
The Potential Effect of Insulin on AChE and Its Interactions with Rivastigmine In Vitro

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
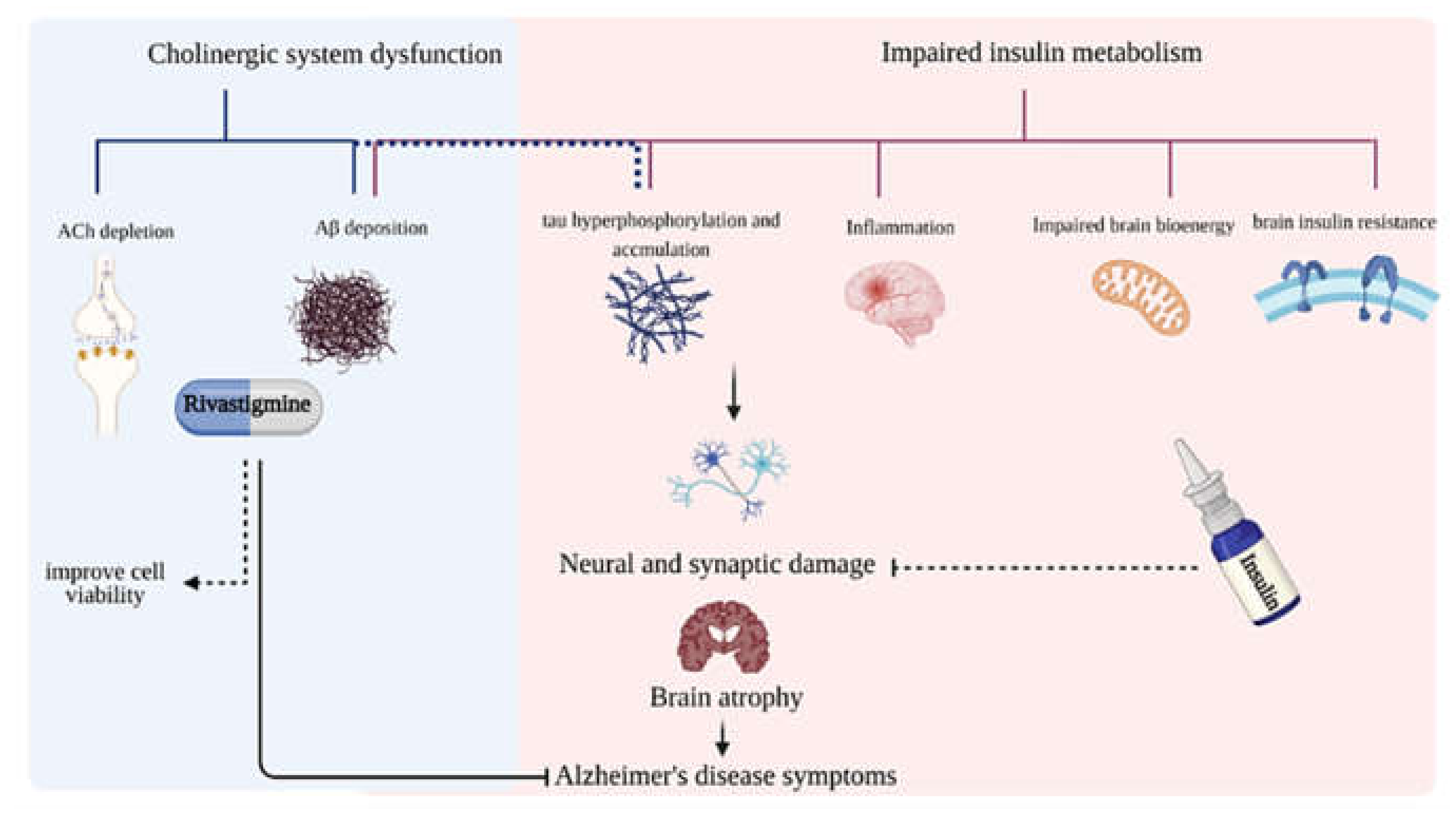
:1. Introduction
2. Results
2.1. Determination of Kinetics for ATCh and Rivastigmine to Study Insulin Effects on AChE Activity
2.2. RSM Studies on the Effect of Different ATCh, Rivastigmine, and Insulin Concentrations on AChE Activity
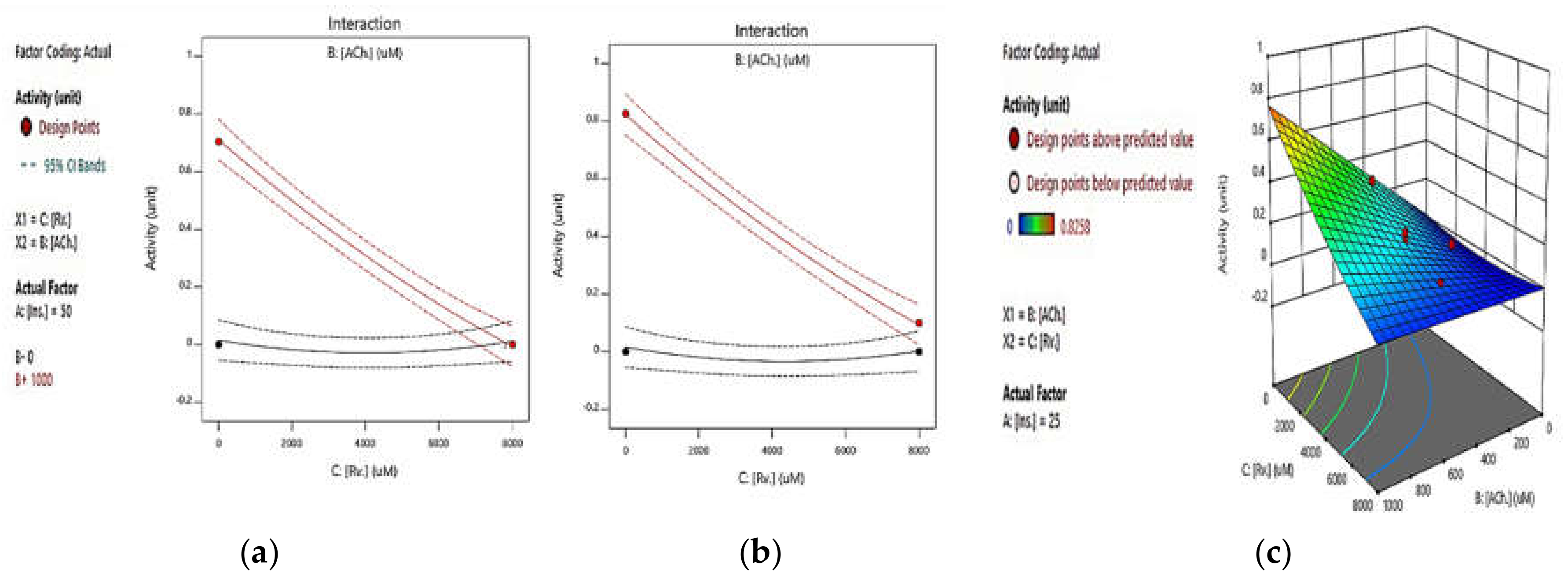
2.2.1. Integrated Effect of Rivastigmine and ATCh
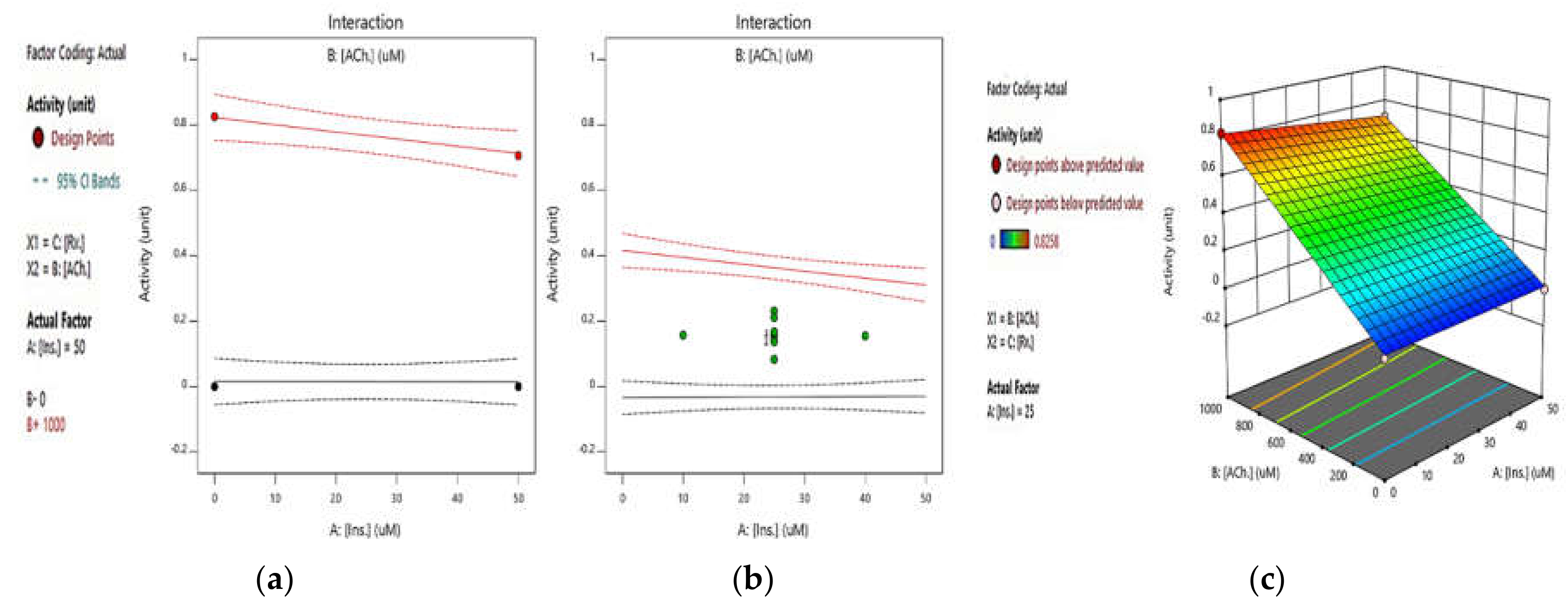
2.2.2. Integrated Effect of Insulin and ATCh
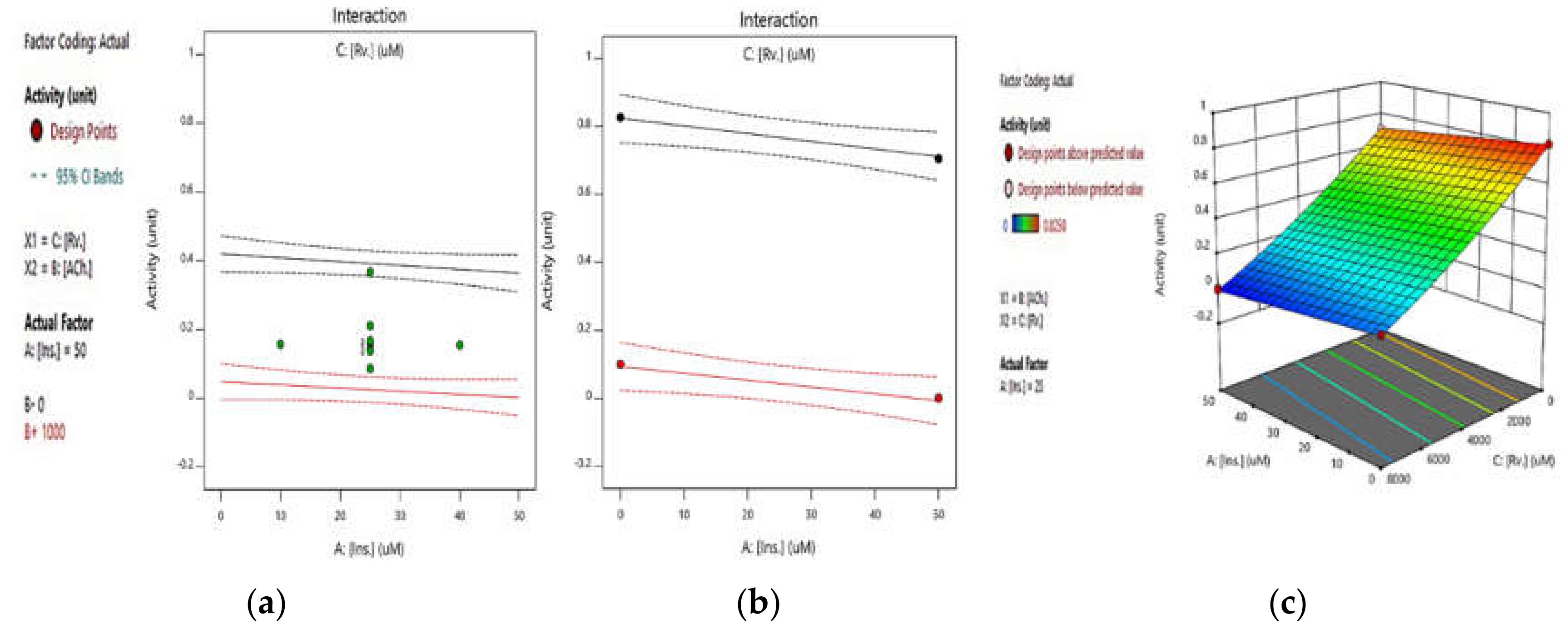
2.2.3. Integrated Effect of Insulin and Rivastigmine
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Determination of Kinetics for ATCh and Rivastigmine to Study Insulin Effect on AChE Activity
4.2.2. RSM Studies on the Effect of Different Atch, Rivastigmine, and Insulin Concentrations on AChE Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: A review. AIMS Neurosci. 2021, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Multi-target drug candidates for multifactorial Alzheimer’s disease: AChE and NMDAR as molecular targets. Mol. Neurobiol. 2021, 58, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Patel, K.; Gold, J.; Hinds, S.; Grossberg, G.T. Recent progress in the pharmacotherapy of Alzheimer’s disease. Drugs Aging 2017, 34, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Polanco, J.C.; Li, C.; Bodea, L.-G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- Bhatt, S.; Puli, L.; Patil, C.R. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug Discov. Today 2020, 26, 794–803. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Hara, Y.; McKeehan, N.; Fillit, H.M. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 2019, 92, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Andreeva, T.V.; Lukiw, W.J.; Rogaev, E.I. Biological Basis for Amyloidogenesis in Alzheimer’s Disease. Biochemistry 2017, 82, 122–139. [Google Scholar] [CrossRef]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef]
- Alzheimer’s Association. Alzheimer’s Association Report (2016 Alzheimer’s disease facts and figures). Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New approaches for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Mamun, A.A.; Jeandet, P.; Aleya, L.; Mansouri, R.A.; Ashraf, G.M.; Mathew, B.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Combination Drug Therapy for the Management of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3272. [Google Scholar] [CrossRef]
- Xiao, G.; Li, Y.; Qiang, X.; Xu, R.; Zheng, Y.; Cao, Z.; Luo, L.; Yang, X.; Sang, Z.; Su, F. Design, synthesis and biological evaluation of 4′-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Luc, M.; Wozniak, M.; Helemejko, M.; Rymaszewska, J. Tackling Alzheimer’s disease: Hypothetical synergism between anti-inflammatory and anti-diabetic agents. Life Sci 2019, 231, 116483. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, B.; Xia, S.; Fang, L.; Gou, S. ROS-responsive and multifunctional anti-Alzheimer prodrugs: Tacrine-ibuprofen hybrids via a phenyl boronate linker. Eur. J. Med. Chem. 2021, 212, 112997. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, M.H.; Nam, G.; Lee, M.; Kang, J.; Song, I.-S.; Choi, M.-K.; Jin, H.K.; Bae, J.-S.; Lim, M.H. A Glycosylated Prodrug to Attenuate Neuroinflammation and Improve Cognitive Deficits in Alzheimer’s Disease Transgenic Mice. Mol. Pharm. 2021, 18, 101–112. [Google Scholar] [CrossRef]
- Dhala, I.; Khan, T.; Prabhu, A. Chimeric Conjugates for Alzheimer’s Disease. Exon Publ. 2020, Ch10, 165–180. [Google Scholar]
- Waseem, R.; Shamsi, A.; Mohammad, T.; Alhumaydhi, F.A.; Kazim, S.N.; Hassan, M.I.; Ahmad, F.; Islam, A. Multispectroscopic and Molecular Docking Insight into Elucidating the Interaction of Irisin with Rivastigmine Tartrate: A Combinational Therapy Approach to Fight Alzheimer’s Disease. ACS Omega 2021, 6, 7910–7921. [Google Scholar] [CrossRef]
- Abdel, M.E.A.E.-F.; Khalil, W.F.; Mohamed, S.M. Effect of Curcumin, Exelon and their Combination on Brain in Alzheimer’s Disease-Induced Rats. J. Adv. Med. Med. Res. 2021, 33, 65–78. [Google Scholar] [CrossRef]
- Zhao, J.; Yin, F.; Ji, L.; Wang, C.; Shi, C.; Liu, X.; Yang, H.; Wang, X.; Kong, L. Development of a Tau-Targeted Drug Delivery System Using a Multifunctional Nanoscale Metal–Organic Framework for Alzheimer’s Disease Therapy. ACS Appl. Mater. Interfaces 2020, 12, 44447–44458. [Google Scholar] [CrossRef]
- Pardridge, W.M. Treatment of Alzheimer’s Disease and Blood–Brain Barrier Drug Delivery. Pharmaceuticals 2020, 13, 394. [Google Scholar] [CrossRef]
- Stavrakov, G.; Philipova, I.; Lukarski, A.; Atanasova, M.; Zheleva, D.; Zhivkova, Z.D.; Ivanov, S.; Atanasova, T.; Konstantinov, S.; Doytchinova, I. Galantamine-Curcumin Hybrids as Dual-Site Binding Acetylcholinesterase Inhibitors. Molecules 2020, 25, 3341. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [Green Version]
- Pakala, R.S.; Brown, K.N.; Preuss, C.V. Cholinergic Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Patel, P.H.; Gupta, V. Rivastigmine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Ray, B.; Maloney, B.; Sambamurti, K.; Karnati, H.K.; Nelson, P.T.; Greig, N.H.; Lahiri, D.K. Rivastigmine modifies the α-secretase pathway and potentially early Alzheimer’s disease. Transl. Psychiatry 2020, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Campos, E.O.; Alvarez, A.; Inestrosa, N.C. Brain acetylcholinesterase promotes amyloid-β-peptide aggregation but does not hydrolyze amyloid precursor protein peptides. Neurochem. Res. 1998, 23, 135–140. [Google Scholar] [CrossRef]
- Inestrosa Cantín, N. Acetylcholinesterase Accelerates Assembly of Amyloid-B-Peptides Into Alzheimer’s Fibrils: Possible Role of the Peripheral Site of the Enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.; Hoffman, H.; Chakkamparambil, B.; Grossberg, G.T. Evaluation of rivastigmine in Alzheimer’s disease. Neurodegener. Dis. Manag. 2021, 11, 35–48. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef]
- De la Monte, S.M. Intranasal insulin therapy for cognitive impairment and neurodegeneration: Current state of the art. Expert Opin. Drug Deliv. 2013, 10, 1699–1709. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A.; Bishnoi, M.; Sah, S.P. Sodium orthovanadate improves learning and memory in intracerebroventricular-streptozotocin rat model of Alzheimer’s disease through modulation of brain insulin resistance induced tau pathology. Brain Res. Bull. 2020, 164, 83–97. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Guillén-Nieto, G.; Rodríguez-Rodríguez, N.; Bringas-Vega, M.L.; García-del-Barco-Herrera, D.; Berlanga-Saez, J.O.; García-Ojalvo, A.; Valdés-Sosa, M.J.; Valdés-Sosa, P.A. Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review. Front. Endocrinol. 2020, 11, 560375. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Tauber, S.C.; Djukic, M.; Gossner, J.; Eiffert, H.; Brück, W.; Nau, R. Sepsis-associated encephalopathy and septic encephalitis: An update. Expert Rev. Anti-Infect. Ther. 2020, 6, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. Effect of insulin and melatonin on acetylcholinesterase activity in the brain of amnesic mice. Behav. Brain Res. 2008, 189, 381–386. [Google Scholar] [CrossRef]
- Nampoothiri, M.; Kumar, N.; Venkata Ramalingayya, G.; Gopalan Kutty, N.; Krishnadas, N.; Mallikarjuna Rao, C. Effect of insulin on spatial memory in aluminum chloride-induced dementia in rats. NeuroReport 2017, 28, 540–544. [Google Scholar] [CrossRef]
- Lakhman, S.; Kaur, G. Effect of alloxan-induced diabetes on acetylcholinesterase activity from discrete areas of rat brain. Neurochem. Int. 1994, 24, 159–163. [Google Scholar] [CrossRef]
- Catalan, R.; Martinez, A.; Mata, F.; Aragones, M. Effect of insulin on acetylcholinesterase activity. Biochem. Biophys. Res. Commun. 1981, 101, 1216–1220. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, L.; Yu, L.; Lin, B.; Hou, Y.; Wu, J.; Huang, Q.; Han, Y.; Guo, L.; Ouyang, Q.; et al. Acetylcholinesterase is associated with apoptosis in β cells and contributes to insulin-dependent diabetes mellitus pathogenesis. Acta Biochim. Biophys. Sin. 2012, 44, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.I.; Zaid, M.A. Insulin-Like Effect Of (–) Epicatechin On Erythrocyte Membrane Acetylcholinesterase Activity In Type 2 Diabetes Mellitus. Clin. Exp. Pharmacol. Physiol. 2001, 28, 776–778. [Google Scholar] [CrossRef]
- Prasasty, V.; Radifar, M.; Istyastono, E. Natural Peptides in Drug Discovery Targeting Acetylcholinesterase. Molecules 2018, 23, 2344. [Google Scholar] [CrossRef] [Green Version]
- Torrent, J.; Vilchez-Acosta, A.; Munoz-Torrero, D.; Trovaslet, M.; Nachon, F.; Chatonnet, A.; Grznarova, K.; Acquatella-Tran Van Ba, I.; Le Goffic, R.; Herzog, L.; et al. Interaction of prion protein with acetylcholinesterase: Potential pathobiological implications in prion diseases. Acta Neuropathol. Commun. 2015, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, A.; Mohammad, T.; Khan, M.S.; Shahwan, M.; Husain, F.M.; Rehman, M.T.; Hassan, M.I.; Ahmad, F.; Islam, A. Unraveling Binding Mechanism of Alzheimer’s Drug Rivastigmine Tartrate with Human Transferrin: Molecular Docking and Multi-Spectroscopic Approach towards Neurodegenerative Diseases. Biomolecules 2019, 9, 495. [Google Scholar] [CrossRef] [Green Version]
- Jamir, K.; Ganguly, R.; Seshagirirao, K. ZCPG, a cysteine protease from Zingiber montanum rhizome exhibits enhanced anti-inflammatory and acetylcholinesterase inhibition potential. Int. J. Biol. Macromol. 2020, 163, 2429–2438. [Google Scholar] [CrossRef]
- Waqar, M.; Batool, S. In silico analysis of binding of neurotoxic venom ligands with acetylcholinesterase for therapeutic use in treatment of Alzheimer’s disease. J. Theor. Biol. 2015, 372, 107–117. [Google Scholar] [CrossRef]
- Arduini, F.; Errico, I.; Amine, A.; Micheli, L.; Palleschi, G.; Moscone, D. Enzymatic Spectrophotometric Method for Aflatoxin B Detection Based on Acetylcholinesterase Inhibition. Anal. Chem. 2007, 79, 3409–3415. [Google Scholar] [CrossRef]
- Zhao, T.; Su, G.; Wang, S.; Zhang, Q.; Zhang, J.; Zheng, L.; Sun, B.; Zhao, M. Neuroprotective Effects of Acetylcholinesterase Inhibitory Peptides from Anchovy (Coilia mystus) against Glutamate-Induced Toxicity in PC12 Cells. J. Agric. Food Chem. 2017, 65, 11192–11201. [Google Scholar] [CrossRef]
- Malomo, S.A.; Aluko, R.E. In vitro acetylcholinesterase-inhibitory properties of enzymatic hemp seed protein hydrolysates. J. Am. Oil Chem. Soc. 2016, 93, 411–420. [Google Scholar] [CrossRef]
- Yu, Z.; Ji, H.; Shen, J.; Kan, R.; Zhao, W.; Li, J.; Ding, L.; Liu, J. Identification and molecular docking study of fish roe-derived peptides as potent BACE 1, AChE, and BChE inhibitors. Food Funct. 2020, 11, 6643–6651. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Kaur, G.; Arora, S.K. Acetylcholinesterase and Na+, K+-ATPase activities in different regions of rat brain during insulin-induced hypoglycemia. Mol. Chem. Neuropathol. 1994, 21, 83–93. [Google Scholar] [CrossRef]
- Baruah, P.; Das, A.; Paul, D.; Chakrabarty, S.; Aguan, K.; Mitra, S. Sulfonylurea Class of Antidiabetic Drugs Inhibit Acetylcholinesterase Activity: Unexplored Auxiliary Pharmacological Benefit toward Alzheimer’s Disease. ACS Pharmacol. Transl. Sci. 2021, 4, 193–205. [Google Scholar] [CrossRef]
- Dubey, S.K.; Lakshmi, K.; Krishna, K.V.; Agrawal, M.; Singhvi, G.; Saha, R.N.; Saraf, S.; Saraf, S.; Shukla, R.; Alexander, A. Insulin mediated novel therapies for the treatment of Alzheimer’s disease. Life Sci. 2020, 249, 117540. [Google Scholar] [CrossRef]
- Mejido, D.C.; Andrade, J.; Vieira, M.N.; Ferreira, S.T.; De Felice, F.G. Insulin and leptin as potential cognitive enhancers in metabolic disorders and Alzheimer’s disease. Neuropharmacology 2020, 171, 108115. [Google Scholar] [CrossRef]
- Doyle, M.E.; Egan, J.M. Pharmacological agents that directly modulate insulin secretion. Pharmacol. Rev. 2003, 55, 105–131. [Google Scholar] [CrossRef]
- Samadian, M.; Gholipour, M.; Hajiesmaeili, M.; Taheri, M.; Ghafouri-Fard, S. The Eminent Role of microRNAs in the Pathogenesis of Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 107. [Google Scholar] [CrossRef]
- Fishwick, K.J.; Rylett, R.J. Insulin regulates the activity of the high-affinity choline transporter CHT. PLoS ONE 2015, 10, e0132934. [Google Scholar] [CrossRef]
- Cao, Y.; Herrero-Nogareda, L.; Cedergreen, N. A comparative study of acetylcholinesterase and general-esterase activity assays using different substrates, in vitro and in vivo exposures and model organisms. Ecotoxicol. Environ. Saf. 2020, 189, 109954. [Google Scholar] [CrossRef]
- Kasteel, E.E.J.; Nijmeijer, S.M.; Darney, K.; Lautz, L.S.; Dorne, J.L.C.M.; Kramer, N.I.; Westerink, R.H.S. Acetylcholinesterase inhibition in electric eel and human donor blood: An in vitro approach to investigate interspecies differences and human variability in toxicodynamics. Arch. Toxicol. 2020, 94, 4055–4065. [Google Scholar] [CrossRef]
- Bezerra, M.A.; Santelli, R.E.; Oliveira, E.P.; Villar, L.S.; Escaleira, L.A. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 2008, 76, 965–977. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jamshidnejad-Tosaramandani, T.; Kashanian, S.; Babaei, M.; Al-Sabri, M.H.; Schiöth, H.B. The Potential Effect of Insulin on AChE and Its Interactions with Rivastigmine In Vitro. Pharmaceuticals 2021, 14, 1136. https://doi.org/10.3390/ph14111136
Jamshidnejad-Tosaramandani T, Kashanian S, Babaei M, Al-Sabri MH, Schiöth HB. The Potential Effect of Insulin on AChE and Its Interactions with Rivastigmine In Vitro. Pharmaceuticals. 2021; 14(11):1136. https://doi.org/10.3390/ph14111136
Chicago/Turabian StyleJamshidnejad-Tosaramandani, Tahereh, Soheila Kashanian, Mahsa Babaei, Mohamed H. Al-Sabri, and Helgi B. Schiöth. 2021. "The Potential Effect of Insulin on AChE and Its Interactions with Rivastigmine In Vitro" Pharmaceuticals 14, no. 11: 1136. https://doi.org/10.3390/ph14111136
APA StyleJamshidnejad-Tosaramandani, T., Kashanian, S., Babaei, M., Al-Sabri, M. H., & Schiöth, H. B. (2021). The Potential Effect of Insulin on AChE and Its Interactions with Rivastigmine In Vitro. Pharmaceuticals, 14(11), 1136. https://doi.org/10.3390/ph14111136