
Design of Topical Ocular Ciprofloxacin Nanoemulsion for the Management of Bacterial Keratitis

Abstract
1. Introduction
2. Results and Discussion
2.1. Screening of Oils
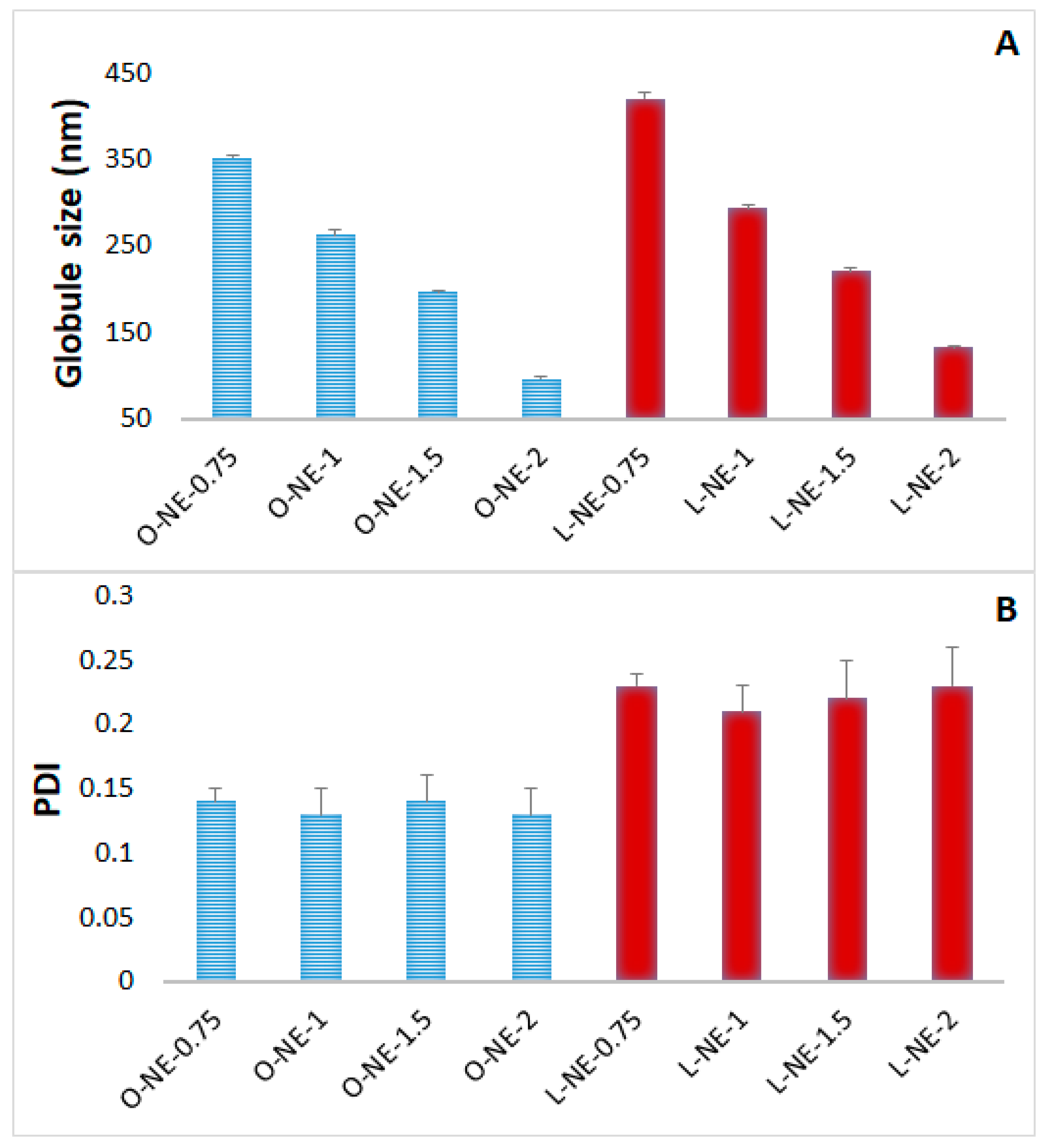
2.2. Preparation and Physical Characterization of CIP-NE Formulations
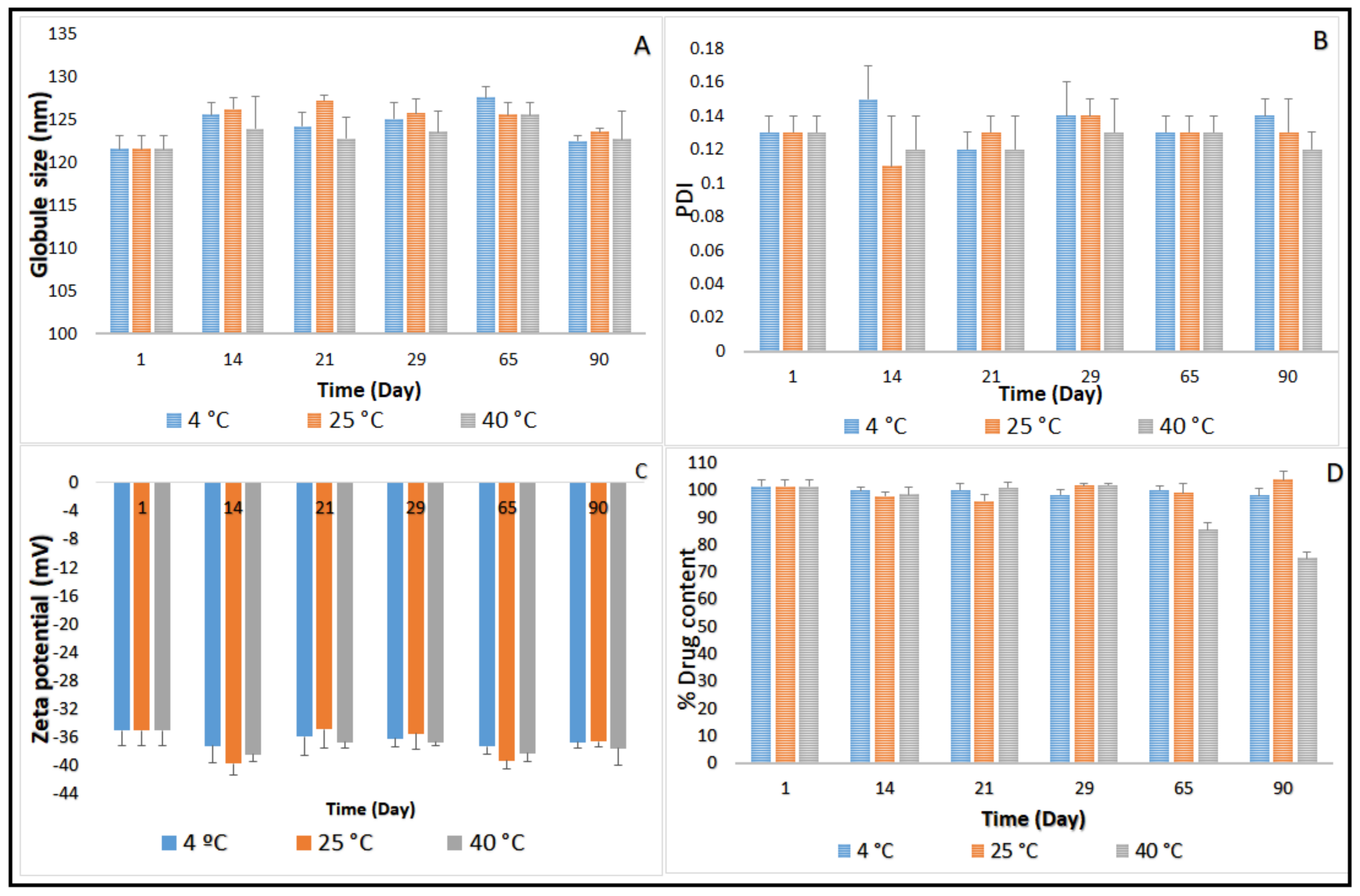
2.3. Stability Studies before Moist Heat Sterilization
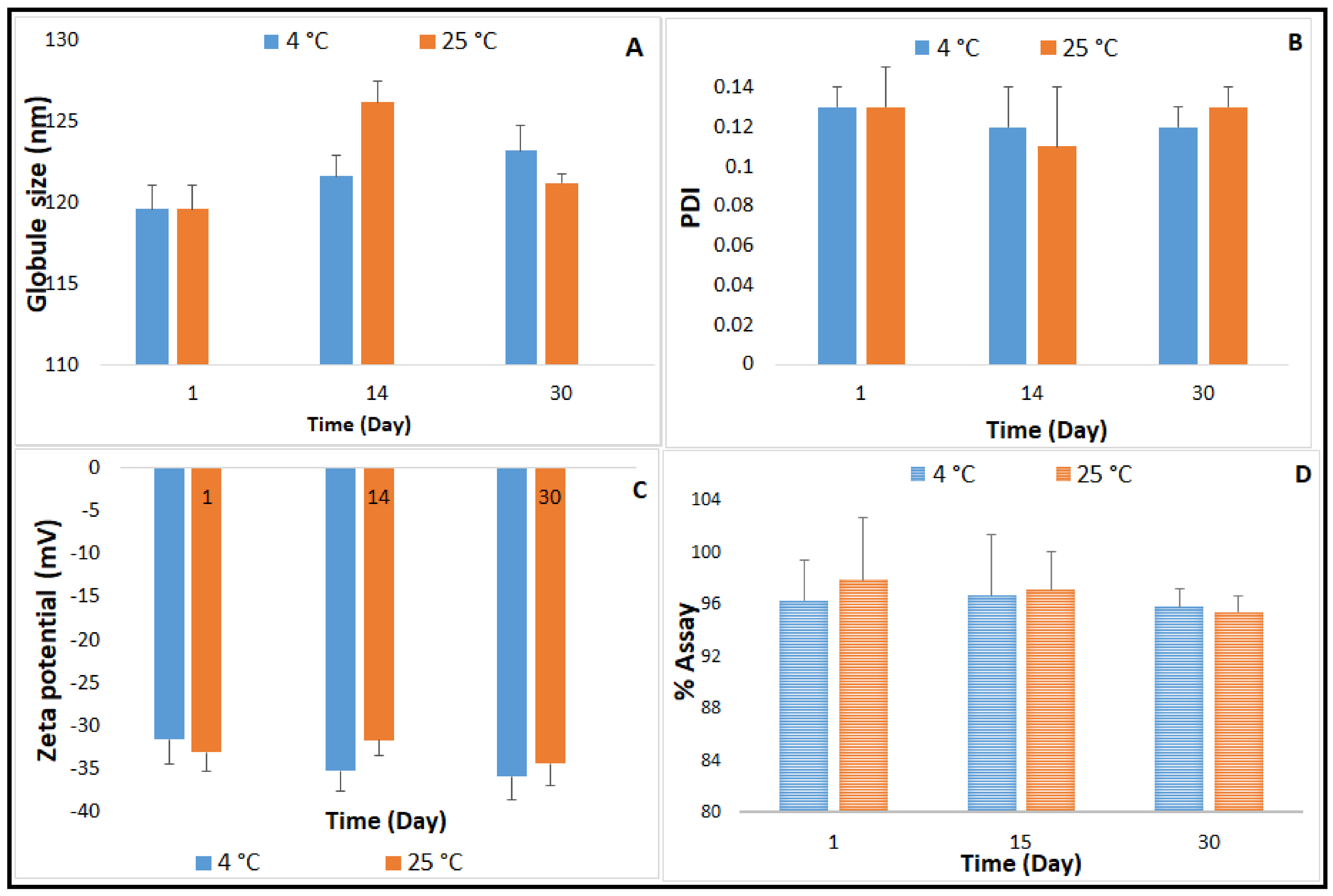
2.4. Autoclavability of the Optimized NE Formulation
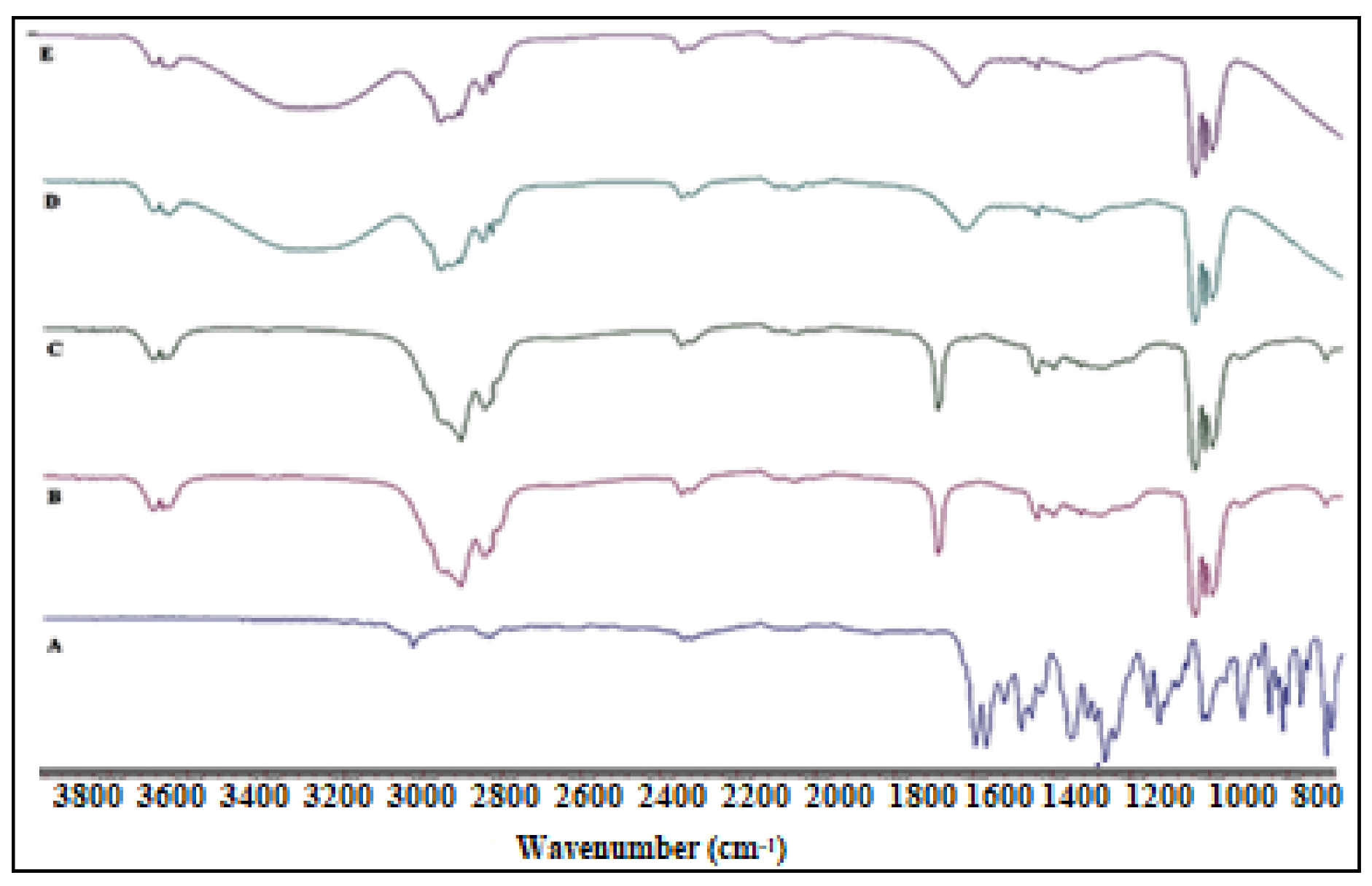
2.5. FTIR Studies
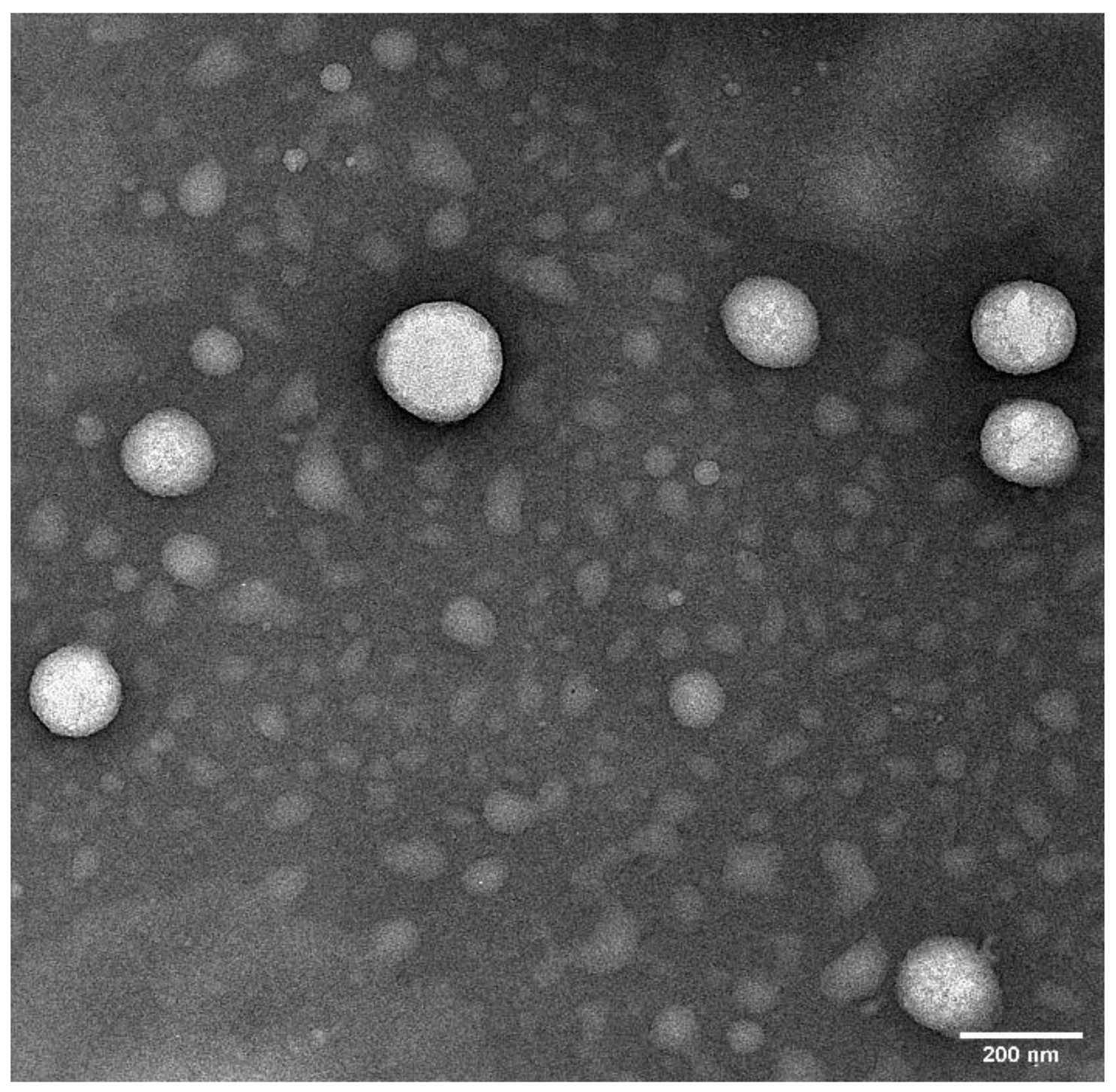
2.6. Transmission Electron Microscopy (TEM)
2.7. In Vitro Release Studies
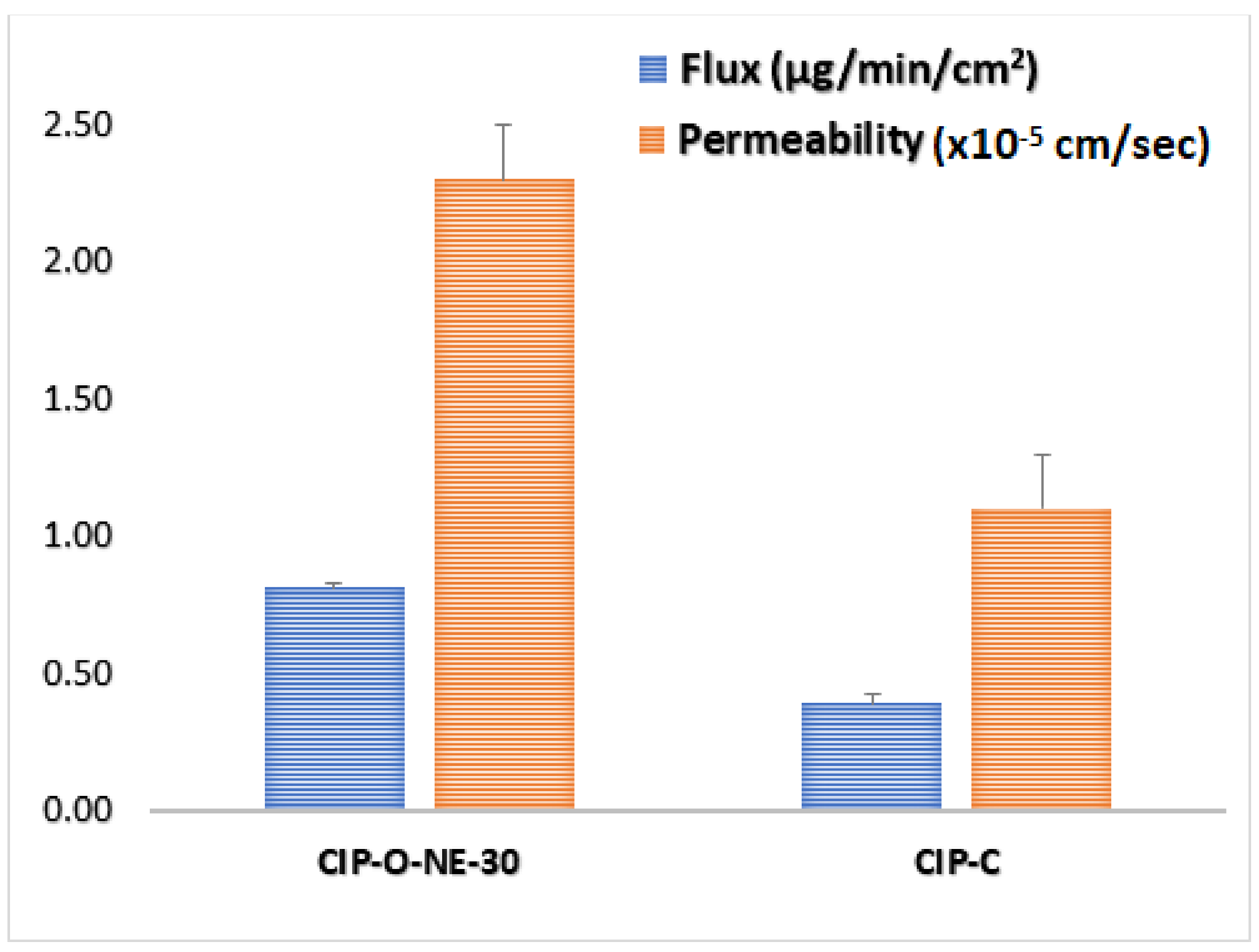
2.8. Ex Vivo Transcorneal Permeation
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Screening of Oils
3.2.2. Quantification of CIP by HPLC
3.2.3. Preparation of CIP-NE Formulations
3.2.4. Control Formulation (CIP-C)
3.2.5. Characterization of CIP-NE
Measurement of Globule Size (GS), Polydispersity Index (PDI), and Zeta Potential (ZP)
Drug Content
Physicochemical Stability
Terminal Moist Heat Sterilization and Stability Assessment of CIP-NE Formulation
3.2.6. Fourier Transform Infrared Spectroscopy (FTIR)
3.2.7. Surface Morphology—Transmission Electron Microscopy (TEM)
3.2.8. In Vitro Release Studies
3.2.9. Ex Vivo Transcorneal Permeation Studies
3.2.10. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pepose, J.S.; Wilhelmus, K.R. Divergent Approaches to the Management of Corneal Ulcers. Am. J. Ophthalmol. 1992, 114, 630–632. [Google Scholar] [CrossRef]
- Jeng, B.H.; Gritz, D.C.; Kumar, A.B.; Holsclaw, D.S.; Porco, T.C.; Smith, S.D.; Whitcher, J.P.; Margolis, T.P.; Wong, I.G. Epidemiology of Ulcerative Keratitis in Northern California. Arch. Ophthalmol. 2010, 128, 1022–1028. [Google Scholar] [CrossRef]
- Teweldemedhin, M.; Gebreyesus, H.; Atsbaha, A.H.; Asgedom, S.W.; Saravanan, M. Bacterial profile of ocular infections: A systematic review. BMC Ophthalmol. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bertino, J.S. Impact of antibiotic resistance in the management of ocular infections: The role of current and future antibiotics. Clin. Ophthalmol. 2009, 3, 507–521. [Google Scholar] [CrossRef]
- Pachigolla, G.; Blomquist, P.; Cavanagh, H.D. Microbial Keratitis Pathogens and Antibiotic Susceptibilities: A 5-Year Review of Cases at an Urban County Hospital in North Texas. Eye Contact Lens Sci. Clin. Pract. 2007, 33, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C.; Jacoby, G.A. Topoisomerase Inhibitors: Fluoroquinolone Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025320. [Google Scholar] [CrossRef] [PubMed]
- Hyndiuk, R.A.; Eiferman, R.A.; Caldwell, D.R.; Rosenwasser, G.O.; Santos, C.I.; Katz, H.R.; Badrinath, S.S.; Reddy, M.K.; Adenis, J.-P.; Klauss, V.; et al. Comparison of Ciprofloxacin Ophthalmic Solution 0.3% to Fortified Tobramycin-Cefazolin in Treating Bacterial Corneal Ulcers. Ophthalmology 1996, 103, 1854–1863. [Google Scholar] [CrossRef]
- Liu, C.; Ji, J.; Li, S.; Wang, Z.; Tang, L.; Cao, W.; Sun, X. Microbiological Isolates and Antibiotic Susceptibilities: A 10-Year Review of Culture-Proven Endophthalmitis Cases. Curr. Eye Res. 2017, 42, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, M. A review of the causes and treatment of bacterial and allergic conjunctivitis. Clin. Ther. 1995, 17, 800–810. [Google Scholar] [CrossRef]
- Balguri, S.P.; Adelli, G.R.; Janga, K.Y.; Bhagav, P.; Majumdar, S. Ocular disposition of ciprofloxacin from topical, PEGylated nanostructured lipid carriers: Effect of molecular weight and density of poly (ethylene) glycol. Int. J. Pharm. 2017, 529, 32–43. [Google Scholar] [CrossRef]
- Youssef, A.; Dudhipala, N.; Majumdar, S. Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis. Pharmaceutics 2020, 12, 572. [Google Scholar] [CrossRef]
- Kowalski, R.P.; Dhaliwal, D.K.; Karenchak, L.M.; Romanowski, E.G.; Mah, F.S.; Ritterband, D.C.; Gordon, Y. Gatifloxacin and moxifloxacin: An in vitro susceptibility comparison to levofloxacin, ciprofloxacin, and ofloxacin using bacterial keratitis isolates. Am. J. Ophthalmol. 2003, 136, 500–505. [Google Scholar] [CrossRef]
- Oliveira, A.D.D.; D’Azevedo, P.A.; Francisco, W.; Höfling-Lima, A.L. In Vitro Activity of Fluoroquinolones against Ocular Bacterial Isolates in São Paulo, Brazil. Cornea 2007, 26, 194–198. [Google Scholar] [CrossRef]
- Chojnacki, M.; Philbrick, A.; Wucher, B.; Reed, J.N.; Tomaras, A.; Dunman, P.M.; Wozniak, R.A.F. Development of a Broad-Spectrum Antimicrobial Combination for the Treatment of Staphylococcus aureus and Pseudomonas aeruginosa Corneal Infections. Antimicrob. Agents Chemother. 2018, 63, 01929-18. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Cholkar, K.; Agrahari, V.; Mitra, A.K. Ocular drug delivery systems: An overview. World J. Pharmacol. 2013, 2, 47–64. [Google Scholar] [CrossRef]
- Achouri, D.; Alhanout, K.; Piccerelle, P.; Andrieu, V. Recent advances in ocular drug delivery. Drug Dev. Ind. Pharm. 2013, 39, 1599–1617. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, P.; Patil, A.; Majumdar, S. Recent advances in topical nano drug-delivery systems for the anterior ocular segment. Ther. Deliv. 2018, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Kansara, V.; Mitra, A.K. Vitreal Pharmacokinetics of Dipeptide Monoester Prodrugs of Ganciclovir. J. Ocul. Pharmacol. Ther. 2006, 22, 231–241. [Google Scholar] [CrossRef]
- Majumdar, S.; Duvvuri, S.; Mitra, A.K. Membrane transporter/receptor-targeted prodrug design: Strategies for human and veterinary drug development. Adv. Drug Deliv. Rev. 2004, 56, 1437–1452. [Google Scholar] [CrossRef]
- Adelli, G.R.; Balguri, S.P.; Bhagav, P.; Raman, V.; Majumdar, S. Diclofenac sodium ion exchange resin complex loaded melt cast films for sustained release ocular delivery. Drug Deliv. 2017, 24, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Tatke, A.; Dudhipala, N.; Janga, K.Y.; Soneta, B.; Avula, B.; Majumdar, S. Melt-Cast Films Significantly Enhance Triamcinolone Acetonide Delivery to the Deeper Ocular Tissues. Pharmaceutics 2019, 11, 158. [Google Scholar] [CrossRef]
- Tamilvanan, S.; Benita, S. The potential of lipid emulsion for ocular delivery of lipophilic drugs. Eur. J. Pharm. Biopharm. 2004, 58, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, R.; Patil, A.; Mehraj, T.; Dudhipala, N.; Majumdar, S. Updates in Ocular Antifungal Pharmacotherapy: Formulation and Clinical Perspectives. Curr. Fungal Infect. Rep. 2019, 13, 45–58. [Google Scholar] [CrossRef]
- Janga, K.Y.; Tatke, A.; Balguri, S.P.; Lamichanne, S.P.; Ibrahim, M.M.; Maria, D.N.; Jablonski, M.M.; Majumdar, S. Ion-sensitive in situ hydrogels of natamycin bilosomes for enhanced and prolonged ocular pharmacotherapy: In vitro permeability, cytotoxicity and in vivo evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Tatke, A.; Dudhipala, N.; Janga, K.Y.; Balguri, S.P.; Avula, B.; Jablonski, M.M.; Majumdar, S. In Situ Gel of Triamcinolone Acetonide-Loaded Solid Lipid Nanoparticles for Improved Topical Ocular Delivery: Tear Kinetics and Ocular Disposition Studies. Nanomaterials 2018, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, M.; Mazumdar, R.; Haque, R. Preparation and assessment of ocular inserts containing sulbactum for controlled drug delivery. J. Drug Deliv. Ther. 2020, 10, 66–71. [Google Scholar] [CrossRef]
- Yukuyama, M.N.; Kato, E.T.M.; Lobenberg, R.; Bou-Chacra, N.A. Challenges and Future Prospects of Nanoemulsion as a Drug Delivery System. Curr. Pharm. Des. 2017, 23, 495–508. [Google Scholar] [CrossRef]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An advanced mode of drug delivery system. 3 Biotech 2015, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A. Chapter 21—Nanoemulsions. In Nanoparticles for Biomedical Applications; Micro and Nano Technologies; Chung, E.J., Leon, L., Rinaldi, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 371–384. ISBN 978-0-12-816662-8. [Google Scholar]
- Fialho, S.L.; Da Silva-Cunha, A. New vehicle based on a microemulsion for topical ocular administration of dexamethasone. Clin. Exp. Ophthalmol. 2004, 32, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Li, N.; Zheng, L.; Tung, C. Studies on the stability of the chloramphenicol in the microemulsion free of alcohols. Eur. J. Pharm. Biopharm. 2006, 62, 288–294. [Google Scholar] [CrossRef]
- Ammar, H.O.; Salama, H.A.; Ghorab, M.; Mahmoud, A.A. Nanoemulsion as a Potential Ophthalmic Delivery System for Dorzolamide Hydrochloride. AAPS Pharm. Sci. Tech. 2009, 10, 808–819. [Google Scholar] [CrossRef]
- Shaker, D.S.; Ishak, R.A.H.; Ghoneim, A.; Elhuoni, M.A. Nanoemulsion: A Review on Mechanisms for the Transdermal Delivery of Hydrophobic and Hydrophilic Drugs. Sci. Pharm. 2019, 87, 17. [Google Scholar] [CrossRef]
- Balguri, S.P.; Adelli, G.R.; Tatke, A.; Janga, K.Y.; Bhagav, P.; Majumdar, S. Melt-Cast Noninvasive Ocular Inserts for Posterior Segment Drug Delivery. J. Pharm. Sci. 2017, 106, 3515–3523. [Google Scholar] [CrossRef]
- Feghhi, M.; Makhmalzadeh, B.S.; Farrahi, F.; Akmali, M.; Hasanvand, N. Anti-microbial Effect and in Vivo Ocular Delivery of Ciprofloxacin-loaded Liposome through Rabbit’s Eye. Curr. Eye Res. 2020, 45, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-K. Liposomes for enhanced bioavailability of water-insoluble drugs: In vivo evidence and recent approaches. Pharmaceutics 2020, 12, 264. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Sarraf, D.; Lee, D.A. The Role of Iontophoresis in Ocular Drug Delivery. J. Ocul. Pharmacol. Ther. 1994, 10, 69–81. [Google Scholar] [CrossRef]
- Rootman, D.S.; Hobden, J.A.; Jantzen, J.A.; Gonzalez, J.R.; O’Callaghan, R.J.; Hill, J.M. Iontophoresis of Tobramycin for the Treatment of Experimental Pseudomonas Keratitis in the Rabbit. Arch. Ophthalmol. 1988, 106, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, F.; Daull, P.; Benita, S.; Buggage, R.; Garrigue, J.-S. Successfully Improving Ocular Drug Delivery Using the Cationic Nanoemulsion, Novasorb. J. Drug Deliv. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cyclosporine A Delivery to the Eye: A Comprehensive Review of Academic and Industrial Efforts|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S0939641116309080?token=796C16FDA68871EECFAFDDB4D2B7C68D1B2626BA99DD049F66098AF71572881DED69449444A99E495FF91ECA063BFA28 (accessed on 25 July 2020).
- Dhahir, R.K.; Al-Nima, A.M.; Al-Bazzaz, F.Y. Nanoemulsions as Ophthalmic Drug Delivery Systems. Turk. J. Pharm. Sci. 2020. [Google Scholar] [CrossRef]
- Lawrence, M.J.; Rees, G.D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2000, 33. [Google Scholar] [CrossRef]
- Narang, A.S.; Delmarre, D.; Gao, D. Stable drug encapsulation in micelles and microemulsions. Int. J. Pharm. 2007, 345, 9–25. [Google Scholar] [CrossRef]
- Ismail, A.; Nasr, M.; Sammour, O. Nanoemulsion as a feasible and biocompatible carrier for ocular delivery of travoprost: Improved pharmacokinetic/pharmacodynamic properties. Int. J. Pharm. 2020, 583, 119402. [Google Scholar] [CrossRef] [PubMed]
- Azmi, N.A.N.; Elgharbawy, A.A.M.; Motlagh, S.R.; Samsudin, N.; Salleh, H.M. Nanoemulsions: Factory for Food, Pharmaceutical and Cosmetics. Process 2019, 7, 617. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Improved anti-hyperlipidemic activity of Rosuvastatin Calcium via lipid nanoparticles: Pharmacokinetic and pharmacodynamic evaluation. Eur. J. Pharm. Biopharm. 2017, 110, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, A.D. Nanoemulsions: Formation, Stability and Applications. Int. J. Res. Sci. Adv. Technol. 2014, 3, 38–43. [Google Scholar]
- Liu, W.; Sun, D.; Li, C.; Liu, Q.; Xu, J. Formation and stability of paraffin oil-in-water nano-emulsions prepared by the emulsion inversion point method. J. Colloid Interface Sci. 2006, 303, 557–563. [Google Scholar] [CrossRef]
- Cappellani, M.R.; Perinelli, D.R.; Pescosolido, L.; Schoubben, A.; Cespi, M.; Cossi, R.; Blasi, P. Injectable nanoemulsions prepared by high pressure homogenization: Processing, sterilization, and size evolution. Appl. Nanosci. 2018, 8, 1483–1491. [Google Scholar] [CrossRef]
- Đorđević, S.M.; Santrač, A.; Cekić, N.D.; Marković, B.D.; Divović, B.; Ilić, T.M.; Savić, M.M.; Savić, S.D. Parenteral nanoemulsions of risperidone for enhanced brain delivery in acute psychosis: Physicochemical and in vivo performances. Int. J. Pharm. 2017, 533, 421–430. [Google Scholar] [CrossRef]
- Narala, A.; Guda, S.; Veerabrahma, K. Lipid Nanoemulsions of Rebamipide: Formulation, Characterization, and In Vivo Evaluation of Pharmacokinetic and Pharmacodynamic Effects. AAPS PharmSciTech 2019, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Peng, H.; Wen, Y.; Li, N. Re-examination of characteristic FTIR spectrum of secondary layer in bilayer oleic acid-coated Fe3O4 nanoparticles. Appl. Surf. Sci. 2010, 256, 3093–3097. [Google Scholar] [CrossRef]
- Sahoo, S.; Chakraborti, C.K.; Mishra, S.C. Qualitative analysis of controlled release ciprofloxacin/carbopol 934 mucoadhesive suspension. J. Adv. Pharm. Technol. Res. 2011, 2, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Laxmi, M.; Bhardwaj, A.; Mehta, S.; Mehta, A. Development and characterization of nanoemulsion as carrier for the en-hancement of bioavailability of artemether. Artif. Cells Nanomed. Biotechnol. 2015, 43, 334–344. [Google Scholar] [CrossRef]
- Dudhipala, N.; Gorre, T. Neuroprotective Effect of Ropinirole Lipid Nanoparticles Enriched Hydrogel for Parkinson’s Disease: In Vitro, Ex Vivo, Pharmacokinetic and Pharmacodynamic Evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Meher, J.G.; Raval, K.; Khan, F.A.; Chaurasia, M.; Jain, N.K.; Chourasia, M.K. Nanoemulsion: Concepts, development and applications in drug delivery. J. Control. Release 2017, 252, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-C.; Qi, H.-P.; Bai, J.-H.; Huang, L.; Cui, H. Effects of Oleic Acid on the Corneal Permeability of Compounds and Evaluation of its Ocular Irritation of Rabbit Eyes. Curr. Eye Res. 2014, 39, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Chetoni, P.; Burgalassi, S.; Najarro, M.; Saettone, M.F. Increased corneal hydration induced by potential ocular penetration enhancers: Assessment by differential scanning calorimetry (DSC) and by desiccation. Int. J. Pharm. 2002, 232, 139–147. [Google Scholar] [CrossRef]
- Clayson, K.; Sandwisch, T.; Ma, Y.; Pavlatos, E.; Pan, X.; Liu, J. Corneal Hydration Control during Ex Vivo Experimentation Using Poloxamers. Curr. Eye Res. 2019, 45, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Dudhipala, N.; Thakkar, R.; Mehraj, T.; Marathe, S.; Gul, W.; ElSohly, M.A.; Murphy, B.; Majumdar, S. Effect of surfactant concentration and sterilization process on intraocular pressure–lowering activity of Δ9-tetrahydrocannabinol-valine-hemisuccinate (NB1111) nanoemulsions. Drug Deliv. Transl. Res. 2020, 1–12. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. Nano-emulsions and Micro-emulsions: Clarifications of the Critical Differences. Pharm. Res. 2010, 28, 978–985. [Google Scholar] [CrossRef]
- Peshkovsky, A.S.; Peshkovsky, S.L.; Bystryak, S. Scalable high-power ultrasonic technology for the production of translucent nanoemulsions. Chem. Eng. Process. Process. Intensif. 2013, 69, 77–82. [Google Scholar] [CrossRef]
- Imre, S.; Dogaru, M.T.; Vari, C.; Muntean, T.; Kelemen, L. Validation of an HPLC method for the determination of ciprofloxacin in human plasma. J. Pharm. Biomed. Anal. 2003, 33, 125–130. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: Characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Deliv. 2016, 23, 395–404. [Google Scholar] [CrossRef]
- Klang, V.; Matsko, N.B.; Valenta, C.; Hofer, F. Electron microscopy of nanoemulsions: An essential tool for characterisation and stability assessment. Micron 2012, 43, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Narendar, D.; Arjun, N.; Someshwar, K.; Rao, Y.M. Quality by design approach for development and optimization of Quetiapine Fumarate effervescent floating matrix tablets for improved oral bioavailability. J. Pharm. Investig. 2016, 46, 253–263. [Google Scholar] [CrossRef]
- Dudhipala, N.; Mohammed, R.P.; Youssef, A.A.A.; Banala, N. Effect of lipid and edge activator concentration on development of Aceclofenac loaded transfersomes gel for transdermal application: In vitro and ex vivo skin permeation. Drug Dev. Ind. Pharm. 2020, 46, 1–28. [Google Scholar] [CrossRef] [PubMed]

| Formulation | Tween® 80 (% w/v) | Formulation | Tween® 80 (% w/v) |
|---|---|---|---|
| O-NE-0.75 | 0.75 | L-NE-0.75 | 0.75 |
| O-NE-1 | 1 | L-NE-1 | 1 |
| O-NE-1.5 | 1.5 | L-NE-1.5 | 1.5 |
| O-NE-2 | 2 | L-NE-2 | 2 |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oil | Solubility | Oil | Solubility |
|---|---|---|---|
| Soybean oil | (−) | Miglyol® 829 | (−) |
| Captex® 355 EP | (−) | Castor oil | (−) |
| Oleic acid | (+) | Labrafac® Lipophile WL 1349 | (+) |
| Sesame oil | (−) | Capryol 90TM | (−) |
| Maisine® CC | (−) | Olive oil | (−) |
| Formulation Composition (% w/v) | CIP-O-NE-10 | CIP-O-NE-20 | CIP-O-NE-30 | CIP-L-NE-10 | CIP-L-NE-20 | CIP-L-NE-30 |
|---|---|---|---|---|---|---|
| Ciprofloxacin | 0.1 | 0.2 | 0.3 | 0.1 | 0.2 | 0.3 |
| Oleic acid | 5 | 5 | 5 | - | - | - |
| Labrafac® Lipophile WL 1349 | - | - | - | 5 | 5 | 5 |
| Tween® 80 | 2 | 2 | 2 | 2 | 2 | 2 |
| Poloxamer 188 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 |
| Glycerin | 2.25 | 2.25 | 2.25 | 2.25 | 2.25 | 2.25 |
| Water up to | 10 mL | 10 mL | 10 mL | 10 mL | 10 mL | 10 mL |
| Formulation | Homogenization Speed (rpm) | With 10 min Sonication | Without Sonication | ||
|---|---|---|---|---|---|
| Globule Size (nm) | PDI | Globule Size (nm) | PDI | ||
| O-NE-2 | 9000 | 225.5 ± 5.1 | 0.15 ± 0.04 | 741.3 ± 60.5 | 0.61 ± 0.09 |
| 10,000 | 187.8 ± 3.7 | 0.11 ± 0.04 | 551.3 ± 75.2 | 0.53 ± 0.04 | |
| 11,000 | 96.3 ± 2.5 | 0.13 ± 0.02 | 395.2 ± 32.7 | 0.43 ± 0.05 | |
| L-NE-2 | 9000 | 283.5 ± 3.6 | 0.22 ± 0.02 | 1098.6 ± 93.3 | 0.92 ± 0.05 |
| 10,000 | 234.7 ± 4.2 | 0.22 ± 0.02 | 760.1 ± 74.5 | 0.71 ± 0.06 | |
| 11,000 | 132.2 ± 1.9 | 0.23 ± 0.03 | 488.2 ± 70.4 | 0.59 ± 0.02 | |
| Formulation | CIP-O-NE-10 | CIP-O-NE-20 | CIP-O-NE-30 | CIP-L-NE-10 | CIP-L-NE-20 | CIP-L-NE-30 |
|---|---|---|---|---|---|---|
| PS (nm) | 134.9 ± 2.9 | 140.5 ± 1.3 | 156.9 ± 1.2 | 156.6 ± 2.3 | 158.9 ± 1.6 | 160.5 ± 1.9 |
| PDI | 0.12 ± 0.07 | 0.15 ± 0.03 | 0.13 ± 0.01 | 0.20 ± 0.02 | 0.23 ± 0.01 | 0.23 ± 0.01 |
| ZP (mV) | −25.13 ± 0.9 | −27.06 ± 0.4 | −29.9 ± 0.55 | −21.9 ± 2.1 | −23.2 ± 1.5 | −24.5 ± 1.2 |
| Drug content (%) | 95.8 ± 1.1 | 97.7 ± 2.6 | 100.0 ± 5.1 | 94.9 ± 2.2 | 95.8 ± 2.1 | 102.1 ± 3.2 |
| Formulation | Flux (µg/min/cm2) | Permeability (×10−5 cm/s) | Folds Enhancement with CIP-C | |
|---|---|---|---|---|
| Flux | P | |||
| CIP-C | 0.39 ± 0.01 | 1.1 ± 0.09 | - | - |
| CIP-O-NE-30 | 0.81 ± 0.03 # | 2.3 ± 0.05 # | 2.1 | 2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssef, A.A.A.; Cai, C.; Dudhipala, N.; Majumdar, S. Design of Topical Ocular Ciprofloxacin Nanoemulsion for the Management of Bacterial Keratitis. Pharmaceuticals 2021, 14, 210. https://doi.org/10.3390/ph14030210
Youssef AAA, Cai C, Dudhipala N, Majumdar S. Design of Topical Ocular Ciprofloxacin Nanoemulsion for the Management of Bacterial Keratitis. Pharmaceuticals. 2021; 14(3):210. https://doi.org/10.3390/ph14030210
Chicago/Turabian StyleYoussef, Ahmed Adel Ali, Chuntian Cai, Narendar Dudhipala, and Soumyajit Majumdar. 2021. "Design of Topical Ocular Ciprofloxacin Nanoemulsion for the Management of Bacterial Keratitis" Pharmaceuticals 14, no. 3: 210. https://doi.org/10.3390/ph14030210
APA StyleYoussef, A. A. A., Cai, C., Dudhipala, N., & Majumdar, S. (2021). Design of Topical Ocular Ciprofloxacin Nanoemulsion for the Management of Bacterial Keratitis. Pharmaceuticals, 14(3), 210. https://doi.org/10.3390/ph14030210