
Advantages of Tyrosine Kinase Anti-Angiogenic Cediranib over Bevacizumab: Cell Cycle Abrogation and Synergy with Chemotherapy

 , , , and
, , , and 
Abstract
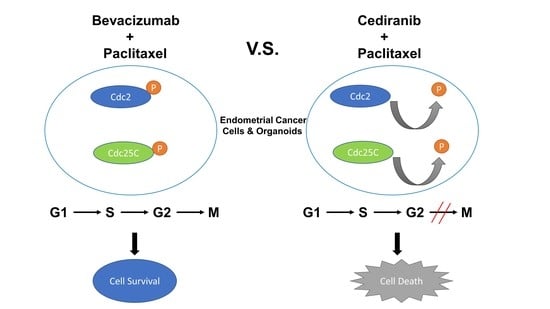
:
1. Introduction
2. Results
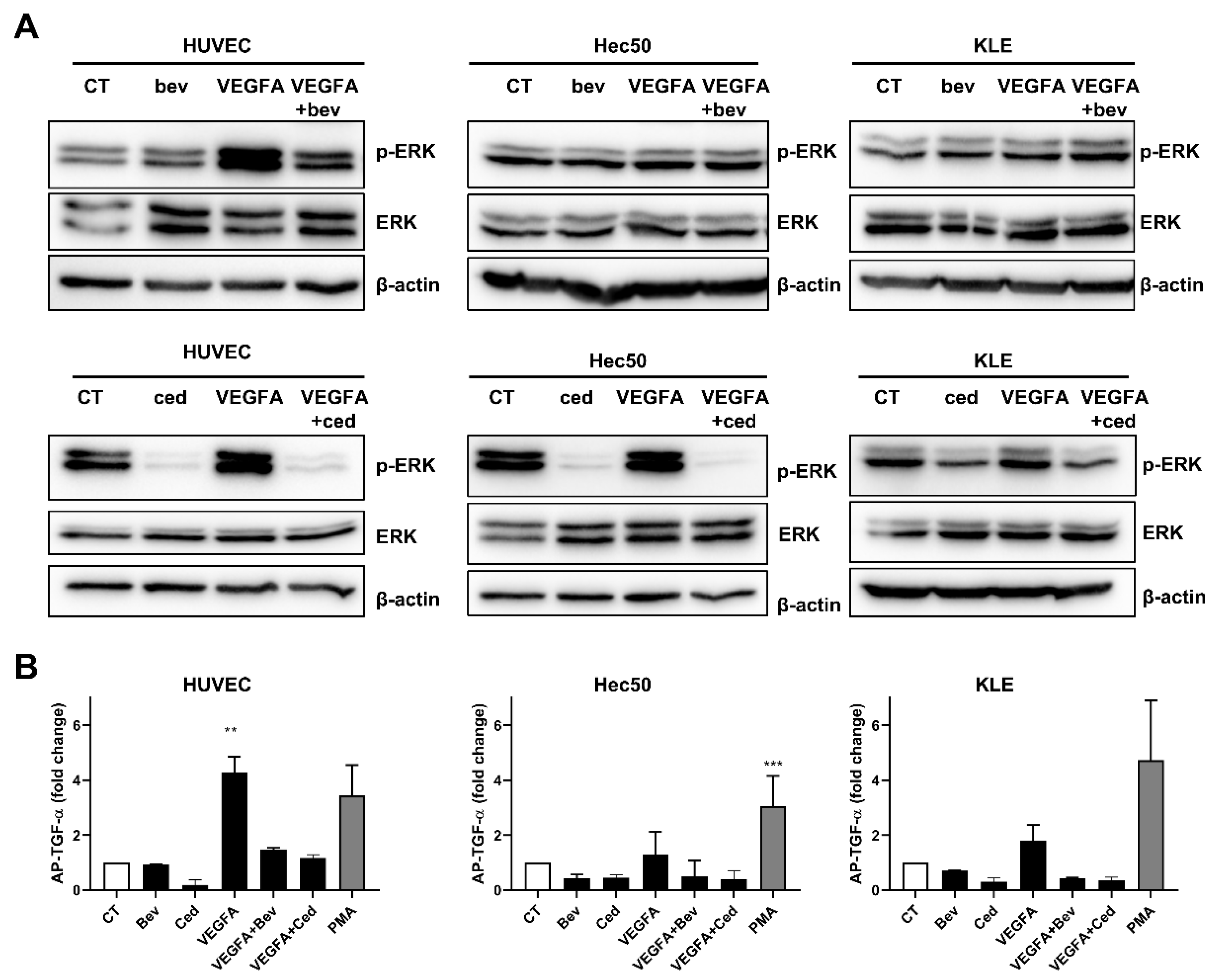
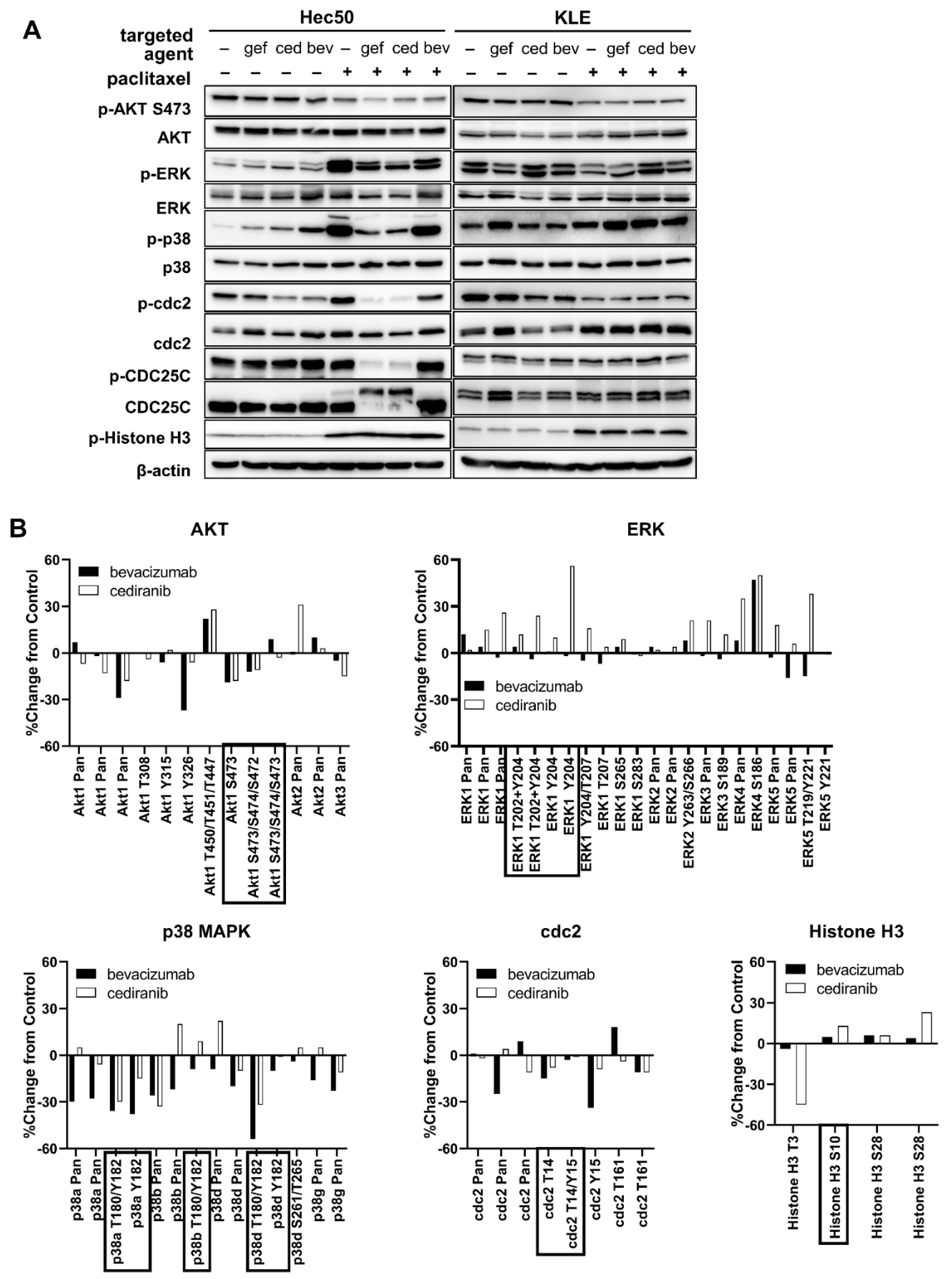
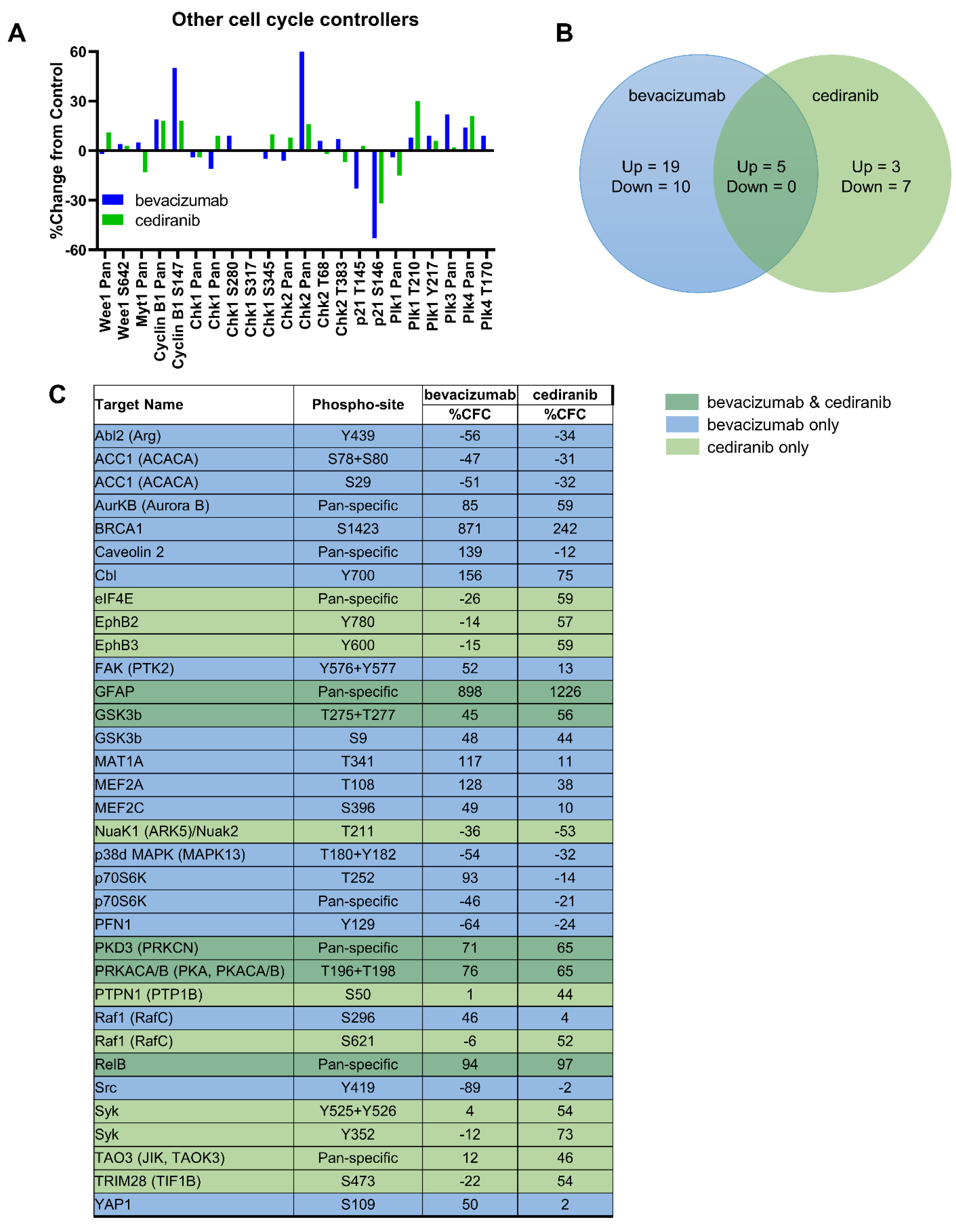
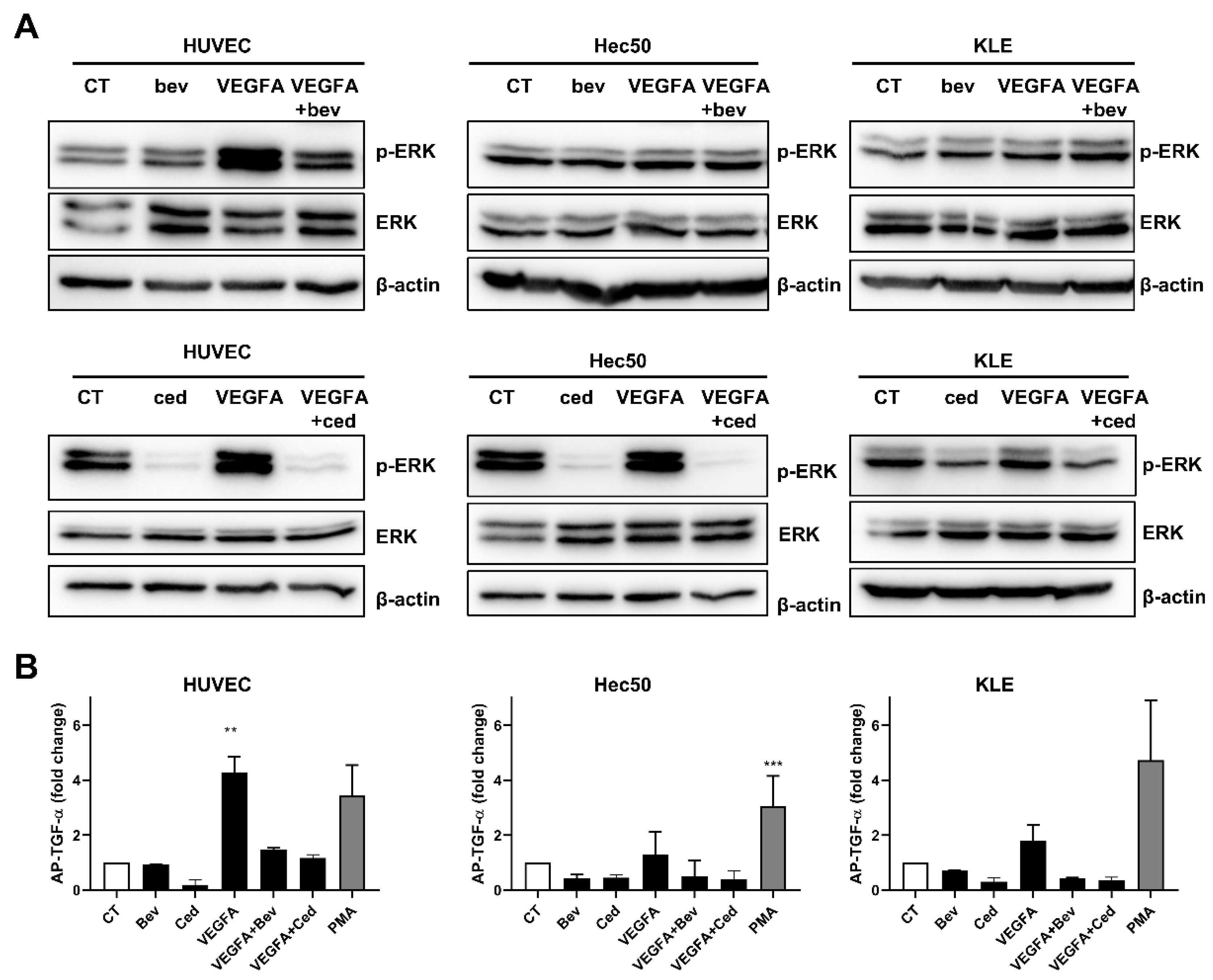
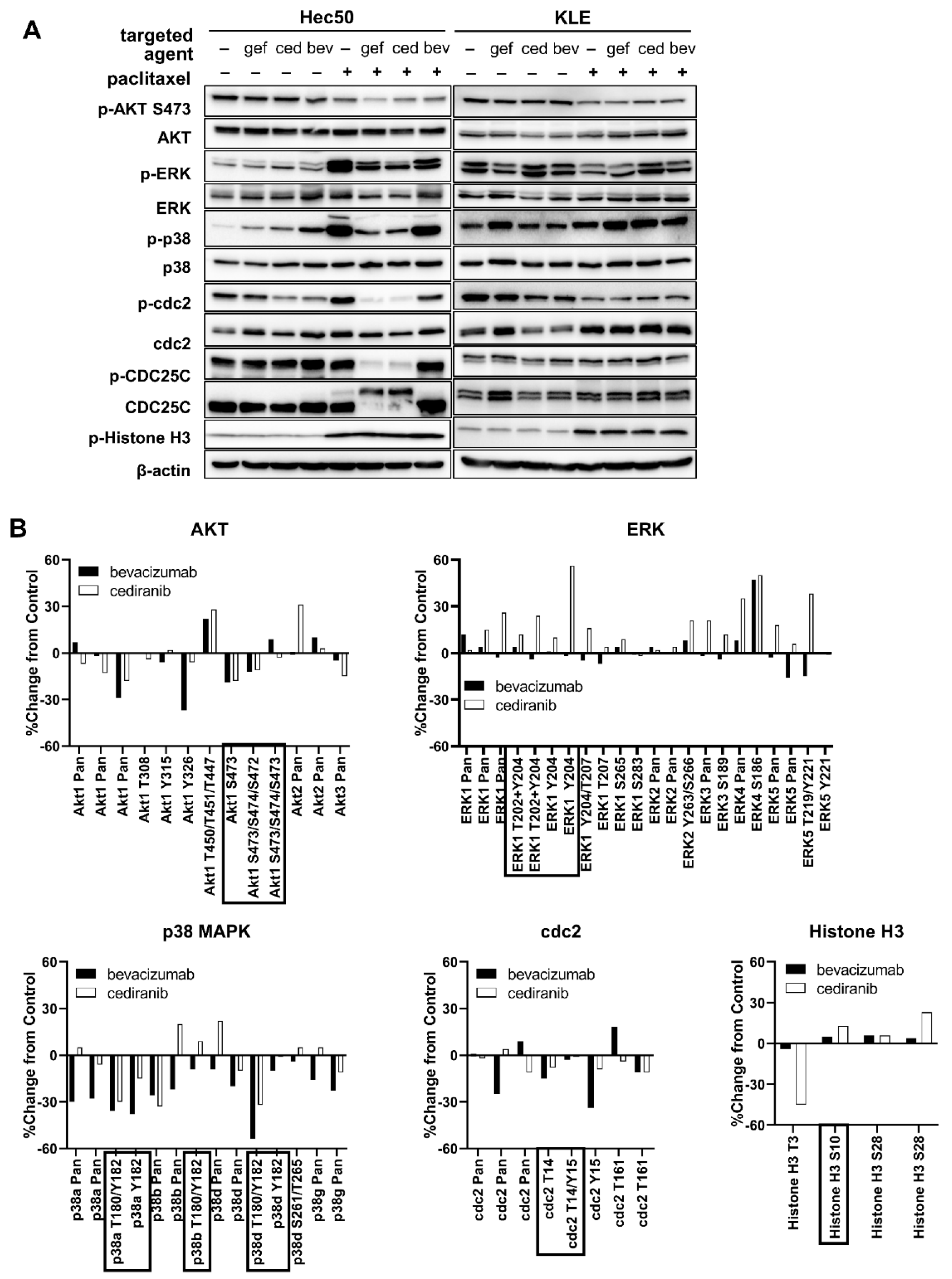
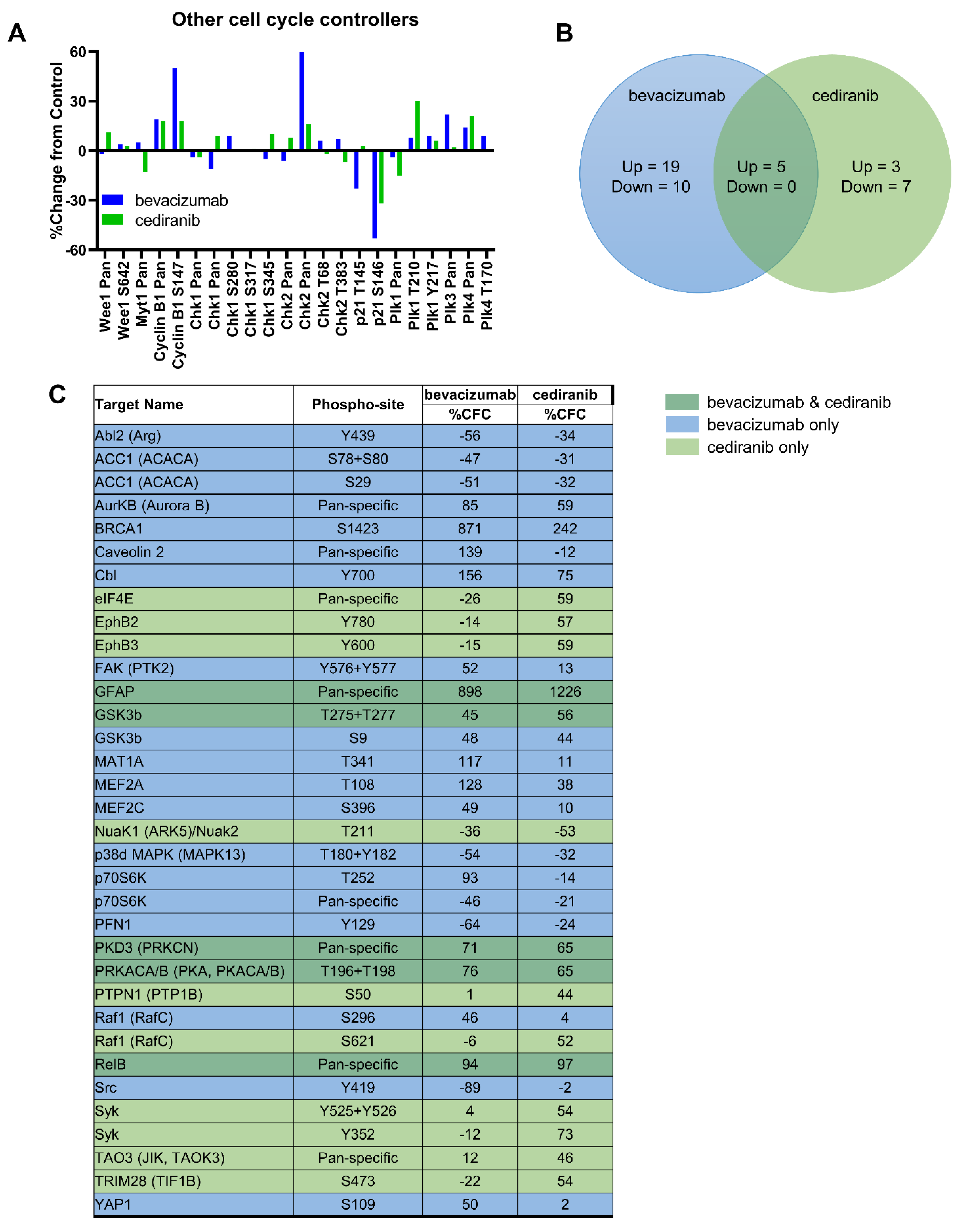
2.1. Tyrosine Kinase Receptor Signaling Pathways Are Downregulated by Cediranib But Not Bevacizumab
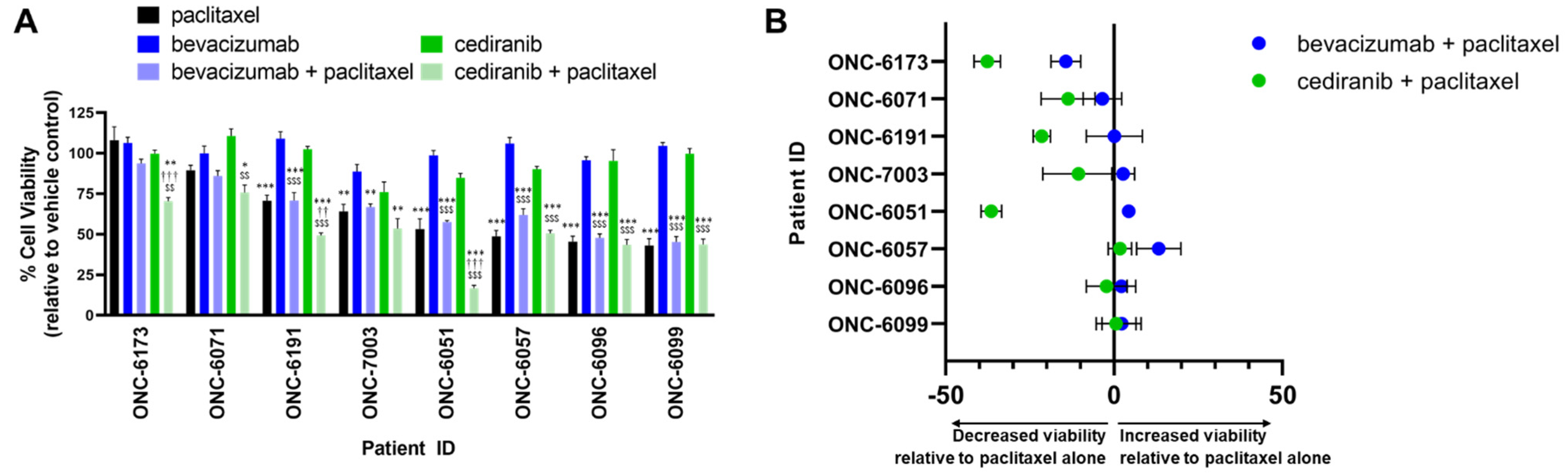
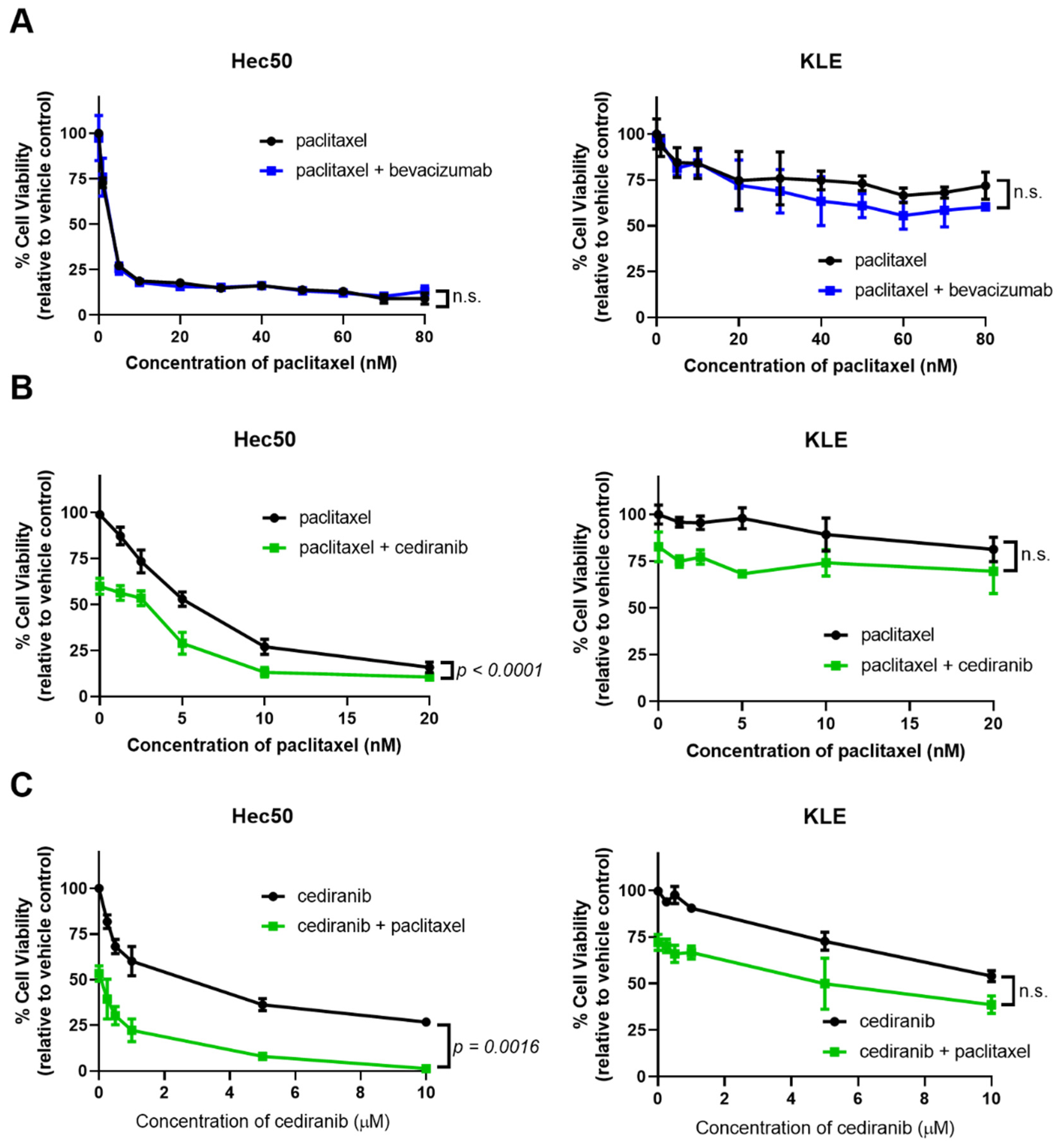
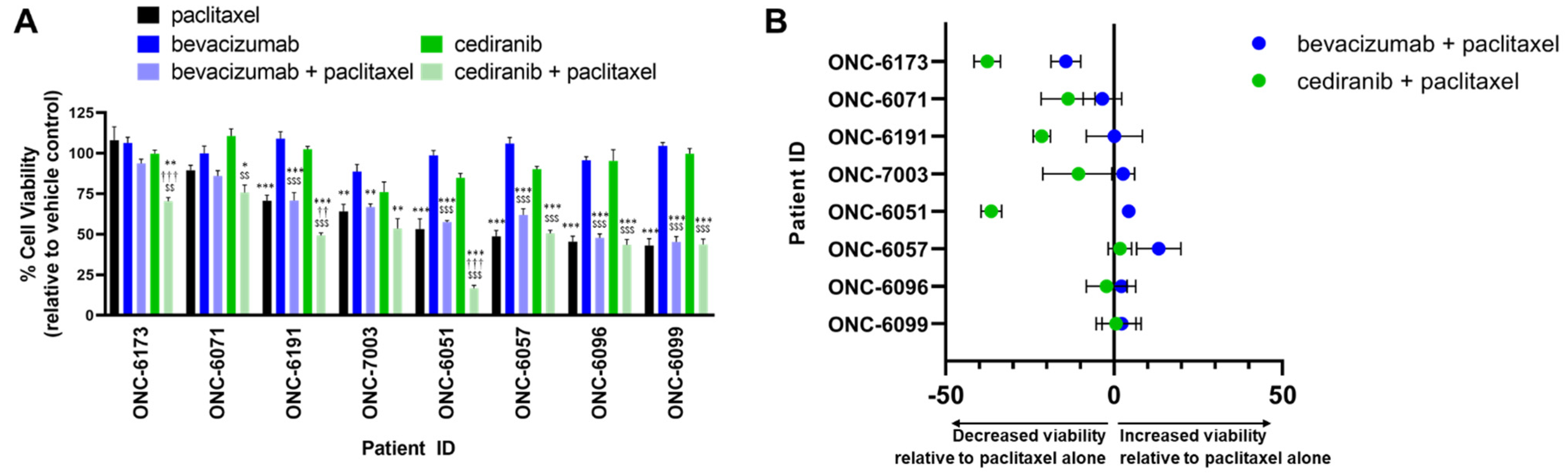
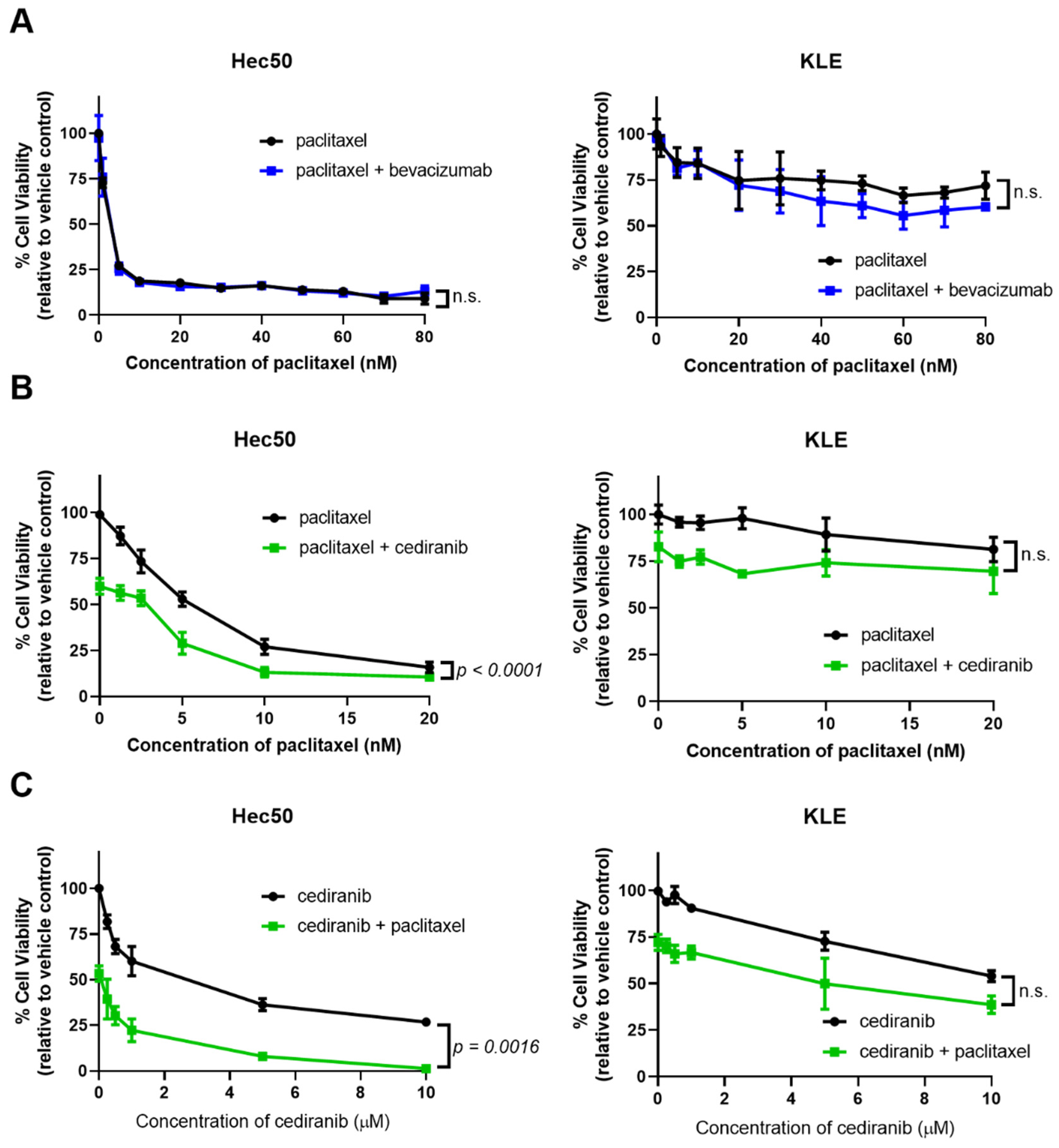
2.2. Cediranib But Not Bevacizumab Synergizes with Chemotherapy
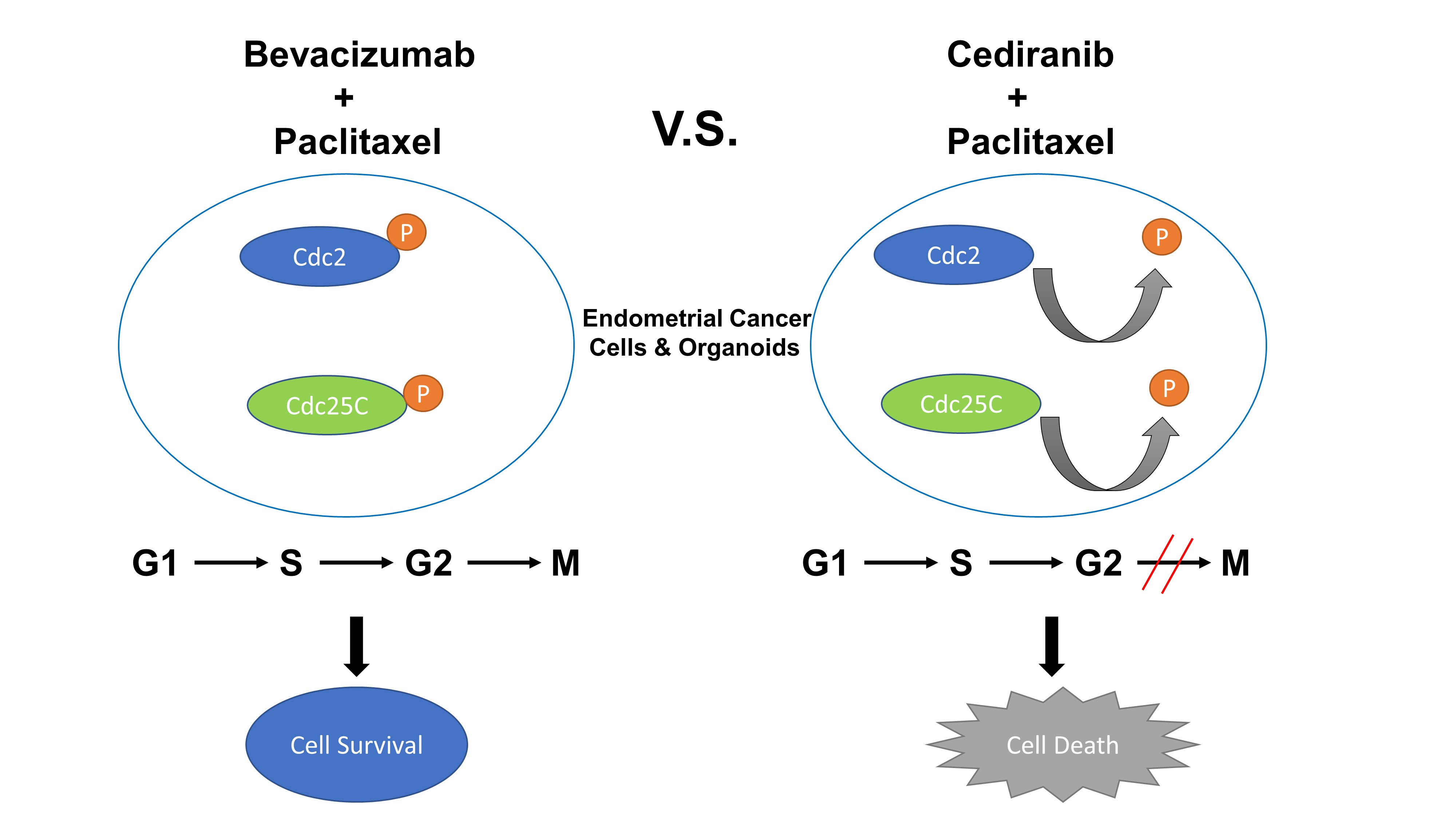
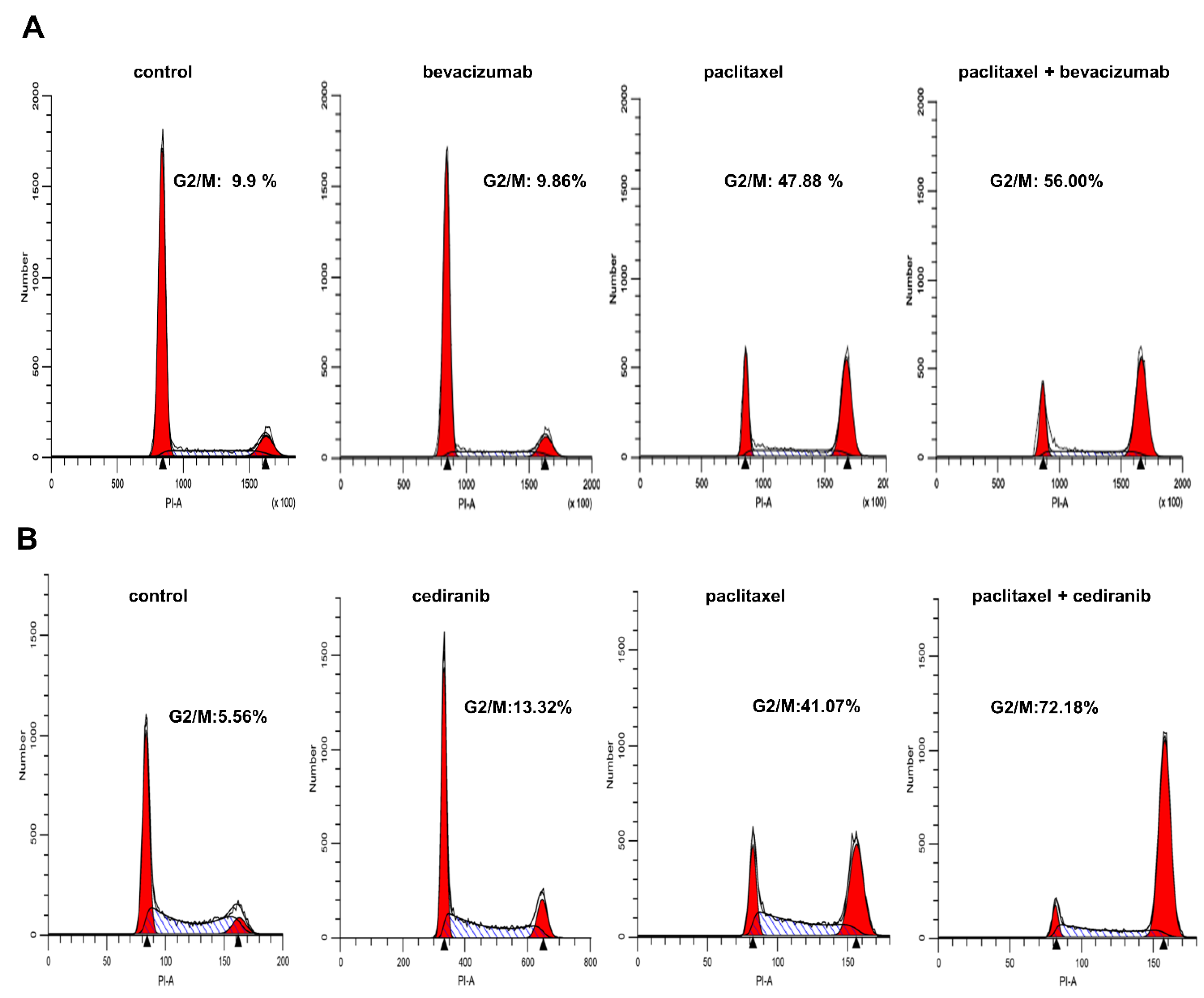
2.3. The Combination of Cediranib But Not Bevacizumab with Paclitaxel Increases the Percentage of Cells in Mitosis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Patient-Derived Organoid Models of Endometrial Cancer
4.3. Cell Culture
4.4. Western Blot
4.5. Cell Viability Assay
4.5.1. Organoids
4.5.2. Cell Lines
4.6. Cell Cycle Analysis
4.7. Shedding Assay
4.8. Phosphoproteomic Microarray
4.9. Reverse Transcription and Quantitative Polymerase Chain Reaction
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cancer Facts and Figures. 2020. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf (accessed on 14 January 2021).
- Sheikh, M.A.; Althouse, A.D.; Freese, K.E.; Soisson, S.; Edwards, R.P.; Welburn, S.; Sukumvanich, P.; Comerci, J.; Kelley, J.; LaPorte, R.E.; et al. USA endometrial cancer projections to 2030: Should we be concerned? Future Oncol. 2014, 10, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.K.; Filiaci, V.L.; Mallen, A.R.; Thiel, K.W.; Devor, E.J.; Moxley, K.; Richardson, D.; Mutch, D.; Secord, A.A.; Tewari, K.S.; et al. Mutated p53 portends improvement in outcomes when bevacizumab is combined with chemotherapy in advanced/recurrent endometrial cancer: An NRG Oncology study. Gynecol. Oncol. 2021, 161, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Sill, M.W.; Darcy, K.M.; Greer, B.; McMeekin, D.S.; Rose, P.G.; Rotmensch, J.; Barnes, M.N.; Hanjani, P.; Leslie, K.K. Phase II trial of bevacizumab in recurrent or persistent endometrial cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2011, 29, 2259–2265. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.A.; Brady, W.E.; Walker, J.L.; Rotmensch, J.; Zhou, X.C.; Kendrick, J.E.; Yamada, S.D.; Schilder, J.M.; Cohn, D.E.; Harrison, C.R.; et al. Phase II trial of combination bevacizumab and temsirolimus in the treatment of recurrent or persistent endometrial carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2013, 129, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Wedge, S.R.; Kendrew, J.; Hennequin, L.F.; Valentine, P.J.; Barry, S.T.; Brave, S.R.; Smith, N.R.; James, N.H.; Dukes, M.; Curwen, J.O.; et al. AZD2171: A highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005, 65, 4389–4400. [Google Scholar] [CrossRef] [Green Version]
- Brave, S.R.; Ratcliffe, K.; Wilson, Z.; James, N.H.; Ashton, S.; Wainwright, A.; Kendrew, J.; Dudley, P.; Broadbent, N.; Sproat, G.; et al. Assessing the activity of cediranib, a VEGFR-2/3 tyrosine kinase inhibitor, against VEGFR-1 and members of the structurally related PDGFR family. Mol. Cancer Ther. 2011, 10, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Bender, D.; Sill, M.W.; Lankes, H.A.; Reyes, H.D.; Darus, C.J.; Delmore, J.E.; Rotmensch, J.; Gray, H.J.; Mannel, R.S.; Schilder, J.M.; et al. A phase II evaluation of cediranib in the treatment of recurrent or persistent endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2015, 138, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Dizon, D.S.; Sill, M.W.; Schilder, J.M.; McGonigle, K.F.; Rahman, Z.; Miller, D.S.; Mutch, D.G.; Leslie, K.K. A phase II evaluation of nintedanib (BIBF-1120) in the treatment of recurrent or persistent endometrial cancer: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol. Oncol. 2014, 135, 441–445. [Google Scholar] [CrossRef] [Green Version]
- Powell, M.A.; Sill, M.W.; Goodfellow, P.J.; Benbrook, D.M.; Lankes, H.A.; Leslie, K.K.; Jeske, Y.; Mannel, R.S.; Spillman, M.A.; Lee, P.S.; et al. A phase II trial of brivanib in recurrent or persistent endometrial cancer: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol. Oncol. 2014, 135, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Nimeiri, H.S.; Oza, A.M.; Morgan, R.J.; Huo, D.; Elit, L.; Knost, J.A.; Wade, J.L., 3rd; Agamah, E.; Vokes, E.E.; Fleming, G.F. A phase II study of sorafenib in advanced uterine carcinoma/carcinosarcoma: A trial of the Chicago, PMH, and California Phase II Consortia. Gynecol. Oncol. 2010, 117, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Sill, M.W.; Lankes, H.A.; Fader, A.N.; Finkler, N.J.; Hoffman, J.S.; Rose, P.G.; Sutton, G.P.; Drescher, C.W.; McMeekin, D.S.; et al. A phase II evaluation of aflibercept in the treatment of recurrent or persistent endometrial cancer: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 127, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, C.; Filiaci, V.; Dizon, D.S.; Carlson, J.W.; Powell, M.A.; Secord, A.A.; Tewari, K.S.; Bender, D.P.; O’Malley, D.M.; Stuckey, A.; et al. A phase II study of frontline paclitaxel/carboplatin/bevacizumab, paclitaxel/carboplatin/temsirolimus, or ixabepilone/carboplatin/bevacizumab in advanced/recurrent endometrial cancer. Gynecol. Oncol. 2018, 150, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.A.; Merritt, W.M.; Coffey, D.; Lin, Y.G.; Patel, P.R.; Broaddus, R.; Nugent, E.; Han, L.Y.; Landen, C.N., Jr.; Spannuth, W.A.; et al. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin. Cancer Res. 2007, 13, 7487–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.; Dai, D.; Pickett, G.; Thiel, K.W.; Korovkina, V.P.; Leslie, K.K. Effects of bevacizumab in mouse model of endometrial cancer: Defining the molecular basis for resistance. Oncol. Rep. 2011, 25, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Decio, A.; Cesca, M.; Bizzaro, F.; Porcu, L.; Bettolini, R.; Ubezio, P.; Taraboletti, G.; Belotti, D.; Giavazzi, R. Cediranib combined with chemotherapy reduces tumor dissemination and prolongs the survival of mice bearing patient-derived ovarian cancer xenografts with different responsiveness to cisplatin. Clin. Exp. Metastasis 2015, 32, 647–658. [Google Scholar] [CrossRef]
- Collins, A.; Miles, G.J.; Wood, J.; MacFarlane, M.; Pritchard, C.; Moss, E. Patient-derived explants, xenografts and organoids: 3-dimensional patient-relevant pre-clinical models in endometrial cancer. Gynecol. Oncol. 2020, 156, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Devor, E.J.; Gonzalez-Bosquet, J.; Thiel, K.W.; Leslie, K.K. Genomic characterization of five commonly used endometrial cancer cell lines. Int. J. Oncol. 2020, 57, 1348–1357. [Google Scholar] [CrossRef]
- Bi, J.; Thiel, K.W.; Litman, J.M.; Zhang, Y.; Devor, E.J.; Newtson, A.M.; Schnieders, M.J.; Gonzalez Bosquet, J.; Leslie, K.K. Characterization of a TP53 somatic variant of unknown function from an ovarian cancer patient using organoid culture and computational modeling. Clin. Obstet. Gynecol. 2020, 63, 109–119. [Google Scholar] [CrossRef]
- Bi, J.; Newtson, A.M.; Zhang, Y.; Devor, E.J.; Samuelson, M.I.; Thiel, K.W.; Leslie, K.K. Successful Patient-Derived Organoid Culture of Gynecologic Cancers for Disease Modeling and Drug Sensitivity Testing. Cancers 2021, 13, 2901. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- Swendeman, S.; Mendelson, K.; Weskamp, G.; Horiuchi, K.; Deutsch, U.; Scherle, P.; Hooper, A.; Rafii, S.; Blobel, C.P. VEGF-A stimulates ADAM17-dependent shedding of VEGFR2 and crosstalk between VEGFR2 and ERK signaling. Circ. Res. 2008, 103, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, S.M.; Bobe, P.; Reiss, K.; Horiuchi, K.; Niu, X.D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Dizon, D.S.; Yang, S.; Wang, X.; Zhu, D.; Thiel, K.W.; Leslie, K.K. Strategies for molecularly enhanced chemotherapy to achieve synthetic lethality in endometrial tumors with mutant p53. Obstet. Gynecol. Int. 2013, 2013, 4916497. [Google Scholar] [CrossRef] [Green Version]
- Ebeid, K.; Meng, X.; Thiel, K.W.; Do, A.V.; Geary, S.M.; Morris, A.S.; Pham, E.L.; Wongrakpanich, A.; Chhonker, Y.S.; Murry, D.J.; et al. Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat. Nanotechnol. 2018, 13, 72–81. [Google Scholar] [CrossRef]
- Meng, X.; Laidler, L.L.; Kosmacek, E.A.; Yang, S.; Xiong, Z.; Zhu, D.; Wang, X.; Dai, D.; Zhang, Y.; Brachova, P.; et al. Induction of mitotic cell death by overriding G2/M checkpoint in endometrial cancer cells with non-functional p53. Gynecol. Oncol. 2013, 128, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shi, X.; Pelech, S. Monitoring Protein Kinase Expression and Phosphorylation in Cell Lysates with Antibody Microarrays. Methods Mol. Biol. 2016, 1360, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Madamsetty, V.S.; Paul, M.K.; Mukherjee, S. Recent Advancements of Nanomedicine towards Antiangiogenic Therapy in Cancer. Int. J. Mol. Sci. 2020, 21, 455. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.W.; Huang, J.; Hu, W.; Yang, X.; Jennings, N.B.; Sehgal, V.; Sohn, B.H.; Han, H.D.; Lee, S.J.; Thanapprapasr, D.; et al. Biologic effects of platelet-derived growth factor receptor alpha blockade in uterine cancer. Clin. Cancer Res. 2014, 20, 2740–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.S.; Secord, A.A. Targeting molecular pathways in endometrial cancer: A focus on the FGFR pathway. Cancer Treat. Rev. 2014, 40, 507–512. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maretzky, T.; Yang, G.; Ouerfelli, O.; Overall, C.M.; Worpenberg, S.; Hassiepen, U.; Eder, J.; Blobel, C.P. Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem. J. 2009, 420, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Primer Sequences | Amplicon | Tm (°C) * |
|---|---|---|---|
| VEGFA | For: GGGCAGAATCATCACGAAGT | 269 bp | 55.0 |
| Rev: AGGAAGCTCATCTCTCCTATGT | 54.7 | ||
| VEGFR1 | For: GGACAGTAGAAAGGGCTTCATC | 251 bp | 55.2 |
| Rev: CAGGGTAACTCCAGGTCATTTG | 55.4 | ||
| VEGFR2 | For: GTGGTCTCTCTGGTTGTGTATG | 235 bp | 55.0 |
| Rev: CCTCCACACTTCTCCATTCTTC | 55.1 | ||
| VEGFR3 | For: CGAAAGTGCATCCACAGAGA | 239 bp | 54.9 |
| Rev: AGAGAGAAGATCTCCCAGAGAAG | 54.9 | ||
| FGFR1 | For: AGGAACTTTTCAAGCTGC | ||
| Rev: CATCATGTACAGCTCGTTG | |||
| FGFR2 | For: ATGAGGAATACTTGGACCTC | ||
| Rev: TTAACACTGCCGTTTATGTG | |||
| FGFR3 | For: GAAGATGCTGAAAGACGATG | ||
| Rev: GCAGGTTGATGATGTTTTTG | |||
| FGFR4 | For: CTGAGGACAATGTGATGAAG | ||
| Rev: CCGTTGCTGGTTTTCTTCTTATAG | |||
| PDGFRA | For: GACTTTCGCCAAAGTGGAGGAG | 121 bp | 58.0 |
| Rev: AGCCACCGTGAGTTCAGAACGC | 62.0 | ||
| PDGFRB | For: TGCAGACATCGAGTCCTCCAAC | 108 bp | 59.0 |
| Rev: GCTTAGCACTGGAGACTCGTTG | 57.8 | ||
| 18S rRNA | For: AACTTTCGATGGTAGTCGCCG | 104 bp | 57.3 |
| Rev: CCTTGGATGTGGTAGCCGTTT | 57.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, J.; Dixit, G.; Zhang, Y.; Devor, E.J.; Losh, H.A.; Newtson, A.M.; Coleman, K.L.; Santillan, D.A.; Maretzky, T.; Thiel, K.W.; et al. Advantages of Tyrosine Kinase Anti-Angiogenic Cediranib over Bevacizumab: Cell Cycle Abrogation and Synergy with Chemotherapy. Pharmaceuticals 2021, 14, 682. https://doi.org/10.3390/ph14070682
Bi J, Dixit G, Zhang Y, Devor EJ, Losh HA, Newtson AM, Coleman KL, Santillan DA, Maretzky T, Thiel KW, et al. Advantages of Tyrosine Kinase Anti-Angiogenic Cediranib over Bevacizumab: Cell Cycle Abrogation and Synergy with Chemotherapy. Pharmaceuticals. 2021; 14(7):682. https://doi.org/10.3390/ph14070682
Chicago/Turabian StyleBi, Jianling, Garima Dixit, Yuping Zhang, Eric J. Devor, Haley A. Losh, Andreea M. Newtson, Kristen L. Coleman, Donna A. Santillan, Thorsten Maretzky, Kristina W. Thiel, and et al. 2021. "Advantages of Tyrosine Kinase Anti-Angiogenic Cediranib over Bevacizumab: Cell Cycle Abrogation and Synergy with Chemotherapy" Pharmaceuticals 14, no. 7: 682. https://doi.org/10.3390/ph14070682
APA StyleBi, J., Dixit, G., Zhang, Y., Devor, E. J., Losh, H. A., Newtson, A. M., Coleman, K. L., Santillan, D. A., Maretzky, T., Thiel, K. W., & Leslie, K. K. (2021). Advantages of Tyrosine Kinase Anti-Angiogenic Cediranib over Bevacizumab: Cell Cycle Abrogation and Synergy with Chemotherapy. Pharmaceuticals, 14(7), 682. https://doi.org/10.3390/ph14070682