
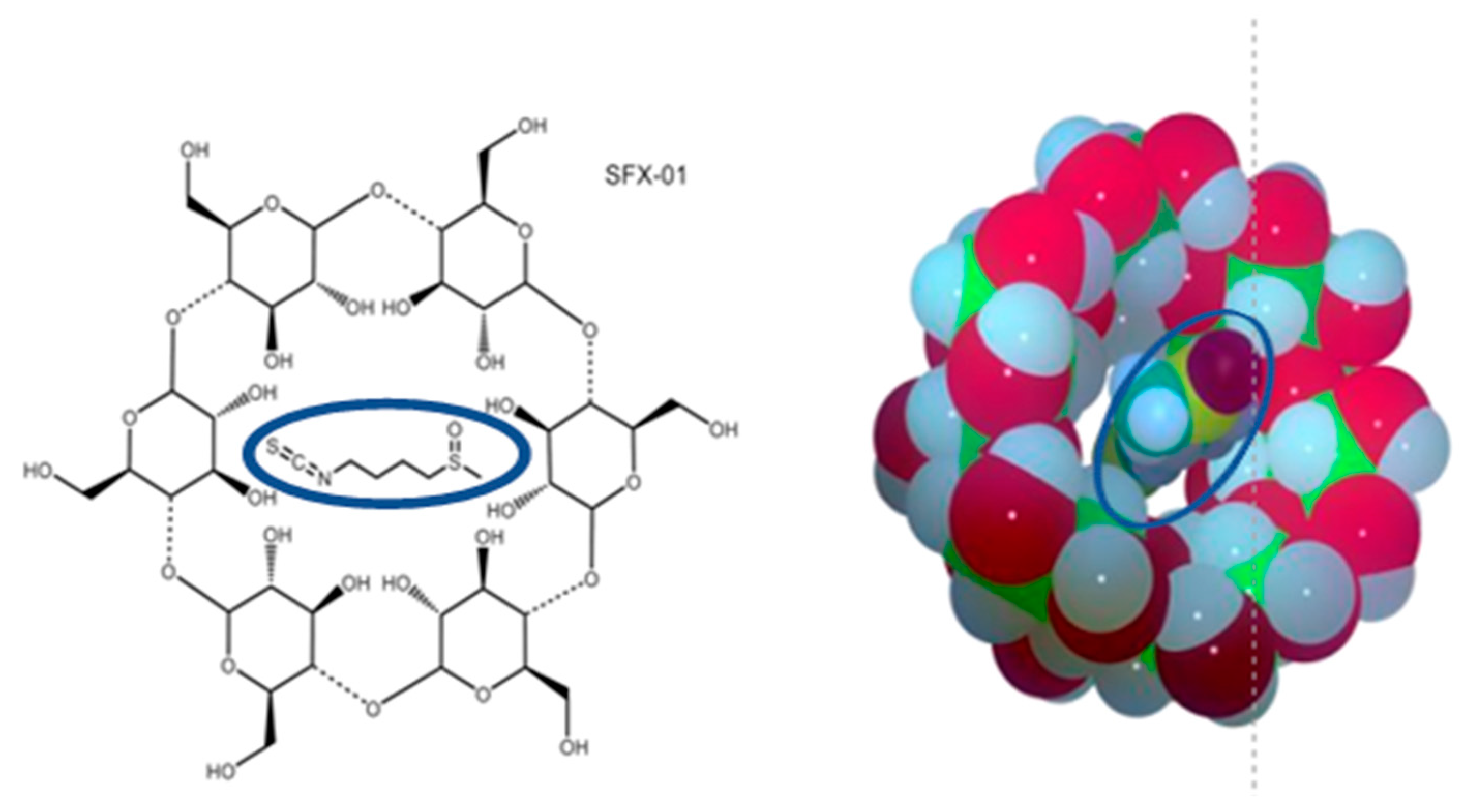
Multiple Antitumor Molecular Mechanisms Are Activated by a Fully Synthetic and Stabilized Pharmaceutical Product Delivering the Active Compound Sulforaphane (SFX-01) in Preclinical Model of Human Glioblastoma


 , ,
, ,  , ,
, , 

Abstract
:1. Introduction
2. Results
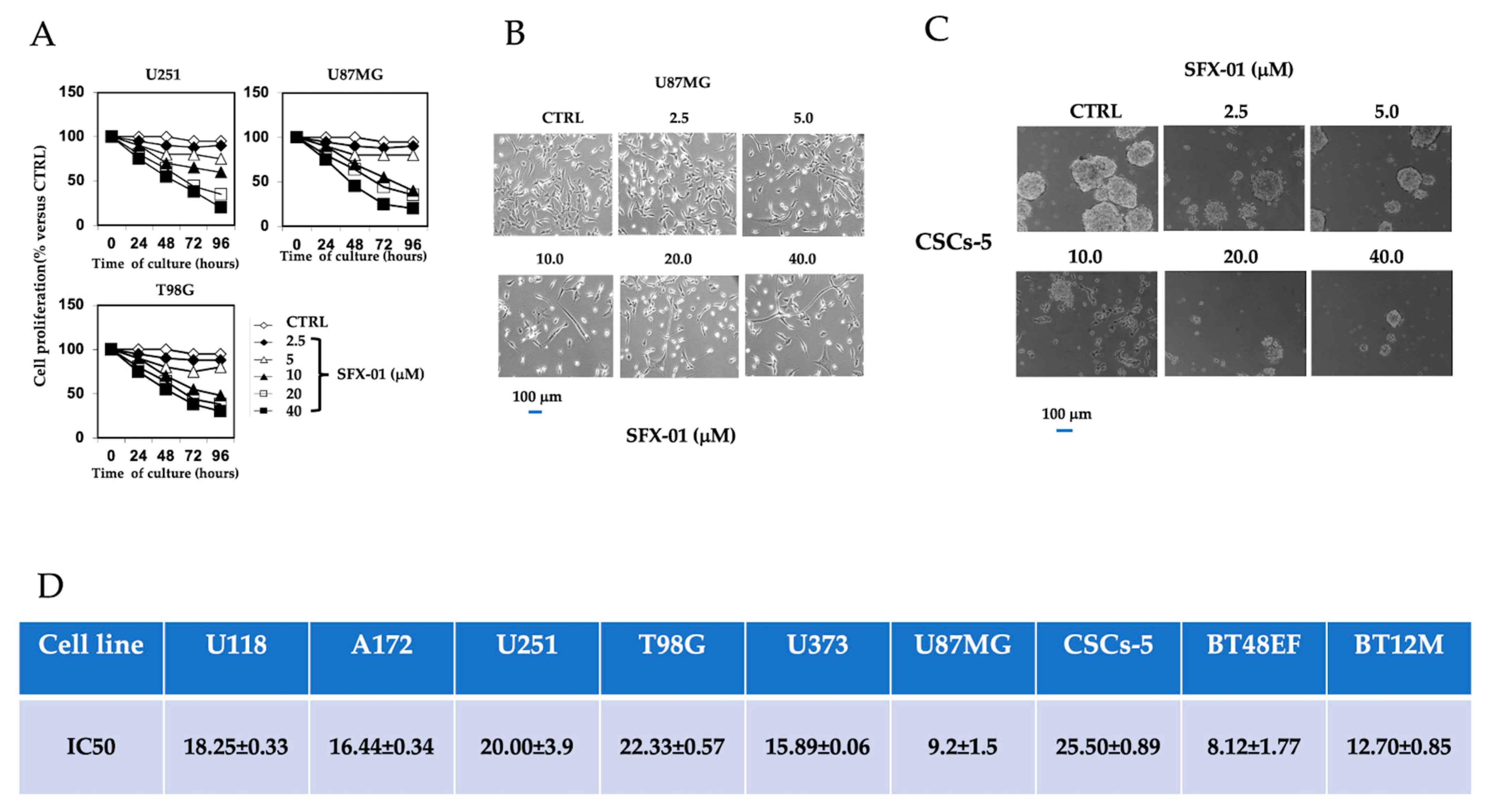
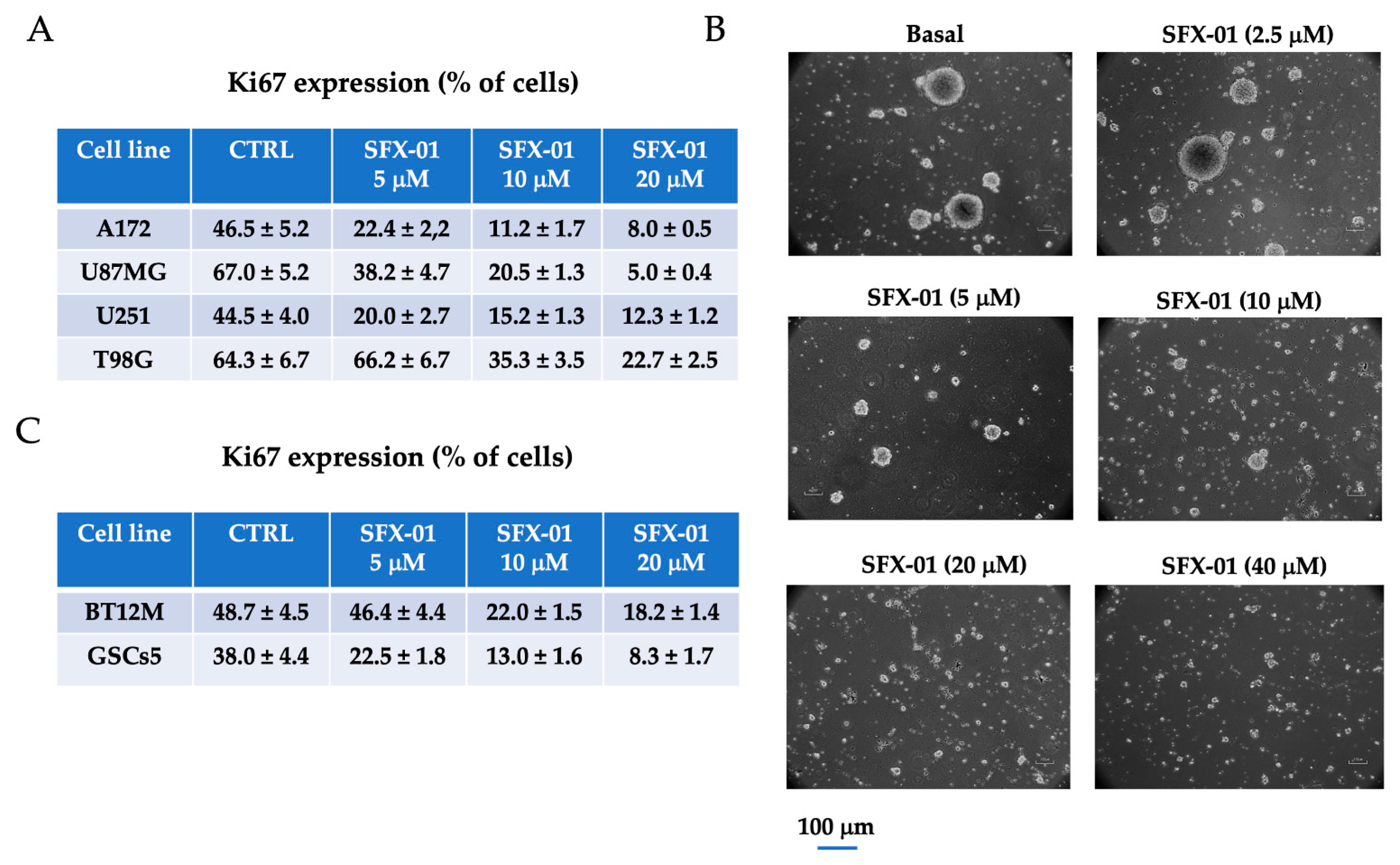
2.1. SFX-01 Inhibited Proliferation of GBM Cells and GICs In Vitro
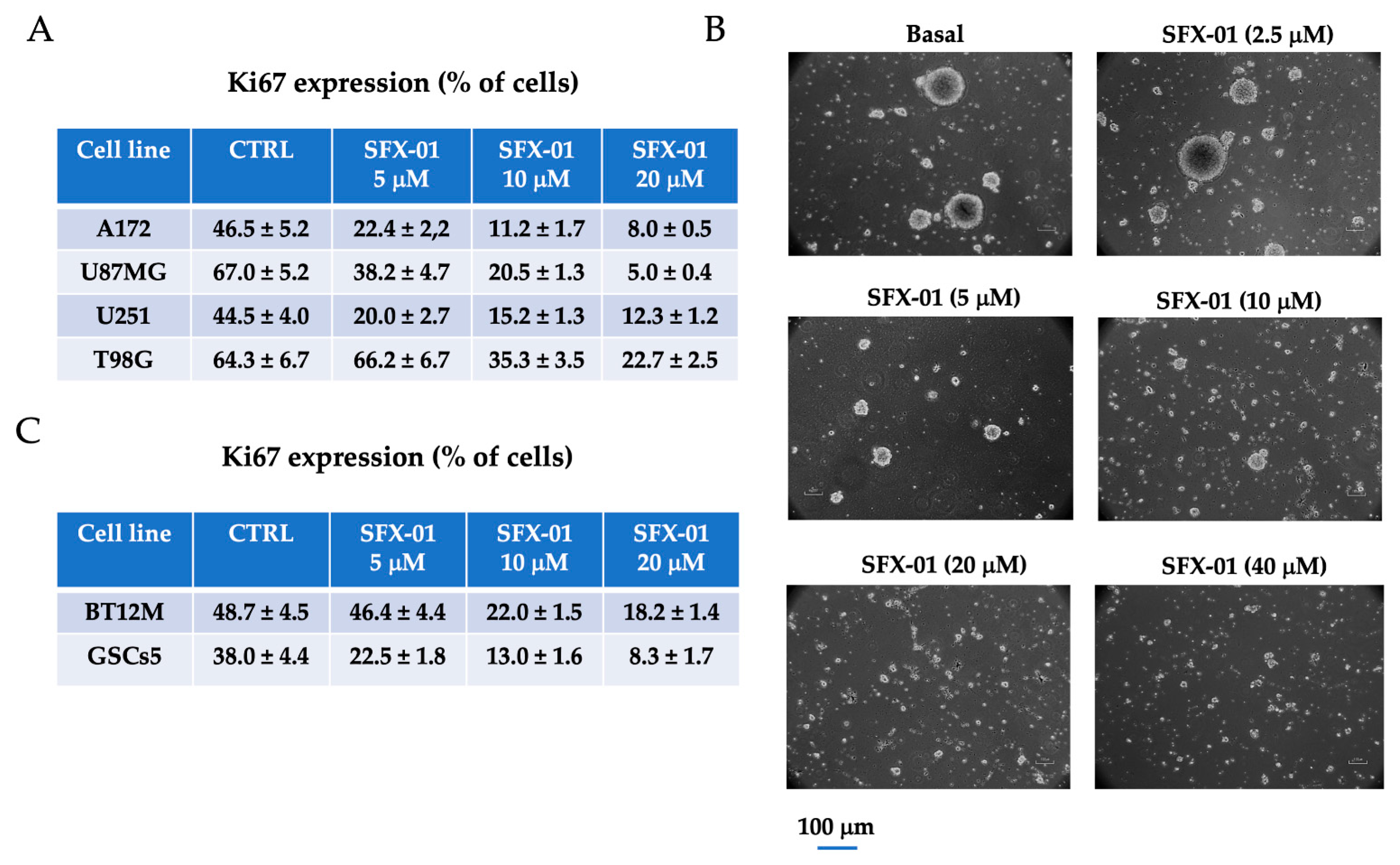
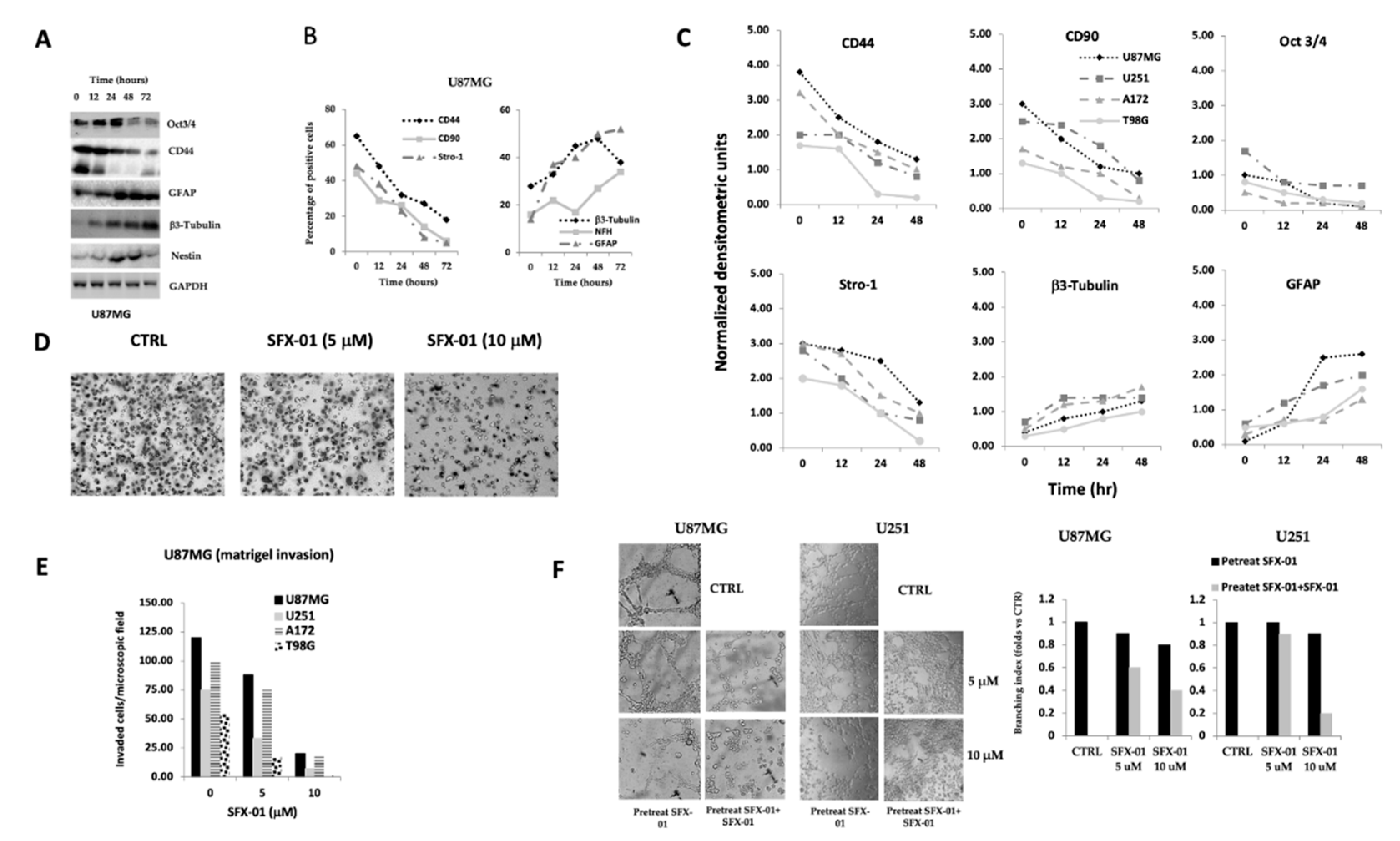
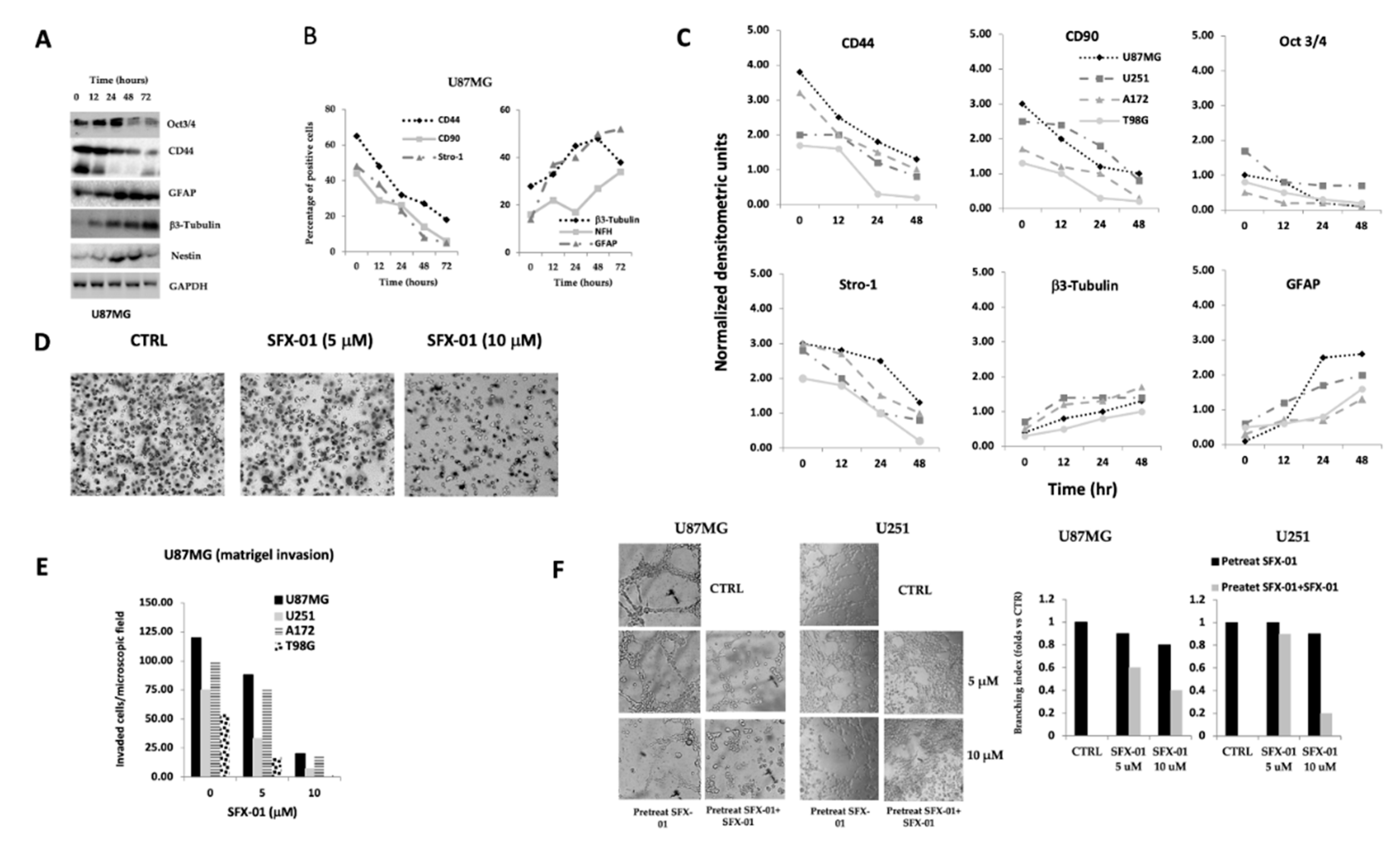
2.2. SFX-01 Reduces Stemness and Malignancy in GBM Cell Lines
2.3. SFX-01 Reduced HIF1α Expression Downregulating Neoangiogenesis
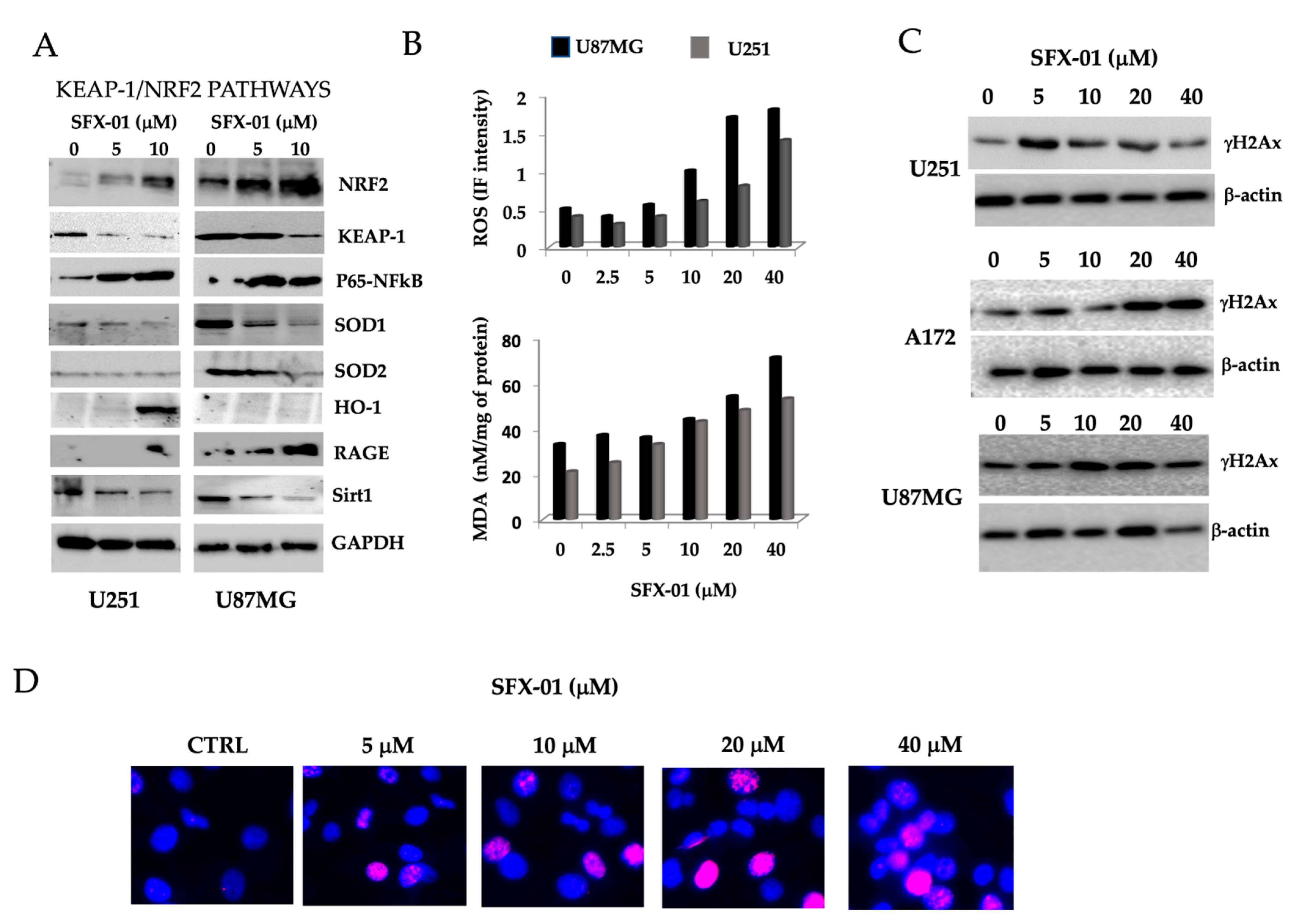
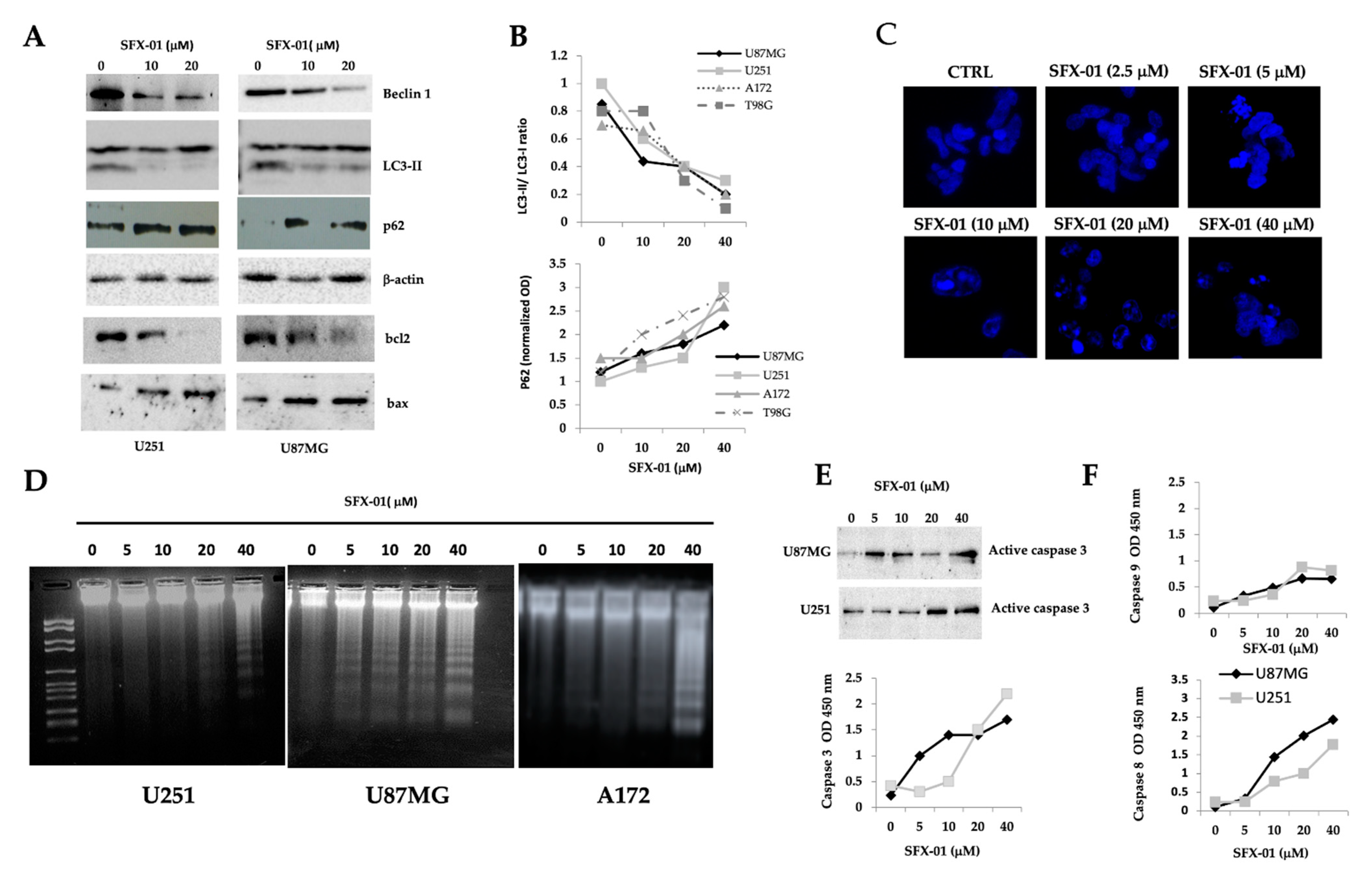
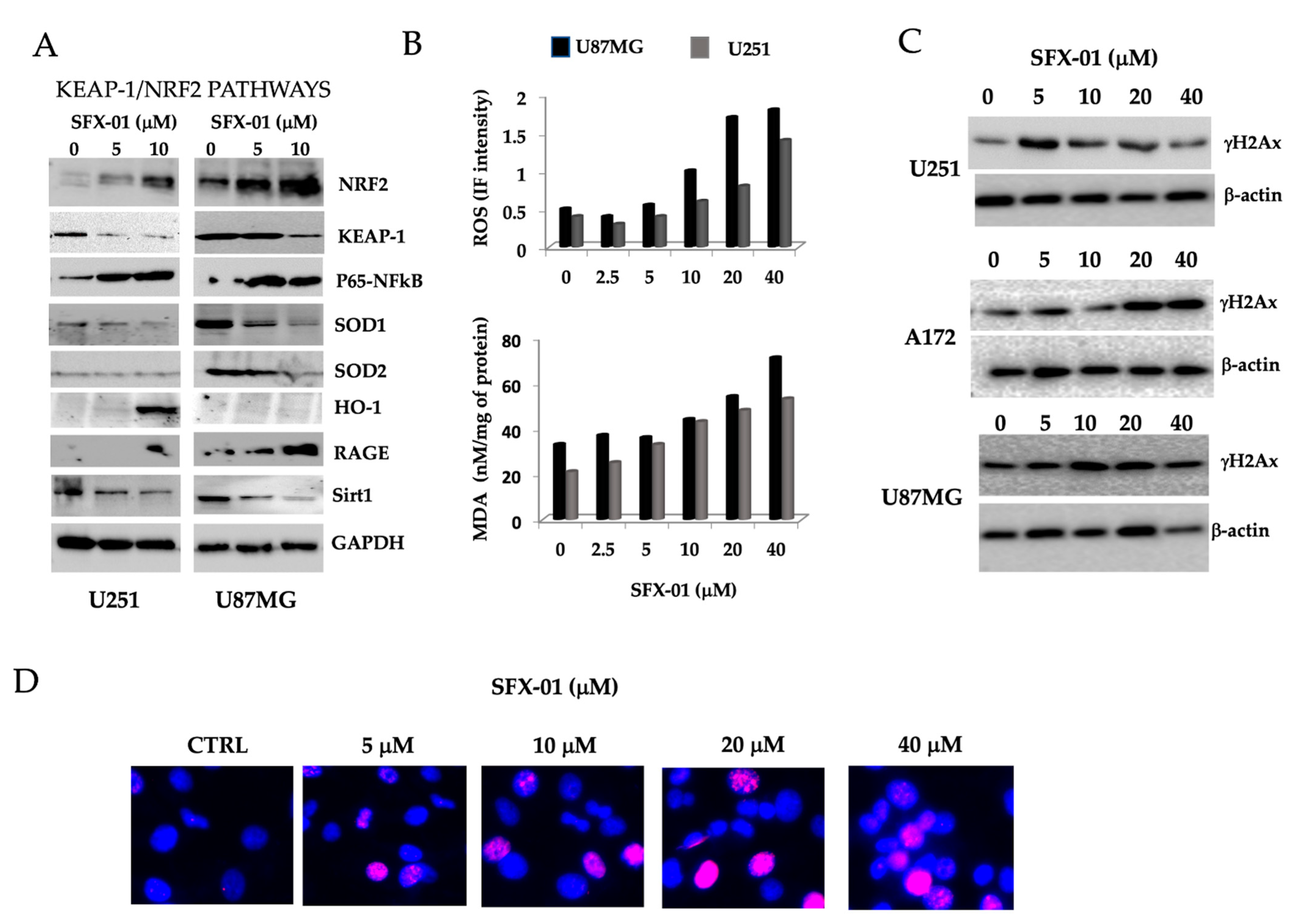
2.4. Sulforaphane Induces Reactive Oxygen Species (ROS), Reduces Basal Autophagy, and Activates Apoptosis-Mediated Cell Death in GBM Cell Lines and GICs
2.5. SFN Induces DNA Laddering and Down-Regulates Autophagic Processes
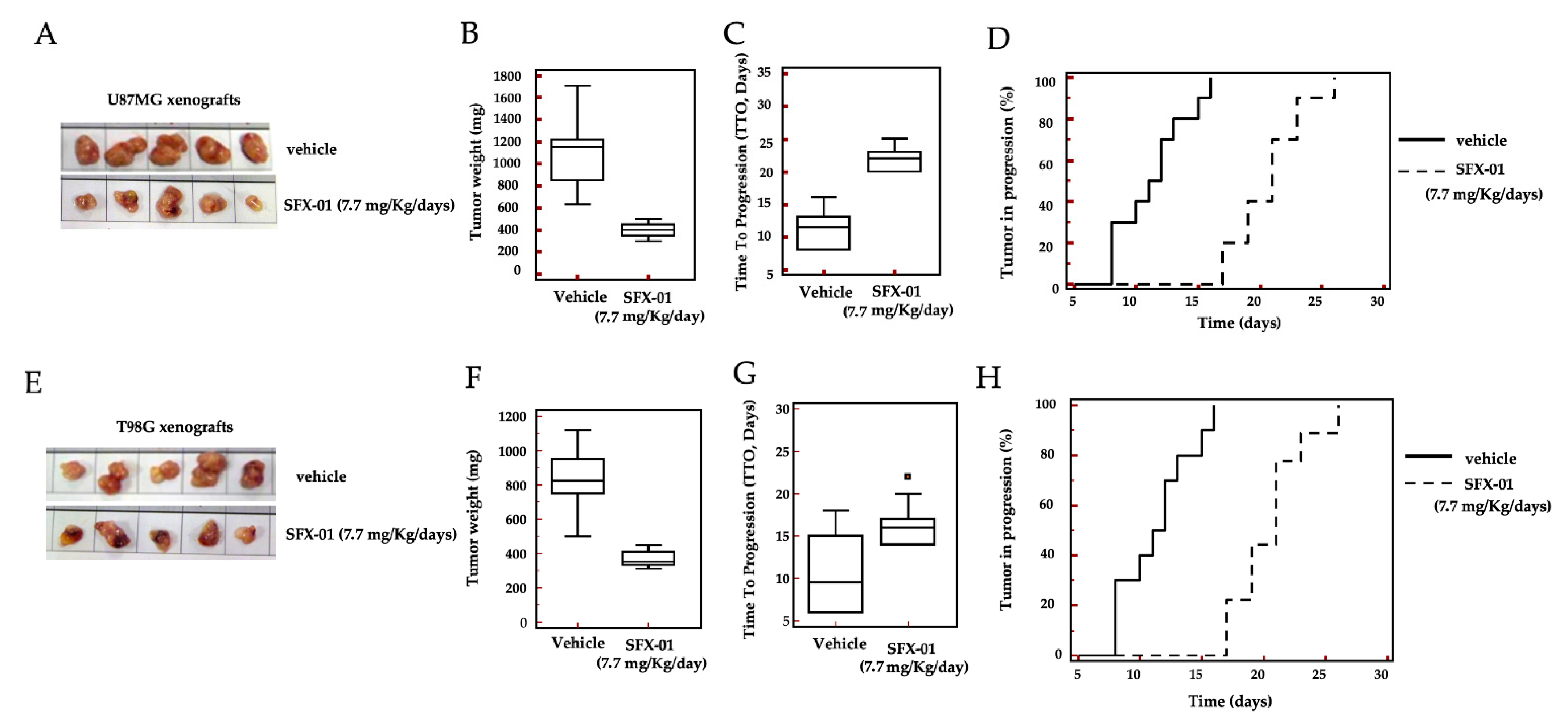
2.6. In Vivo Administration of SFX-01 in U87MG and T98G Subcutaneous Xenografts
2.7. Immunohistochemistry Analyses
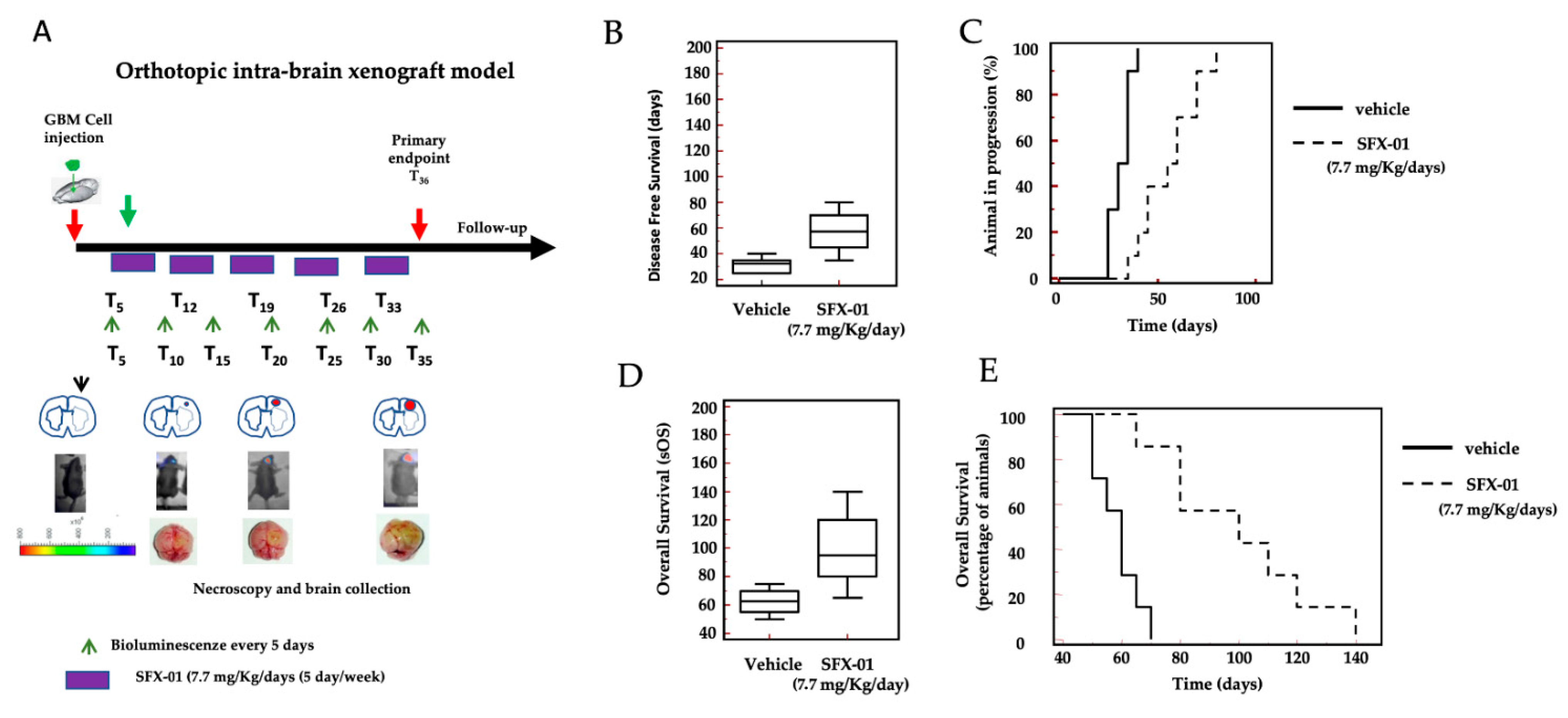
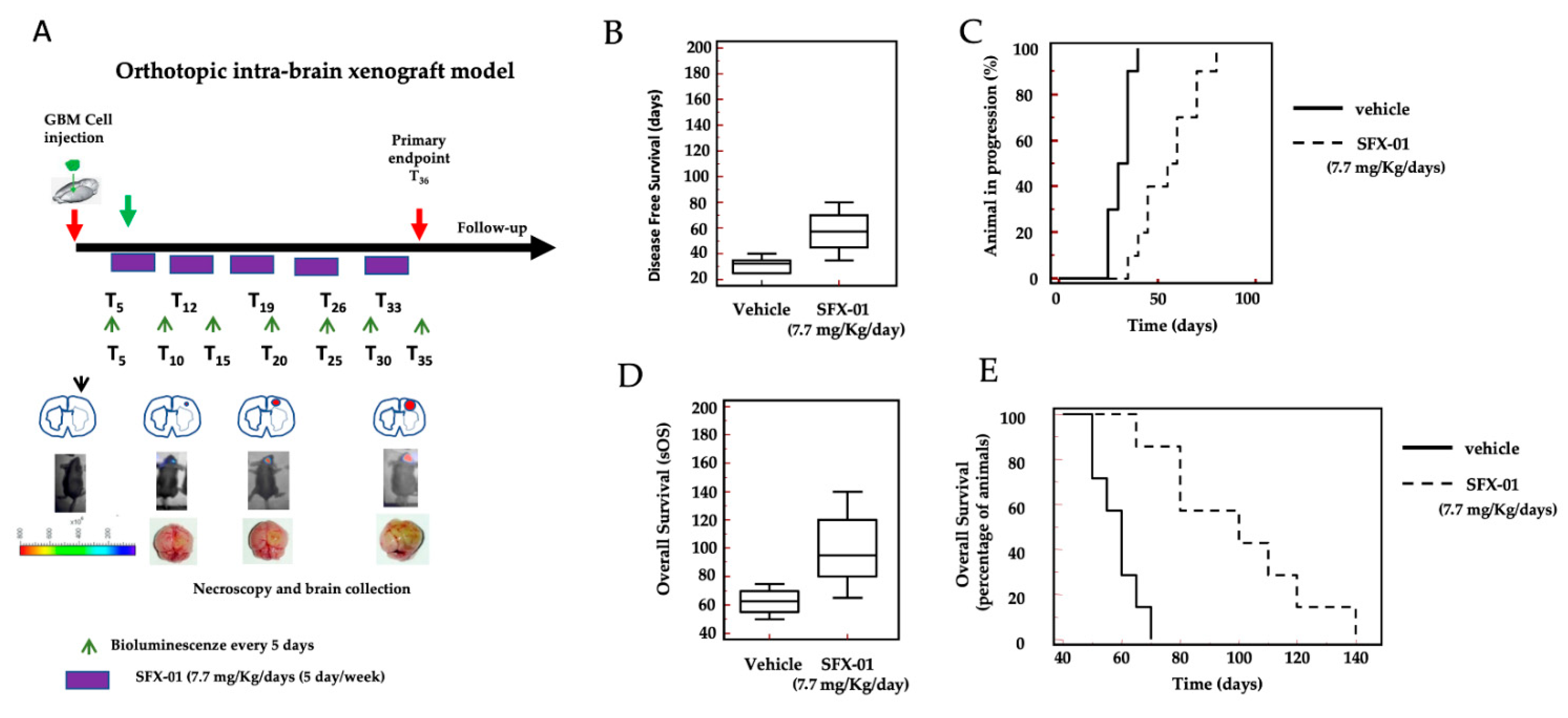
2.8. SFX-01 Increases Disease-Free Survival (DFS) and Overall Survival (OS) in Orthotopic Intra-Brain Tumors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Cell Proliferation Analyses
4.4. Neurosphere Growth
4.5. Analysis of Lipid Peroxidation
4.6. Detection of Intracellular ROS
4.7. Cell Lysate, Western Blot, and Akt/mTOR Enzymatic Analyses
4.8. Immunofluorescence (IF) Studies
4.9. FACS Analyses
4.10. Migration and Invasion Assays
4.11. Conditioned Medium Preparation, Vasculogenic Mimicry, and Endothelial Tubule Formation
4.12. Animal Experiments: Subcutaneous Xenograft Model
4.13. HDAC Activity in Subcutaneously Xenografted Tumors
4.14. Immunohistochemical Analyses
4.15. Orthotopic Intracranial Model
4.16. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Guo, S.; Zhang, X.; Mo, Y.; Ke, S.; Duan, C. Optimal treatment strategy for adult patients with newly diagnosed glioblastoma: A systematic review and network meta-analysis. Neurosurg. Rev. 2021, 44, 1943–1955. [Google Scholar] [CrossRef]
- Available online: https://www.guidelinecentral.com/summaries/glioblastoma (accessed on 1 October 2021).
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ Necrosis in Glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wu, S.; Chen, X.; Ye, Y.; Weng, Y.; Pan, Y.; Chen, Z.; Chen, L.; Qiu, X.; Qiu, S. Characterization of hypoxia signature to evaluate the tumor immune microenvironment and predict prognosis in glioma groups. Front. Oncol. 2020, 10, 796. [Google Scholar] [CrossRef]
- Valtorta, S.; Salvatore, D.; Rainone, P.; Belloli, S.; Bertoli, G.; Moresco, R.M. Molecular and cellular complexity of glioma. Focus on tumour microenvironment and the use of molecular and imaging biomarkers to overcome treatment resistance. Int. J. Mol. Sci. 2020, 21, 5631. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Winkler, F.; Elia, A.E.H. New directions in the treatment of glioblastoma. Semin. Neurol. 2018, 38, 50–61. [Google Scholar] [CrossRef]
- Taga, T.; Tabu, K. Glioma progression and recurrence involving maintenance and expansion strategies of glioma stem cells by organizing self-advantageous niche microenvironments. Inflamm. Regen. 2020, 40, 1–5. [Google Scholar] [CrossRef]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef]
- Steponaitis, G.; Tamasauskas, A. Mesenchymal and proneural subtypes of glioblastoma disclose branching based on gsc associated signature. Int. J. Mol. Sci. 2021, 22, 4964. [Google Scholar] [CrossRef]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal variation in drug and radiation response among glio-ma-initiating cells is linked to proneural-mesenchymal transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef]
- Guohao, H.; Wang, T.; Huang, G.; Clive, D.C.; Da Costa, C.; Li, H.; Lv, S.; Li, N. Rediscovering potential molecular targets for glioma therapy through the analysis of the cell of origin, microenvironment, and metabolism. Curr. Cancer Drug Targets 2021, 21, 1. [Google Scholar] [CrossRef]
- Papale, M.; Buccarelli, M.; Mollinari, C.; Russo, M.A.; Pallini, R.; Ricci-Vitiani, L.; Tafani, M. hypoxia, inflammation and necrosis as determinants of glioblastoma cancer stem cells progression. Int. J. Mol. Sci. 2020, 21, 2660. [Google Scholar] [CrossRef] [PubMed]
- Sattiraju, A.; Sai, K.K.S.; Mintz, A. Glioblastoma stem cells and their microenvironment. Neurobiol. Essent. Fat. Acids 2017, 1041, 119–140. [Google Scholar] [CrossRef]
- Xiao, K.; Tan, J.; Yuan, J.; Peng, G.; Long, W.; Su, J.; Xiao, Y.; Xiao, Q.; Wu, C.; Qin, C.; et al. Prognostic value and immune cell infiltration of hypoxic phenotype-related gene signatures in glioblastoma microenvironment. J. Cell. Mol. Med. 2020, 24, 13235–13247. [Google Scholar] [CrossRef]
- Angel, I.; Kerman, O.P.; Rousso-Noori, L.; Friedmann-Morvinski, D. Tenascin C promotes cancer cell plasticity in mesenchymal glioblastoma. Oncogene 2020, 39, 6990–7004. [Google Scholar] [CrossRef]
- Jung, E.; Osswald, M.; Ratliff, M.; Dogan, H.; Xie, R.; Weil, S.; Hoffmann, D.C.; Kurz, F.T.; Kessler, T.; Heiland, S.; et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Mattei, V.; Santilli, F.; Martellucci, S.; Monache, S.D.; Fabrizi, J.; Colapietro, A.; Angelucci, A.; Festuccia, C. The importance of tumor stem cells in glioblastoma resistance to therapy. Int. J. Mol. Sci. 2021, 22, 3863. [Google Scholar] [CrossRef]
- Garcia-Oliveira, P.; Otero, P.; Pereira, A.; Chamorro, F.; Carpena, M.; Echave, J.; Fraga-Corral, M.; Simal-Gandara, J.; Prieto, M. Status and challenges of plant-anticancer compounds in cancer treatment. Pharmaceuticals 2021, 14, 157. [Google Scholar] [CrossRef]
- Colapietro, A.; Mancini, A.; Vitale, F.; Martellucci, S.; Angelucci, A.; Llorens, S.; Mattei, V.; Gravina, G.L.; Alonso, G.L.; Festuccia, C. Crocetin extracted from saffron shows antitumor effects in models of human glioblastoma. Int. J. Mol. Sci. 2020, 21, 423. [Google Scholar] [CrossRef] [PubMed]
- Colapietro, A.; Yang, P.; Rossetti, A.; Mancini, A.; Vitale, F.; Martellucci, S.; Conway, T.L.; Chakraborty, S.; Marampon, F.; Mattei, V.; et al. The botanical drug pbi-05204, a supercritical co2 extract of nerium oleander, inhibits growth of human glioblastoma, reduces akt/mtor activities, and modulates gsc cell-renewal properties. Front. Pharmacol. 2020, 11, 1438. [Google Scholar] [CrossRef]
- Nandini, D.B.; Rao, R.S.; Deepak, B.S.; Reddy, P.B. Sulforaphane in broccoli: The green chemoprevention!! Role in cancer prevention and therapy. J. Oral Maxillofac. Pathol. 2020, 24, 405. [Google Scholar] [CrossRef]
- Pham, N.-A.; Jacobberger, J.; Schimmer, A.D.; Cao, P.; Gronda, M.; Hedley, D.W. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol. Cancer Ther. 2004, 3, 21. [Google Scholar]
- Wang, L.; Tian, Z.; Yang, Q.; Li, H.; Guan, H.; Shi, B.; Hou, P.; Ji, M. Sulforaphane inhibits thyroid cancer cell growth and invasiveness through the reactive oxygen species-dependent pathway. Oncotarget 2015, 6, 25917–25931. [Google Scholar] [CrossRef]
- Karmakar, S.; Weinberg, M.; Banik, N.; Patel, S.; Ray, S. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience 2006, 141, 1265–1280. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Sun, Q.; Ji, X.; Zhu, L.; Cong, Z.; Zhou, Y.; Liu, H.; Zhou, M. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 2013, 13, 380. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Ciccarese, F.; Ciminale, V. Escaping death: Mitochondrial redox homeostasis in cancer cells. Front. Oncol. 2017, 7, 117. [Google Scholar] [CrossRef]
- Rodrigues-Silva, E.; Siqueira-Santos, E.S.; Ruas, J.S.; Ignarro, R.S.; Figueira, T.R.; Rogério, F.; Castilho, R.F. Evaluation of mitochondrial respiratory function in highly glycolytic glioma cells reveals low ADP phosphorylation in relation to oxidative capacity. J. Neuro-Oncol. 2017, 133, 519–529. [Google Scholar] [CrossRef]
- Shen, S.-C.; Wu, M.-S.; Lin, H.-Y.; Yang, L.-Y.; Chen, Y.-H.; Chen, Y.-C. Reactive oxygen species-dependent nitric oxide production in reciprocal interactions of glioma and microglial cells. J. Cell. Physiol. 2014, 229, 2015–2026. [Google Scholar] [CrossRef]
- Ma, L.; Liu, J.; Zhang, X.; Qi, J.; Yu, W.; Gu, Y. p38 MAPK-dependent Nrf2 induction enhances the resistance of glioma cells against TMZ. Med. Oncol. 2015, 32, 69. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Ganapathy, S.; Srivastava, R.K. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin. Cancer Res. 2008, 14, 6855–6866. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Volti, G.L.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme oxygenase-1 in central nervous system malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef] [PubMed]
- Misiewicz, I.; Skupinska, K.; Kasprzycka-Guttman, T. Differential response of human healthy lympho-blastoid and CCRF-SB leukemia cells to sulforaphane and its two analogues: 2-oxohexyl isothiocyanate and alissi. Pharmacol. Rep. 2007, 59, 80–87. [Google Scholar] [PubMed]
- Ge, M.; Zhang, L.; Cao, L.; Xie, C.; Li, X.; Li, Y.; Meng, Y.; Chen, Y.; Wang, X.; Chen, J.; et al. Sulforaphane inhibits gastric cancer stem cells via suppressing sonic hedgehog pathway. Int. J. Food Sci. Nutr. 2019, 70, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Simões, B.M.; Santiago-Gómez, A.; Chiodo, C.; Moreira, T.; Conole, D.; Lovell, S.; Alferez, D.; Eyre, R.; Spence, K.; Sarmiento-Castro, A.; et al. Targeting STAT3 signaling using stabilised sulforaphane (SFX-01) inhibits endocrine resistant stem-like cells in ER-positive breast cancer. Oncogene 2020, 39, 4896–4908. [Google Scholar] [CrossRef]
- Lellahi, S.M.; Rosenlund, I.A.; Hedberg, A.; Kiær, L.T.; Mikkola, I.; Knutsen, E.; Perander, M. The long noncoding RNA NEAT1 and nuclear paraspeckles are up regulated by the transcription factor HSF1 in the heat shock response. J. Biol. Chem. 2018, 293, 18965–18976. [Google Scholar] [CrossRef]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef]
- Available online: https://www.ema.europa.eu/en/cyclodextrins (accessed on 1 October 2021).
- Leung, E.; Wright, E.; Baguley, B.C. Effect of SFX-01 on proliferation of human glioblastoma cell lines. BioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Howell, S.J.; Campone, M.; Corte’, J.; Duhoux, F.P.; Ross, S.; Morris, T.; Franklin, S. Final results of the STEM trial: SFX-01 in the treatment and evaluation of ER1 Her2- metastatic breast cancer (mBC). Ann. Oncol. 2019, 122, 30. [Google Scholar]
- Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2015-004851-28/results (accessed on 1 October 2021).
- Emens, L.; Esteva, F.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.-B.; Im, S.-A.; Wang, Y.; Mani, A.; Shah, J.; et al. Overall survival (OS) in KATE2, a phase II study of programmed death ligand 1 (PD-L1) inhibitor atezolizumab (atezo)+trastuzumab emtansine (T-DM1) vs. placebo (pbo)+T-DM1 in previously treated HER2+ advanced breast cancer (BC). Ann. Oncol. 2019, 104, 30. [Google Scholar] [CrossRef]
- Liang, Q.; Guan, G.; Li, X.; Wei, C.; Wu, J.; Cheng, P.; Wu, A.; Cheng, W. Profiling pro-neural to mesenchymal transition identifies a lncRNA signature in glioma. J. Transl. Med. 2020, 18, 1–16. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Chen, Y.; Zhang, Q.; Chen, F.; Xi, S.; Long, Y.; Zhang, J.; Cai, H.; Ke, C.; Wang, J.; et al. Association between glioblastoma cell-derived vessels and poor prognosis of the patients. Cancer Commun. 2020, 40, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, F.; Liu, J.; Huang, Z.; Zheng, Y.; Deng, S.; Liu, Y.; Wang, J.; Sun, X. Dual role of WNT5A in promoting endothelial differentiation of glioma stem cells and angiogenesis of glioma derived endothelial cells. Oncogene 2021, 40, 5081–5094. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, C.; Montermini, L.; Meehan, B.; Brisson, A.R.; Tan, S.; Choi, D.; Nakano, I.; Rak, J. Molecular subtypes and differentiation programmes of glioma stem cells as determinants of extracellular vesicle profiles and endothelial cell-stimulating activities. J. Extracell. Vesicles 2018, 7, 1490144. [Google Scholar] [CrossRef]
- Jeon, Y.K.; Yoo, D.R.; Jang, Y.H.; Jang, S.Y.; Nam, M.J. Sulforaphane induces apoptosis in human hepatic cancer cells through inhibition of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase4, mediated by hypoxia inducible factor-1-dependent pathway. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2011, 1814, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Q.; Xie, Y.-K.; Wu, Y.; Li, L.-L.; Liu, Y.; Miao, X.-B.; Liu, Q.-Z.; Yao, K.-T.; Xiao, G.-H. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget 2017, 8, 12067–12080. [Google Scholar] [CrossRef]
- Kaminski, B.M.; Weigert, A.; Brüne, B.; Schumacher, M.; Wenzel, U.; Steinhilber, D.; Stein, J.; Ulrich, S. Sulforaphane potentiates oxaliplatin-induced cell growth inhibition in colorectal cancer cells via induction of different modes of cell death. Cancer Chemother. Pharmacol. 2011, 67, 1167–1178. [Google Scholar] [CrossRef]
- Bose, C.; Awasthi, S.; Sharma, R.; Beneš, H.; Hauer-Jensen, M.; Boerma, M.; Singh, S.P. Sulforaphane potentiates anticancer effects of doxorubicin and attenuates its cardiotoxicity in a breast cancer model. PLoS ONE 2018, 13, e0193918. [Google Scholar] [CrossRef]
- Lan, F.; Yang, Y.; Han, J.; Wu, Q.; Yu, H.; Yue, X. Sulforaphane reverses chemo-resistance to temozolomide in glioblastoma cells by NF-κB-dependent pathway downregulating MGMT expression. Int. J. Oncol. 2015, 48, 559–568. [Google Scholar] [CrossRef]
- Naumann, P.; Liermann, J.; Fortunato, F.; Schmid, T.; Weber, K.-J.; Debus, J.; Combs, S.E. Sulforaphane enhances irradiation effects in terms of perturbed cell cycle progression and increased DNA damage in pancreatic cancer cells. PLoS ONE 2017, 12, e0180940. [Google Scholar] [CrossRef]
- Ganai, S.A.; Srinivasan, P.; Rajamanikandan, S.; Shah, B.A.; Mohan, S.; Gani, M.; Padder, B.A.; Qadri, R.A.; Bhat, M.A.; Baba, Z.A.; et al. Delineating binding potential, stability of Sulforaphane-N-acetyl-cysteine in the active site of histone deacetylase 2 and testing its cytotoxicity against distinct cancer lines through stringent molecular dynamics, DFT and cell-based assays. Chem. Biol. Drug Des. 2021, 98, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, Y.; Jiang, Y.; Wang, K.; Wang, X.; Zhou, D.; Wang, Y.; Yu, R.; Zhou, X. YAP promotes autophagy and progression of gliomas via upregulating HMGB. J. Exp. Clin. Cancer Res. 2021, 40, 1–15. [Google Scholar] [CrossRef]
- Fernández-García, P.; Rosselló, C.A.; Rodríguez-Lorca, R.; Beteta-Göbel, R.; Fernández-Díaz, J.; Lladó, V.; Busquets, X.; Escribá, P.V. The opposing contribution of SMS1 and SMS2 to glioma progression and their value in the therapeutic response to 2OHOA. Cancers 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.D.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, 1601. [Google Scholar] [CrossRef]
- Kansanen, E.; Linna-Kuosmanen, S.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Hać, A.; Brokowska, J.; Rintz, E.; Bartkowski, M.; Węgrzyn, G.; Herman-Antosiewicz, A. Mechanism of selective anticancer activity of isothiocyanates relies on differences in DNA damage repair between cancer and healthy cells. Eur. J. Nutr. 2019, 59, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Hartmann, R.; Schröter, F.; Suwala, A.K.; Maciaczyk, D.; Krüger, A.C.; Willbold, D.; Kahlert, U.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Ambati, R.; Gundamaraju, R. Exploring the crosstalk between inflammation and epithelial-mesenchymal transition in cancer. Mediat. Inflamm. 2021, 2021, 1–13. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’Er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. the impact of the tumor microenvironment on the properties of glioma stem-like cells. Front. Oncol. 2017, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Hess, A.R.; Seftor, R.E. Vasculogenic mimicry and tumour-cell plasticity: Lessons from mel-anoma. Nat. Rev. Cancer. 2003, 3, 411–421. [Google Scholar] [CrossRef]
- Folberg, R.; Arbieva, Z.; Moses, J.; Hayee, A.; Sandal, T.; Kadkol, S.; Lin, A.Y.; Valyi-Nagy, K.; Setty, S.; Leach, L.; et al. Tumor cell plasticity in uveal melanoma: Microenvironment directed dampening of the invasive and metastatic genotype and phenotype accompanies the generation of vasculogenic mimicry patterns. Am. J. Pathol. 2006, 169, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Rezabakhsh, A.; Montazersaheb, S.; Nabat, E.; Hassanpour, M.; Montaseri, A.; Malekinejad, H.; Movassaghpour, A.A.; Rahbarghazi, R.; Garjani, A. Effect of hydroxychloroquine on oxidative/nitrosative status and angiogenesis in endothelial cells under high glucose condition. BioImpacts 2017, 7, 219–226. [Google Scholar] [CrossRef]
- Torsvik, A.; Stieber, D.; Enger, P.Ø.; Golebiewska, A.; Molven, A.; Svendsen, A.; Westermark, B.; Niclou, S.P.; Olsen, T.K.; Enger, M.C.; et al. U-251 revisited: Genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014, 3, 812–824. [Google Scholar] [CrossRef]
- Luchman, H.A.; Stechishin, O.D.; Dang, N.H.; Blough, M.D.; Chesnelong, C.; Kelly, J.J.; Nguyen, S.A.; Chan, J.A.; Weljie, A.M.; Cairncross, J.G.; et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro-Oncology 2012, 14, 184–191. [Google Scholar] [CrossRef]
- Mendiburu-Elicabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef]
- Bass, J.I.F.; Reece-Hoyes, J.S.; Walhout, A.J. Gene-centered yeast one-hybrid assays. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef] [PubMed]
- Koeplinger, K.A.; Mildner, A.M.; Leone, J.W.; Wheeler, J.S.; Heinrikson, R.L.; Tomasselli, A.G. Caspase 8: An efficient method for large-scale autoactivation of recombinant procaspase 8 by matrix adsorption and characterization of the active enzyme. Protein Expr. Purif. 2000, 18, 378–387. [Google Scholar] [CrossRef]
- Talanian, R.V.; Quinlan, C.; Trautz, S.; Hackett, M.C.; Mankovich, J.A.; Banach, D.; Ghayur, T.; Brady, K.D.; Wong, W.W. Substrate specificities of caspase family proteases. J. Biol. Chem. 1997, 272, 9677–9682. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Vitale, F.; Cristiano, L.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The small molecule ephrin receptor inhibitor, Glpg1790, reduces renewal capabilities of cancer stem cells, showing anti-tumour efficacy on preclinical glioblastoma models. Cancers 2019, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Pompili, S.; Vitale, F.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The brain penetrating and dual TORC1/TORC2 inhibitor, RES529, elicits anti-glioma activity and enhances the therapeutic effects of anti-angiogenetic compounds in preclinical murine models. Cancers 2019, 11, 1604. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.abcam.com/protocols (accessed on 1 October 2021).
- Gravina, G.L.; Mancini, A.; Muzi, P.; Ventura, L.; Biordi, L.; Ricevuto, E.; Pompili, S.; Mattei, C.; Di Cesare, E.; Jannini, E.A.; et al. CXCR4 pharmacogical inhibition reduces bone and soft tissue metastatic burden by affecting tumor growth and tumorigenic potential in prostate cancer preclinical models. Prostate 2015, 75, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Monache, S.D.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravina, G.L.; Giorgio, C.; Mancini, A.; Pellegrini, C.; Colapietro, A.; Monache, S.D.; Maturo, M.G.; Sferra, R.; Chiodelli, P.; et al. UniPR1331, a small molecule targeting Eph/ephrin interaction, prolongs survival in glioblastoma and potentiates the effect of antiangiogenic therapy in mice. Oncotarget 2018, 9, 24347–24363. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Mancini, A.; Colapietro, A.; Gravina, G.L.; Vitale, F.; Marampon, F.; Monache, S.D.; Pompili, S.; Cristiano, L.; Vetuschi, A.; et al. The first-in-class alkylating deacetylase inhibitor molecule tinostamustine shows antitumor effects and is synergistic with radiotherapy in preclinical models of glioblastoma. J. Hematol. Oncol. 2018, 11, 1–19. [Google Scholar] [CrossRef]
- Gravina, G.L.; Marampon, F.; Giusti, I.; Carosa, E.; Di Sante, S.; Ricevuto, E.; Dolo, V.; Tombolini, V.; Jannini, E.A.; Festuccia, C. Differential effects of PXD101 (belinostat) on androgen-dependent and androgen-independent prostate cancer models. Int. J. Oncol. 2012, 40, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; Muzi, P.; Pomante, R.; Ventura, L.; Vessella, R.L.; Vicentini, C.; Bologna, M. Bicalutamide increases phospho-Akt levels through Her2 in patients with prostate cancer. Endocr.-Relat. Cancer 2007, 14, 601–611. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U87MG | |||||||
|---|---|---|---|---|---|---|---|
| Groups | Tumor Weight (mg ± SE) | Statistics | TTP (Days) | Statistics | HR | CI 95% | Statistics |
| 1. CTRL | 1095 ± 111 | 13.7 ± 1.8 | |||||
| 2. SFX-01 | 400 ± 21 | p < 0.001 | 21.6 ± 1.0 | p < 0.001 | 4.45 | 1.51 to 15.22 | p < 0.0001 |
| T98G | |||||||
| 3. CTRL | 819 ± 65 | 13.2 ± 0.7 | |||||
| 4. SFX-01 | 368 ±16 | p < 0.0001 | 17.0 ± 0.5 | p < 0.001 | 2.98 | 1.09 to 8.20 | p < 0.001 |
| Groups | Vessel Count (CD31) | Ki67 Percentage | Apoptosis (% of Tunel Positive Cells) | Necrosis (% of Necrotic Area) | HIF1α (Arbitrary Score) | Phospho-(ser473)-Akt (Arbitrary Score) | HDAC Activity OD405nm/µg Protein | p-Stat3 (Arbitrary Score) |
|---|---|---|---|---|---|---|---|---|
| U87MG Xenografts | ||||||||
| 1. CTRL | 30.8 ± 5.0 | 35.5 ± 4.0 | <2 | 15.0 ± 5.5 | 6 (3 × 2) | 9 (3 × 3) | 1.687 ± 0.244 | 6 (2 × 3) |
| 2. SFX-01 (50 mg/Kg/day) | 18.0 ± 4.0 * | 21.5 ± 0.5 ** | 15.5 ± 5.5 ** | 10.0 ± 5.0 | 2 (2 × 1) | 2 (1 × 2) | 0.557 ± 0.188 | 1 (1 × 1) |
| T98G Xenografts | ||||||||
| 3. CTRL | 22.2 ± 2.4 | 20.7 ± 4.0 | <2 | 8.0 ± 4.0 | 4 (2 × 2) | 6 (2 × 3) | 1.332 ±0.330 | 6 (2 × 3) |
| 4. SFX-01 (50 mg/Kg/day) | 17.0 ± 2.0 * | 8.0 ± 2.5 | 8.5 ± 2.0 | 15.0 ± 3.5 | 2 (1 × 2) | 1 (1 × 1) | 0.233 ± 0.088 | 1 (1 × 1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colapietro, A.; Rossetti, A.; Mancini, A.; Martellucci, S.; Ocone, G.; Pulcini, F.; Biordi, L.; Cristiano, L.; Mattei, V.; Delle Monache, S.; et al. Multiple Antitumor Molecular Mechanisms Are Activated by a Fully Synthetic and Stabilized Pharmaceutical Product Delivering the Active Compound Sulforaphane (SFX-01) in Preclinical Model of Human Glioblastoma. Pharmaceuticals 2021, 14, 1082. https://doi.org/10.3390/ph14111082
Colapietro A, Rossetti A, Mancini A, Martellucci S, Ocone G, Pulcini F, Biordi L, Cristiano L, Mattei V, Delle Monache S, et al. Multiple Antitumor Molecular Mechanisms Are Activated by a Fully Synthetic and Stabilized Pharmaceutical Product Delivering the Active Compound Sulforaphane (SFX-01) in Preclinical Model of Human Glioblastoma. Pharmaceuticals. 2021; 14(11):1082. https://doi.org/10.3390/ph14111082
Chicago/Turabian StyleColapietro, Alessandro, Alessandra Rossetti, Andrea Mancini, Stefano Martellucci, Giuseppe Ocone, Fanny Pulcini, Leda Biordi, Loredana Cristiano, Vincenzo Mattei, Simona Delle Monache, and et al. 2021. "Multiple Antitumor Molecular Mechanisms Are Activated by a Fully Synthetic and Stabilized Pharmaceutical Product Delivering the Active Compound Sulforaphane (SFX-01) in Preclinical Model of Human Glioblastoma" Pharmaceuticals 14, no. 11: 1082. https://doi.org/10.3390/ph14111082
APA StyleColapietro, A., Rossetti, A., Mancini, A., Martellucci, S., Ocone, G., Pulcini, F., Biordi, L., Cristiano, L., Mattei, V., Delle Monache, S., Marampon, F., Gravina, G. L., & Festuccia, C. (2021). Multiple Antitumor Molecular Mechanisms Are Activated by a Fully Synthetic and Stabilized Pharmaceutical Product Delivering the Active Compound Sulforaphane (SFX-01) in Preclinical Model of Human Glioblastoma. Pharmaceuticals, 14(11), 1082. https://doi.org/10.3390/ph14111082