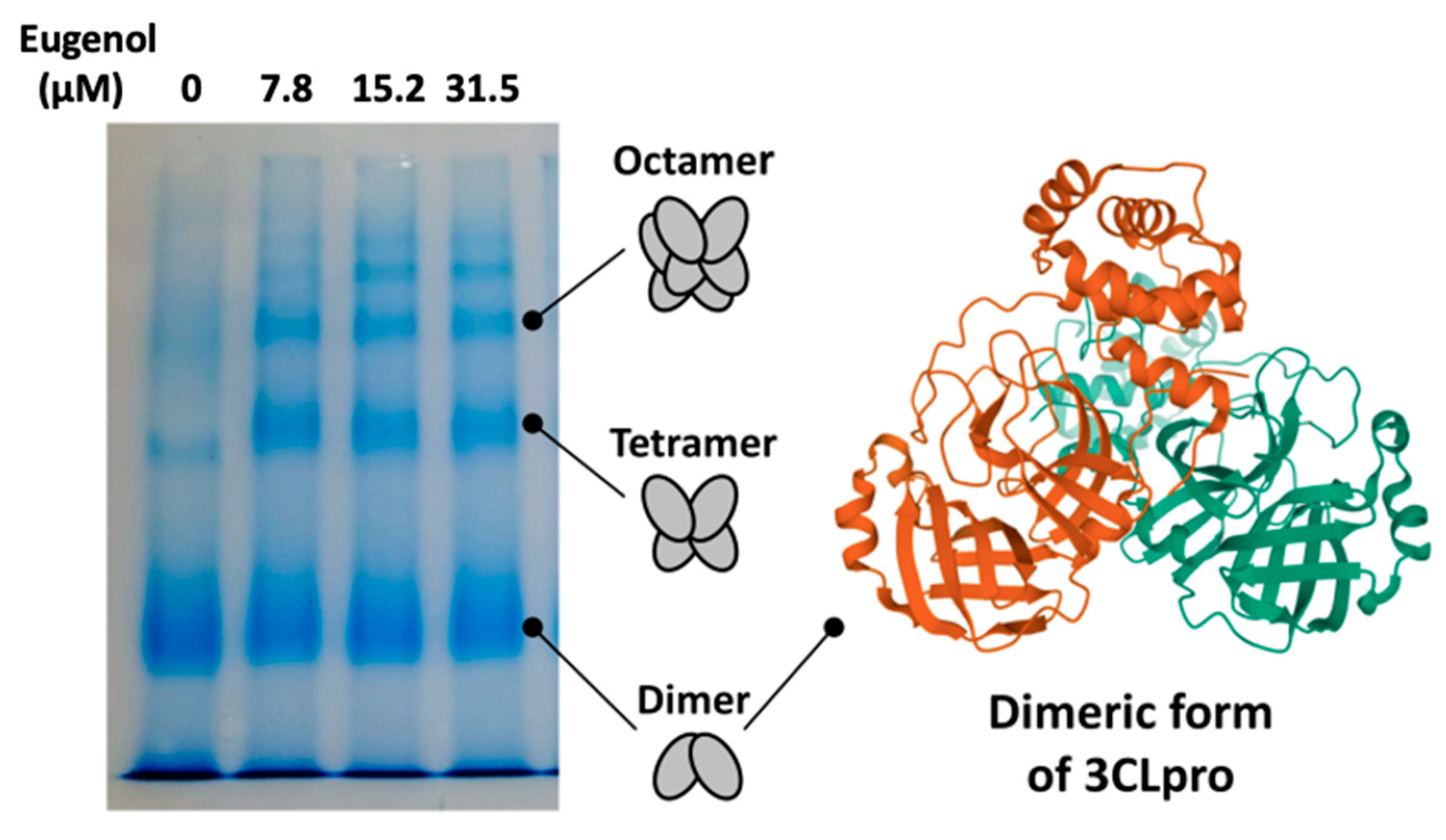
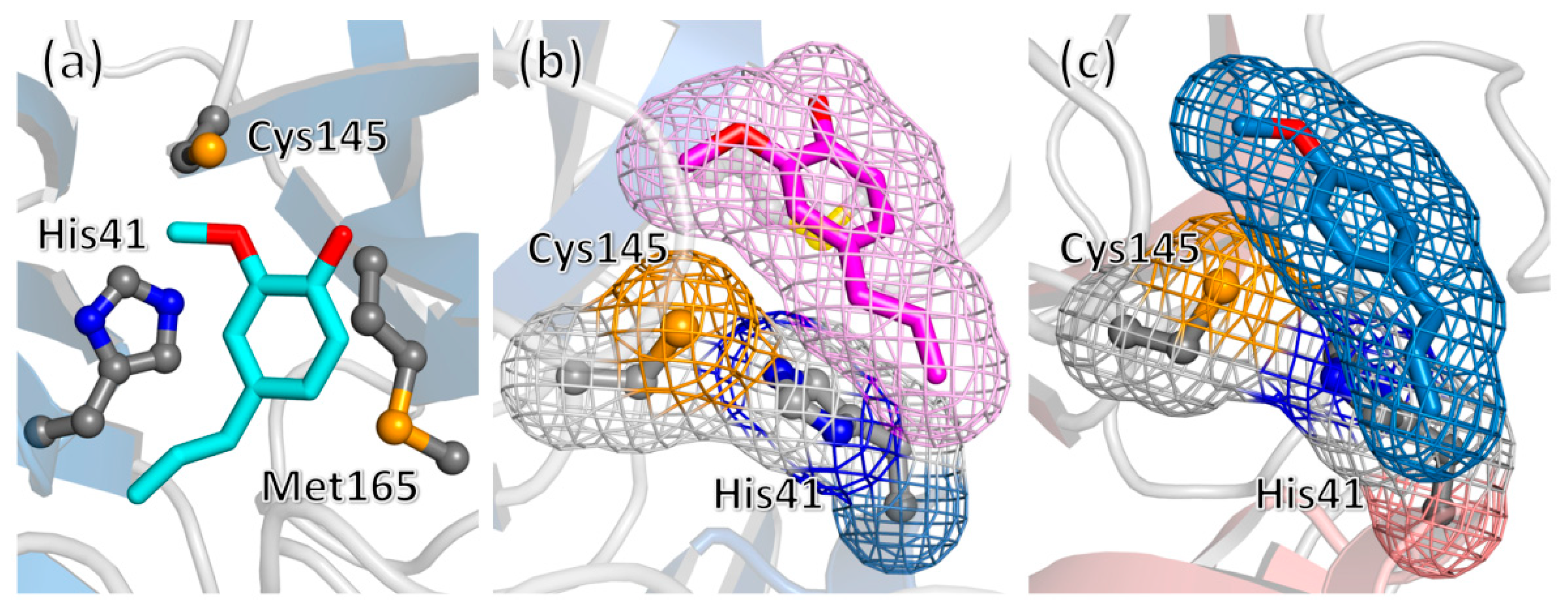
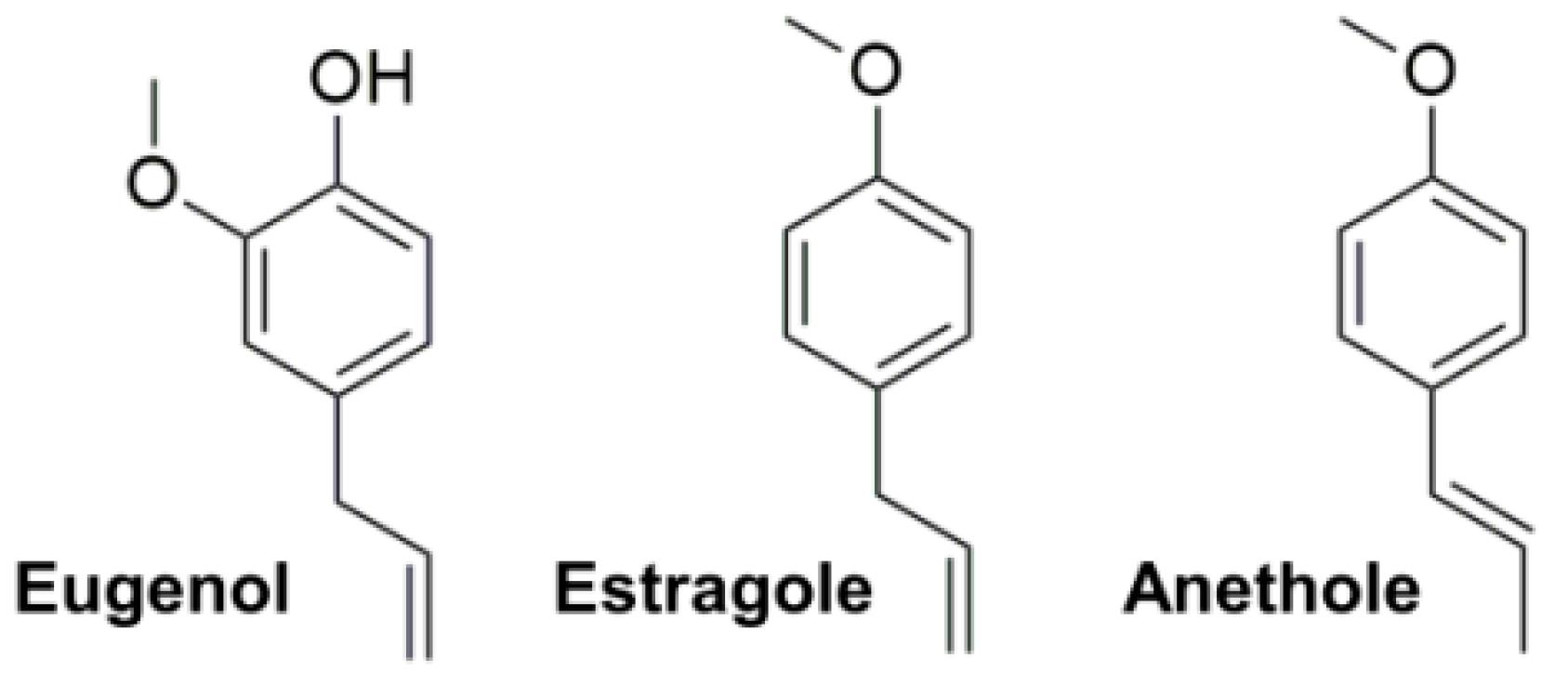
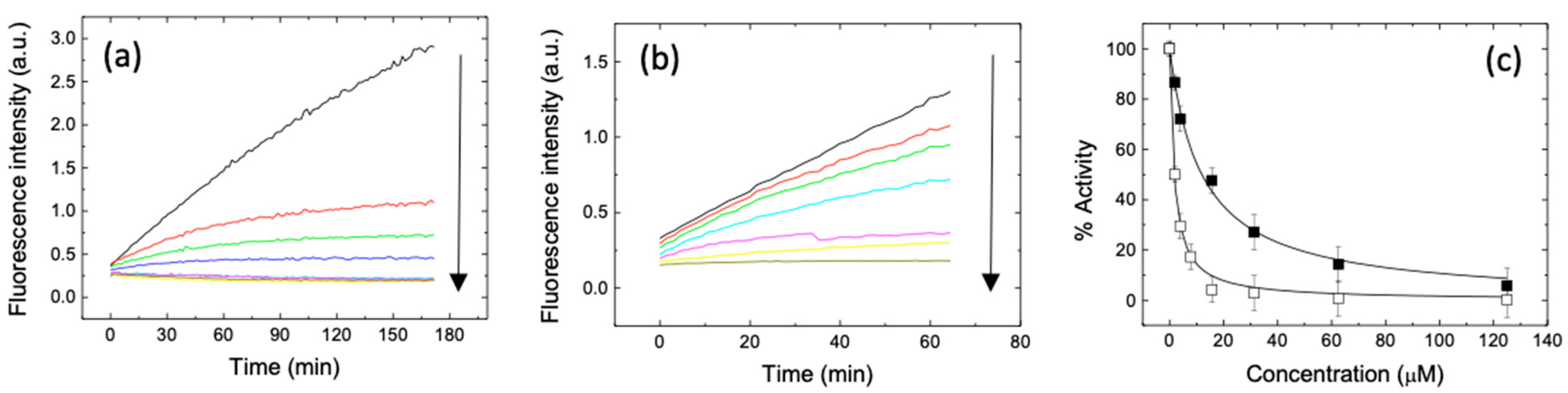
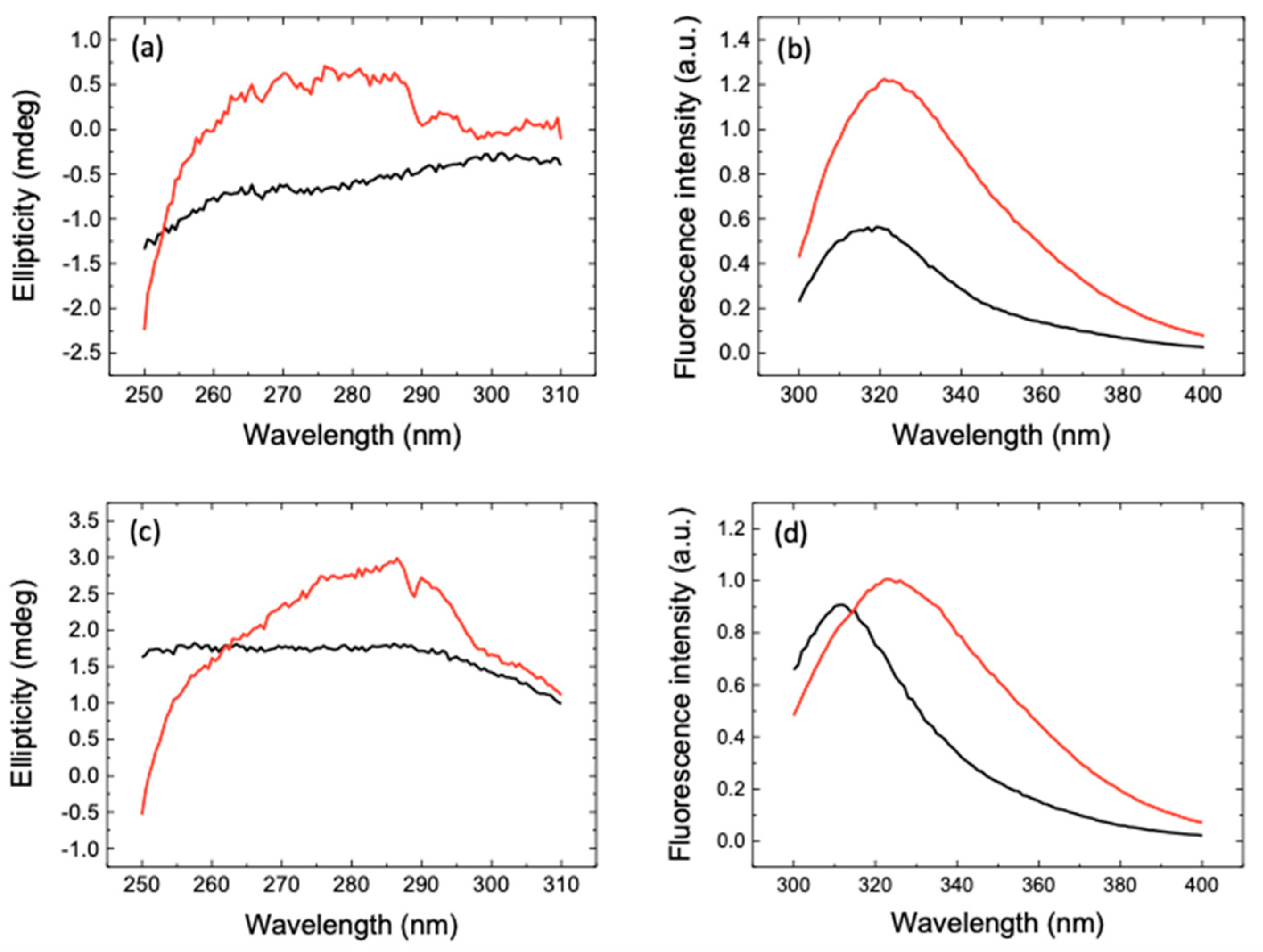
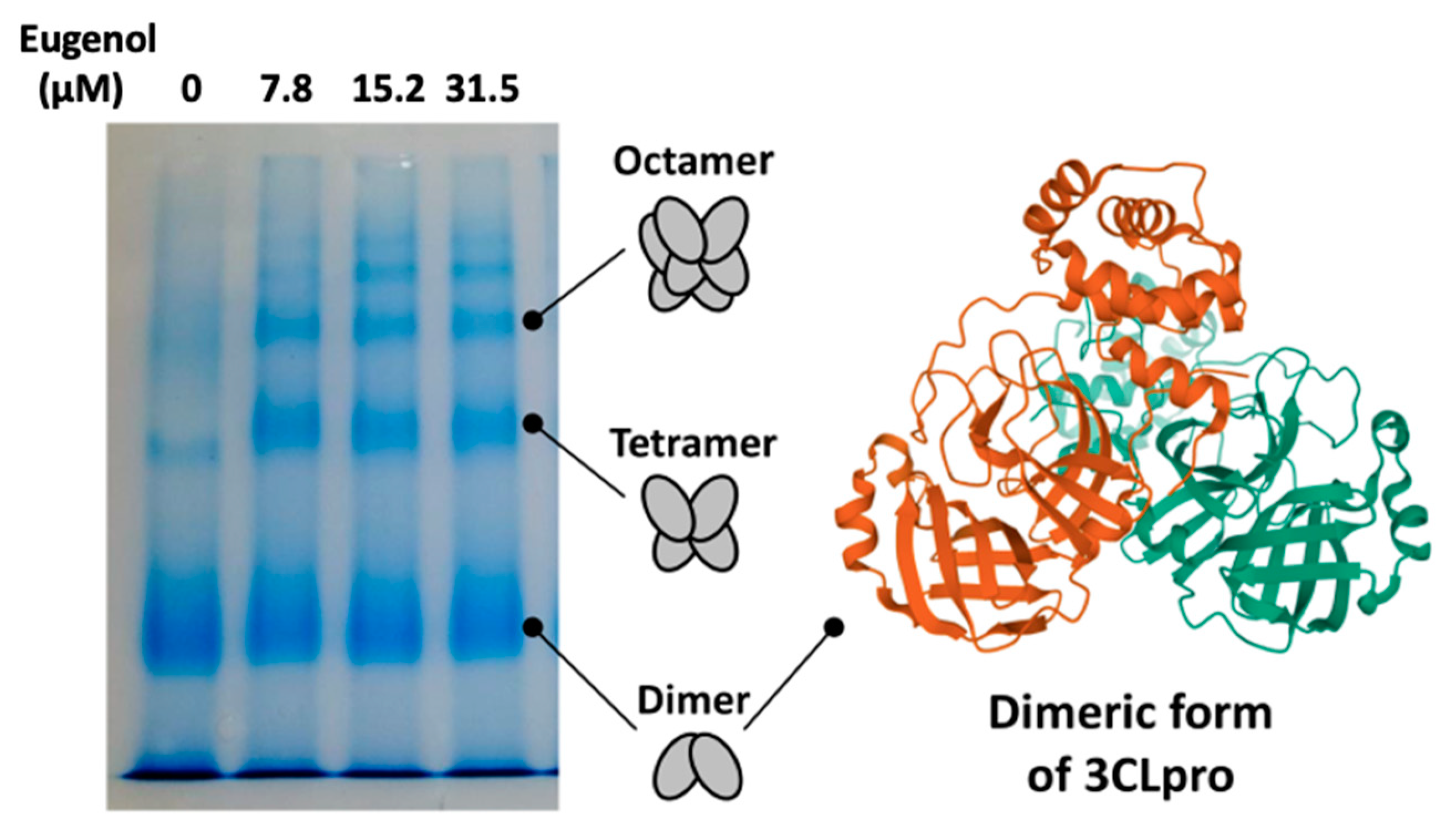
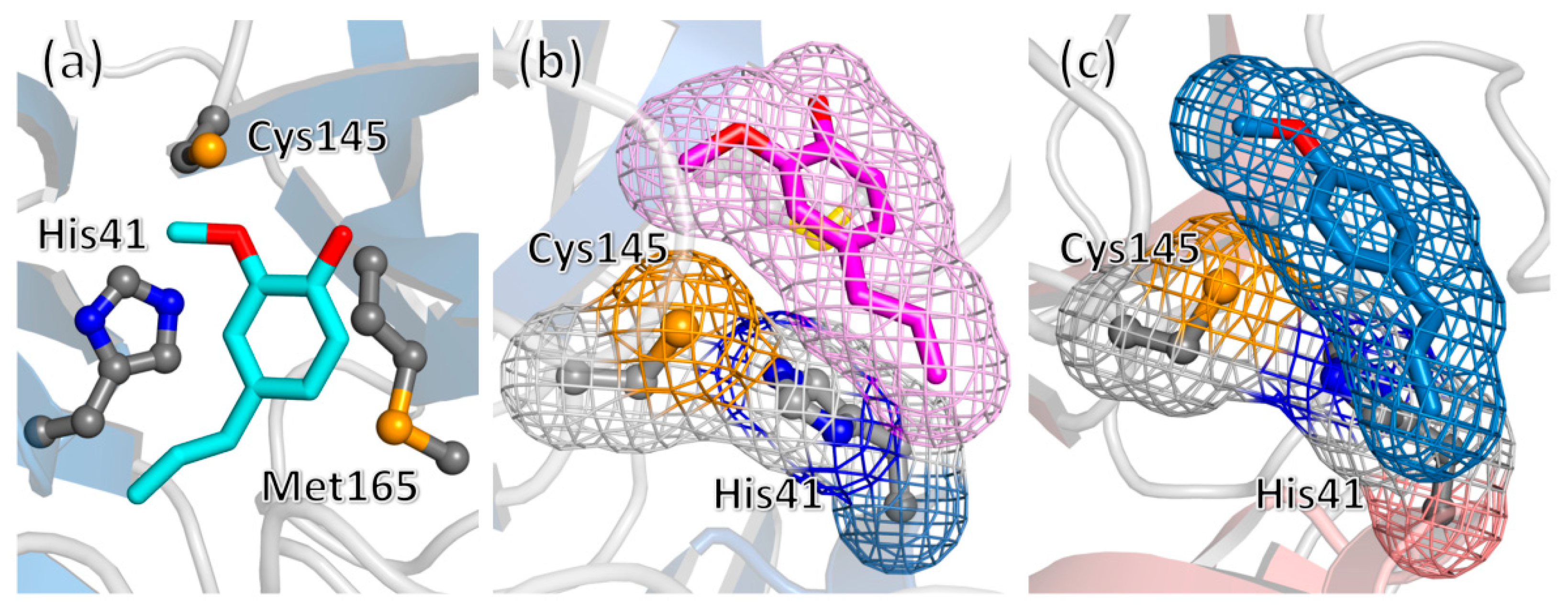
2. Results and Discussion
3. Materials and Methods
3.1. Chemical Compounds
3.2. CLpro Expression and Purification
3.3. CLpro Catalytic Activity
3.4. Inhibition Assay
3.5. Spectroscopy: Circular Dichroism and Emission Fluorescence
3.6. PAGE Native Electrophoresis
3.7. Molecular Docking
3.8. Molecular Dynamics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rubin, D.; Chan-Tack, K.; Farley, J.; Sherwat, A. FDA approval of remdesivir—A step in the right direction. N. Engl. J. Med. 2020, 383, 2598–2600. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Chodera, J.; Lee, A.A.; London, N.; von Delft, F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 2020, 12, 581. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Identification of potential binders of the main protease 3CL pro of the COVID-19 via structure-based ligand design and molecular modeling. Chem. Phys. Lett. 2020, 750, 137489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.-S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzyme Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xie, H.; Ke, C.Q.; Hu, H.C.; Gao, M.N.; et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Abian, O.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Vega, S.; Reyburn, H.T.; Rizzuti, B.; Velazquez-Campoy, A. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin is a low micromolar inhibitor of SARS-CoV-2 main protease 3CLpro: Implications for drug design of quercetin analogs. Biomedicines 2021, 9, 375. [Google Scholar] [CrossRef]
- Du, A.; Zheng, R.; Disoma, C.; Li, S.; Chen, Z.; Li, S.; Liu, P.; Zhou, Y.; Shen, Y.; Liu, S.; et al. Epigallocatechin-3-gallate, an active ingredient of traditional chinese medicines, inhibits the 3CLpro activity of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 176, 1–12. [Google Scholar] [CrossRef]
- Xiao, T.; Cui, M.; Zheng, C.; Wang, M.; Sun, R.; Gao, D.; Bao, J.; Ren, S.; Yang, B.; Lin, J.; et al. Myricetin inhibits SARS-CoV-2 Viral replication by targeting Mpro and ameliorates pulmonary inflammation. Front. Pharmacol. 2021, 12, 669642. [Google Scholar] [CrossRef] [PubMed]
- Gossen, J.; Albani, S.; Hanke, A.; Joseph, B.P.; Bergh, C.; Kuzikov, M.; Costanzi, E.; Manelfi, C.; Storici, P.; Gribbon, P.; et al. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from activity-based compound library screening guided by analysis of protein dynamics. ACS Pharmacol. Transl. Sci. 2021, 4, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Ying, M.; Cremades, N.; Velazquez-Campoy, A.; Scherer, T.; Thöny, B.; Sancho, J.; Martinez, A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremades, N.; Velazquez-Campoy, A.; Martínez-Julvez, M.; Neira, J.L.; Perez-Dorado, I.; Hermoso, J.; Jimenez, P.; Lanas, A.; Hoffman, P.S.; Sancho, J. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chem. Biol. 2009, 4, 928–938. [Google Scholar] [CrossRef]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velazquez-Campoy, A.; Iovanna, J.L.; Abian, O. Identification of a drug targeting an intrinsically disordered protein involved in pancreatic adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velazquez-Campoy, A.; Abian, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Mangiavacchi, F.; Kula-Pacurar, A.; Menichetti, E.; Bagnoli, L.; Rosati, O.; Marini, F.; Fonseca, S.F.; Abenante, L.; Alves, D.; Dabrowska, A.; et al. Seleno-functionalization of quercetin improves the non-covalent inhibition of Mpro and its antiviral activity in cells against SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 7048. [Google Scholar] [CrossRef]
- Atkinson, R.G. Phenylpropenes: Occurrence, distribution, and biosynthesis in fruit. J. Agric. Food Chem. 2018, 66, 2259–2272. [Google Scholar] [CrossRef]
- Rivera-Perez, A.; Lopez-Ruiz, R.; Romero-Gonzalez, R.; Garrido Frenich, A. A new strategy based on gas chromatography-high resolution mass spectrometry (GC-HRMS-Q-Orbitrap) for the determination of alkenylbenzenes in pepper and its varieties. Food Chem. 2020, 321, 126727. [Google Scholar] [CrossRef]
- Ahad, A.; Aqil, M.; Ali, A. The application of anethole, menthone, and eugenol in transdermal penetration of valsartan: Enhancement and mechanistic investigation. Pharm. Biol. 2016, 54, 1042–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhong, N.; Xue, F.; Kang, X.; Ren, X.; Chen, J.; Jin, C.; Lou, Z.; Xia, B. Three-dimensional domain swapping as a mechanism to lock the active conformation in a super-active octamer of SARS-CoV main protease. Protein Cell 2010, 1, 371–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestrini, L.; Belhaj, N.; Comez, L.; Gerelli, Y.; Lauria, A.; Libera, V.; Mariani, P.; Marzullo, P.; Ortore, M.G.; Palumbo Piccionello, A.; et al. The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Sci. Rep. 2021, 11, 9283. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Pant, S.; Zhang, Q.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. [Google Scholar] [CrossRef] [PubMed]
- Puttaswamy, H.; Gowtham, H.G.; Ojha, M.D.; Yadav, A.; Choudhir, G.; Raguraman, V.; Kongkham, B.; Selvaraju, K.; Shareef, S.; Gehlot, P.; et al. In silico studies evidenced the role of structurally diverse plant secondary metabolites in reducing SARS-CoV-2 pathogenesis. Sci. Rep. 2020, 10, 20584. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, L.; Zhang, X.; Zhang, Q.; Yang, Z.; Liu, Y.; Wei, S.; Liu, W. Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front. Mol. Biosci. 2020, 7, 556481. [Google Scholar] [CrossRef] [PubMed]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Sellamuthu, R. Eugenol. In Encyclopedia of Toxicology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 539–541. [Google Scholar]
- Mohammadi Nejad, S.; Özgüne, H.; Başaran, N. Pharmacological and toxicological properties of eugenol. Turk. J. Pharm. Sci. 2017, 14, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courrèges, M.C. In vitro and in vivo activity of eugenol on human herpesvirus. Phytother. Res. 2000, 14, 495–500. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Ioele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by molecular docking of homoisoflavones from Leopoldia comosa as ligands of estrogen receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, A.; Rizzuti, B.; Guzzi, R. Stereoselective and domain-specific effects of ibuprofen on the thermal stability of human serum albumin. Eur. J. Pharm. Sci. 2018, 112, 122–131. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzuti, B.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Vega, S.; Grande, F.; Conforti, F.; Abian, O.; Velazquez-Campoy, A. Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds. Pharmaceuticals 2021, 14, 892. https://doi.org/10.3390/ph14090892
Rizzuti B, Ceballos-Laita L, Ortega-Alarcon D, Jimenez-Alesanco A, Vega S, Grande F, Conforti F, Abian O, Velazquez-Campoy A. Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds. Pharmaceuticals. 2021; 14(9):892. https://doi.org/10.3390/ph14090892
Chicago/Turabian StyleRizzuti, Bruno, Laura Ceballos-Laita, David Ortega-Alarcon, Ana Jimenez-Alesanco, Sonia Vega, Fedora Grande, Filomena Conforti, Olga Abian, and Adrian Velazquez-Campoy. 2021. "Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds" Pharmaceuticals 14, no. 9: 892. https://doi.org/10.3390/ph14090892
APA StyleRizzuti, B., Ceballos-Laita, L., Ortega-Alarcon, D., Jimenez-Alesanco, A., Vega, S., Grande, F., Conforti, F., Abian, O., & Velazquez-Campoy, A. (2021). Sub-Micromolar Inhibition of SARS-CoV-2 3CLpro by Natural Compounds. Pharmaceuticals, 14(9), 892. https://doi.org/10.3390/ph14090892