
NbX: Machine Learning-Guided Re-Ranking of Nanobody–Antigen Binding Poses


Abstract
:1. Introduction
2. Results and Discussion
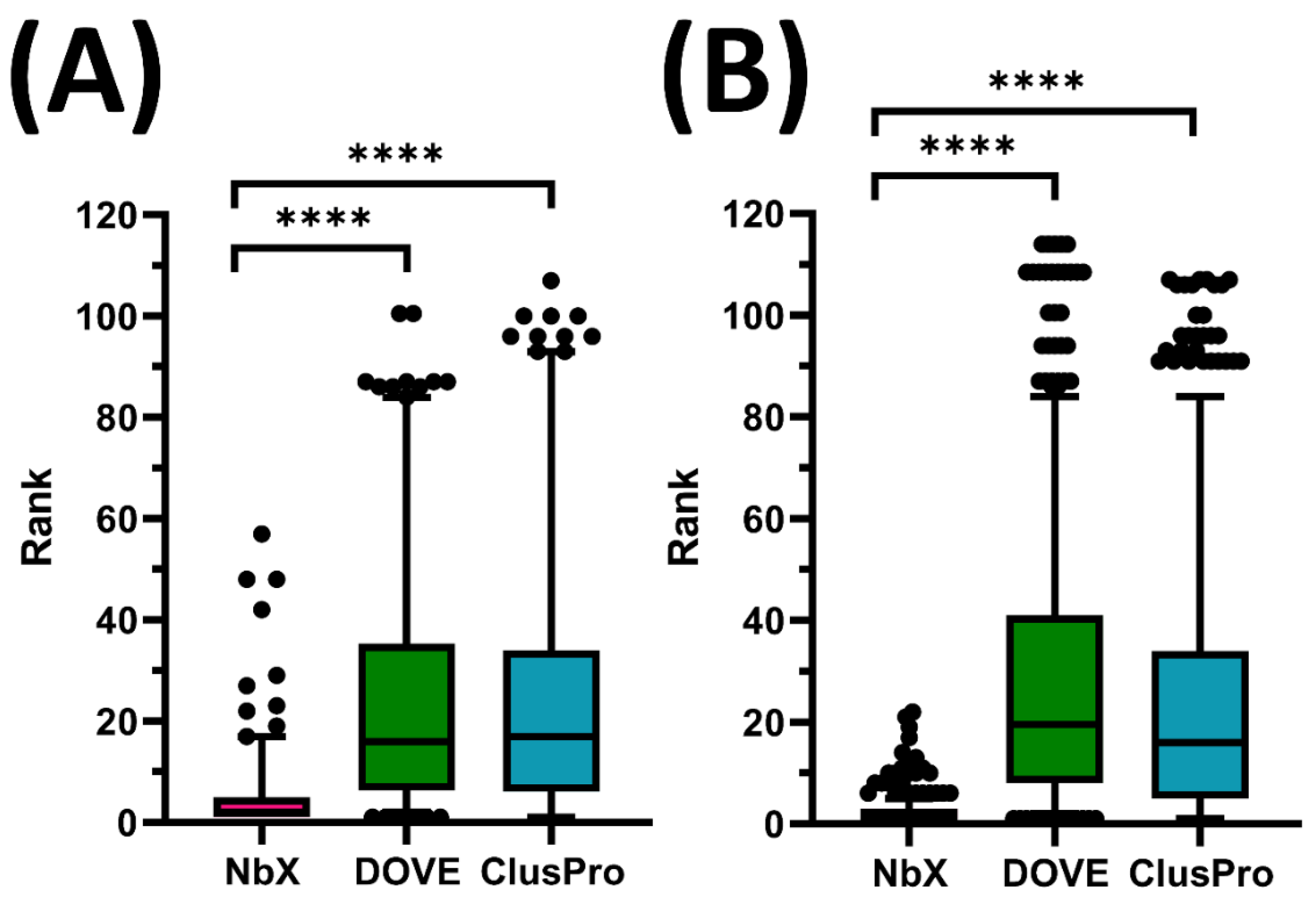
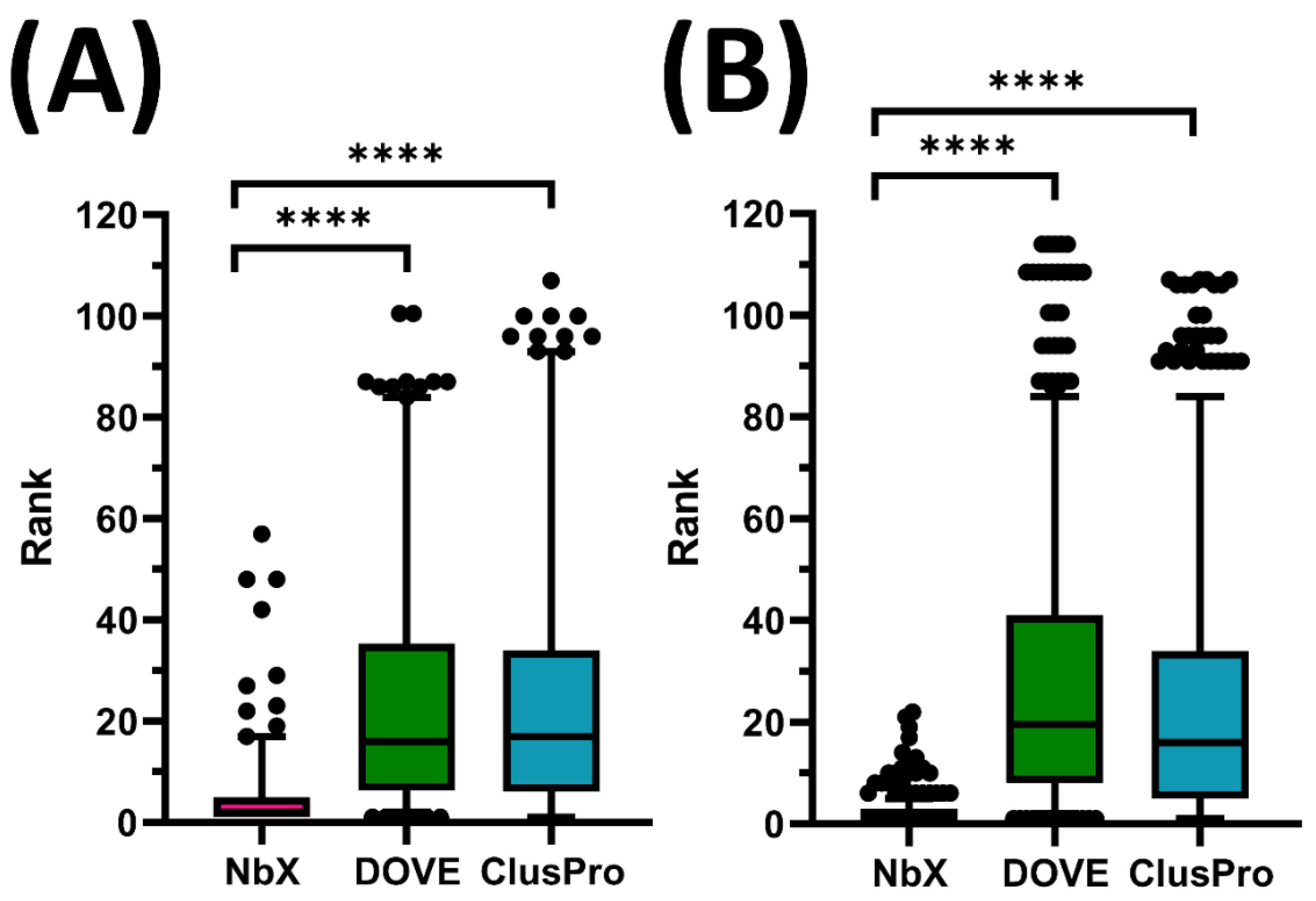
2.1. Benchmarking with DOVE and ClusPro
2.2. NbX Was Better at Prioritizing Docking Solutions than Determining Absolute Binding Feasibility
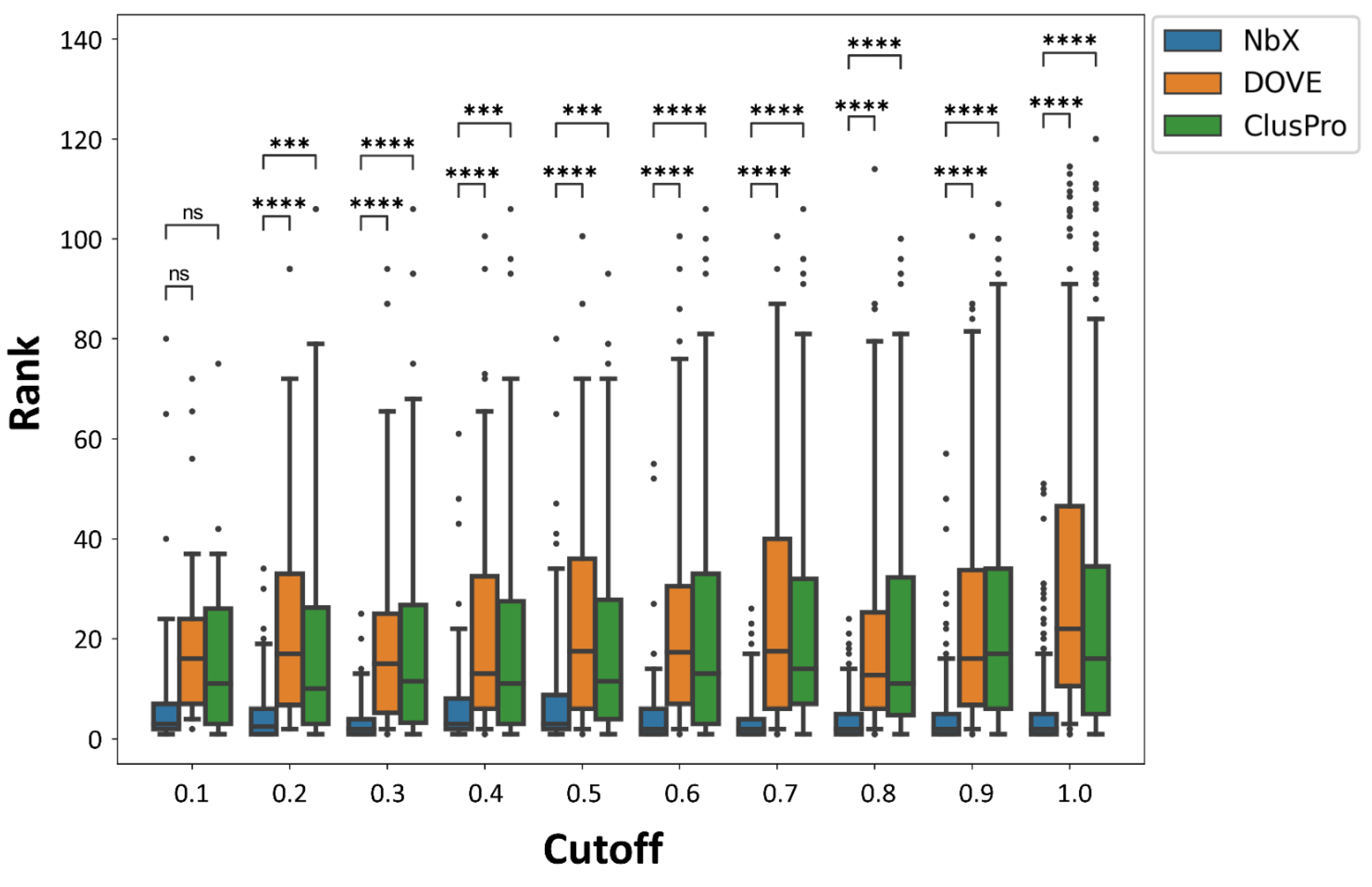
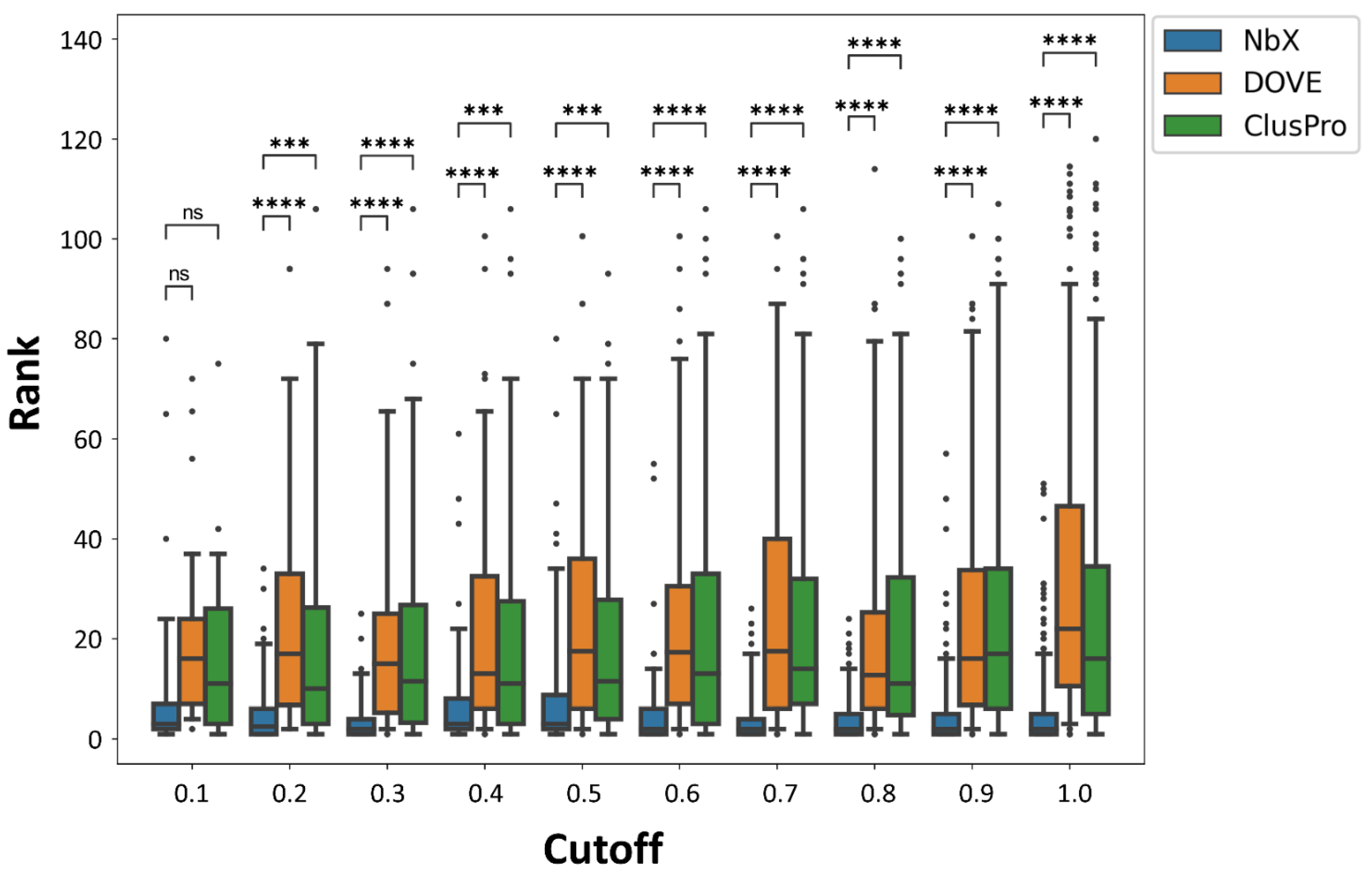
2.3. Re-Ranking Performance of NbX Was Insensitive to a Substantial Decrease in the Size of the Training Dataset
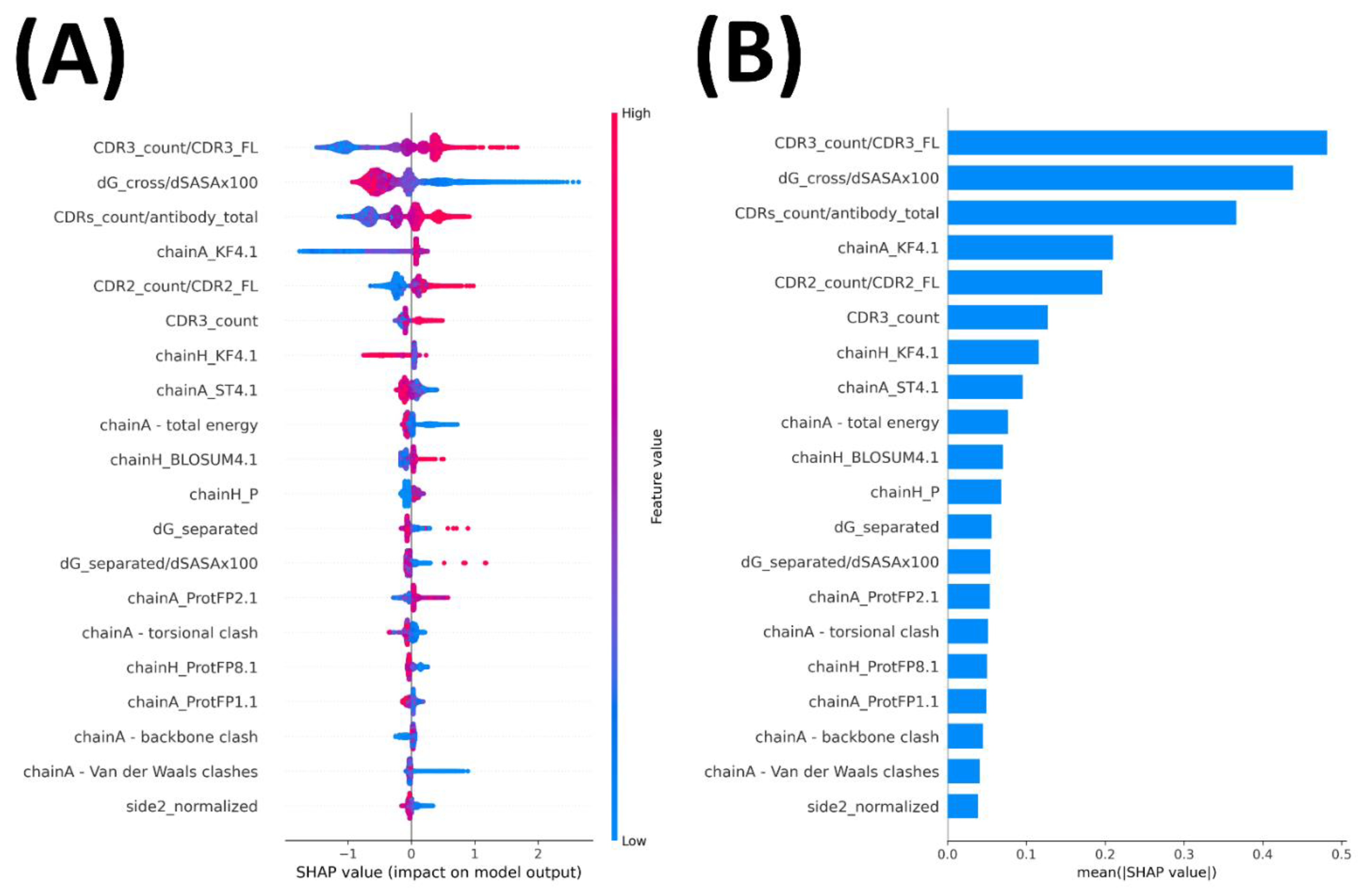
2.4. Important Features Contributed to Prediction Performance
3. Materials and Methods
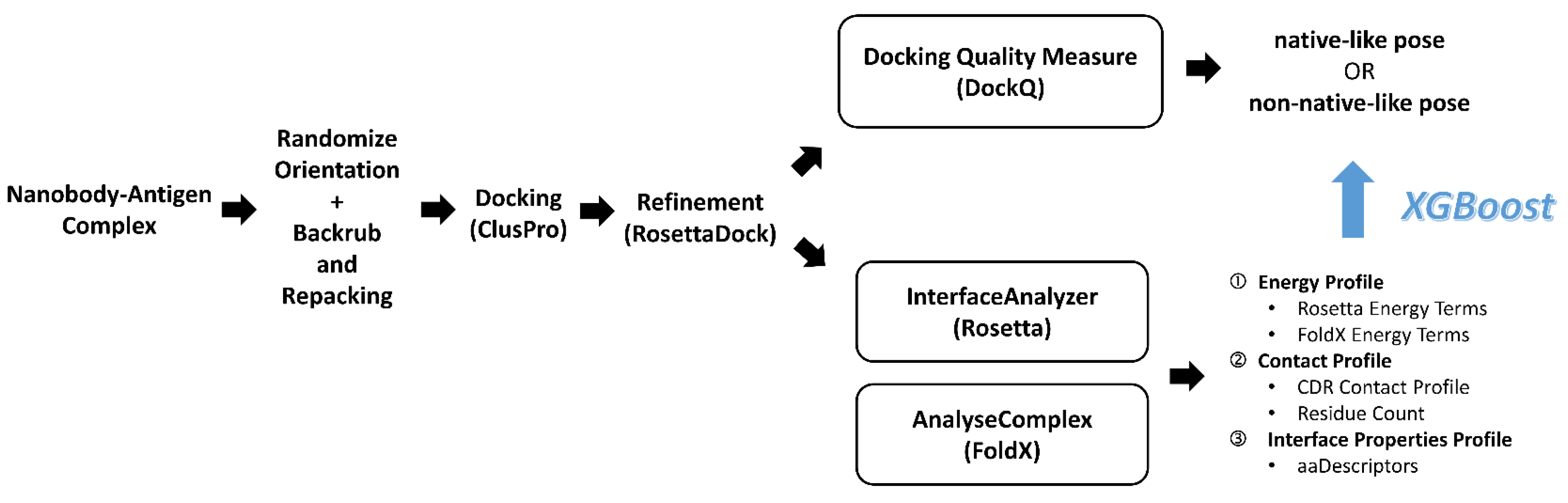
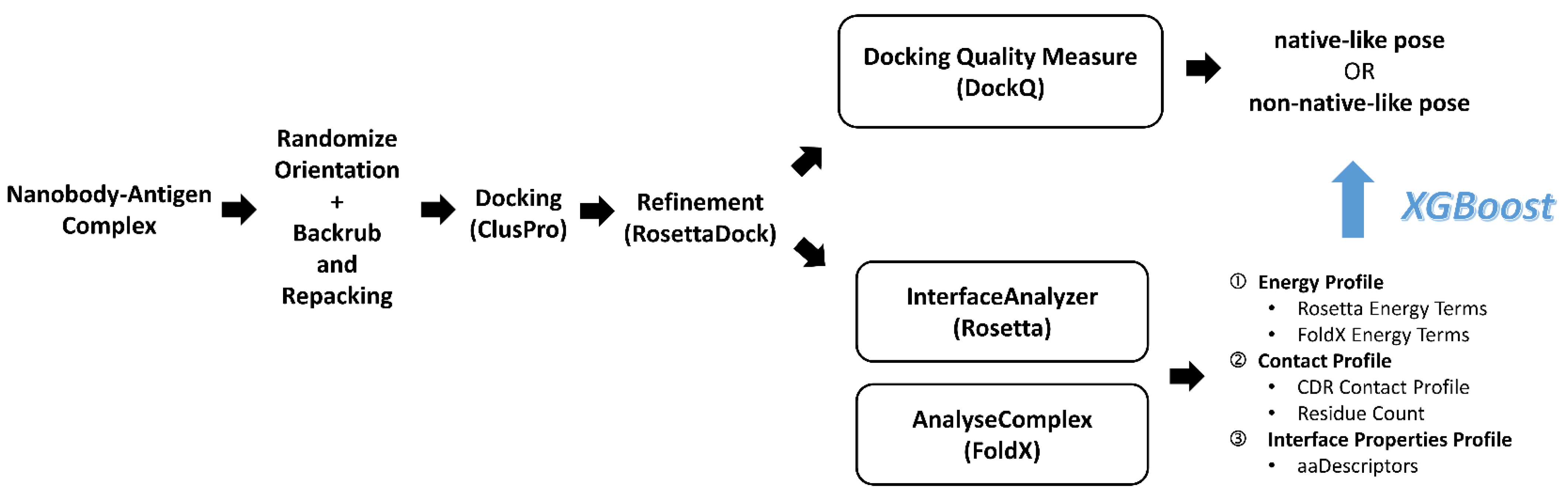
3.1. Data Collection and Cleaning
3.2. Rigid-Body Orientation, Backbone and Side Chain Randomization
3.3. ClusPro Pose Generation and Refinement
3.4. Pose Classification by Docking Quality Assessment
3.5. Feature Set Preparation
3.6. Model Selection and Training and Test Set Partition
3.7. Re-Ranking Method and Benchmarking
3.8. Feature Importance Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norman, R.A.; Ambrosetti, F.; Bonvin, A.M.J.J.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational approaches to therapeutic antibody design: Established methods and emerging trends. Brief. Bioinform. 2020, 21, 1549–1567. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Sircar, A.; Gray, J.J. SnugDock: Paratope structural optimization during antibody–antigen docking compensates for errors in antibody homology models. PLoS Comput. Biol. 2010, 6, e1000644. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetti, F.; Olsen, T.H.; Olimpieri, P.P.; Jiménez-García, B.; Milanetti, E.; Marcatilli, P. ProABC-2: PRediction Of AntiBody Contacts v2 and its application to information-driven docking. Bioinformatics 2020, 36, 5107–5108. [Google Scholar] [CrossRef]
- Krawczyk, K.; Baker, T.; Shi, J.; Deane, C.M. Antibody i-Patch prediction of the antibody binding site improves rigid local antibody–antigen docking. Protein Eng. Des. Sel. 2013, 26, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunik, V.; Ashkenazi, S.; Ofran, Y. Paratome: An online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012, 40, W521–W524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberis, E.; Velickovic, P.; Sormanni, P.; Vendruscolo, M.; Liò, P. Parapred: Antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics 2018, 34, 2944–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, K.; Liu, X.; Baker, T.; Shi, J.; Deane, C.M. Improving B-cell epitope prediction and its application to global antibody–antigen docking. Bioinformatics 2014, 30, 2288–2294. [Google Scholar] [CrossRef]
- Eismann, S.; Townshend, R.J.L.; Thomas, N.; Jagota, M.; Jing, B.; Dror, R.O. Hierarchical, rotation-equivariant neural networks to select structural models of protein complexes. Proteins 2021, 89, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Jung, Y.; Renaud, N.; Honavar, V.; Bonvin, A.M.J.J.; Xue, L.C. iScore: A novel graph kernel-based function for scoring protein–protein docking models. Bioinformatics 2020, 36, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanemura, K.A.; Pei, J.; Merz, K.M., Jr. Refinement of pairwise potentials via logistic regression to score protein–protein interactions. Proteins 2020, 88, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Buchanan, A.; Taddese, B.; Deane, C.M. DLAB—Deep learning methods for structure-based virtual screening of antibodies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, X.; Terashi, G.; Christoffer, C.W.; Zhu, M.; Kihara, D. Protein docking model evaluation by 3D deep convolutional neural networks. Bioinformatics 2020, 36, 2113–2118. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, Y. Energy-based graph convolutional networks for scoring protein docking models. Proteins 2020, 88, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Akbal-Delibas, B.; Farhoodi, R.; Pomplun, M.; Haspel, N. Accurate refinement of docked protein complexes using evolutionary information and deep learning. J. Bioinform. Comput. Biol. 2016, 14, 1642002. [Google Scholar] [CrossRef] [Green Version]
- Kingsley, L.J.; Esquivel-Rodríguez, J.; Yang, Y.; Kihara, D.; Lill, M.A. Ranking protein–protein docking results using steered molecular dynamics and potential of mean force calculations. J. Comput. Chem. 2016, 37, 1861–1865. [Google Scholar] [CrossRef] [Green Version]
- Degiacomi, M.T. Coupling molecular dynamics and deep learning to mine protein conformational space. Structure 2019, 27, 1034–1040.e3. [Google Scholar] [CrossRef] [Green Version]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodolà, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2020, 17, 184–192. [Google Scholar] [CrossRef]
- Nadaradjane, A.A.; Guerois, R.; Andreani, J. Protein–protein docking using evolutionary information. Methods Mol. Biol. 2018, 1764, 429–447. [Google Scholar]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins 2007, 67, 1078–1086. [Google Scholar] [CrossRef]
- Lu, H.; Lu, L.; Skolnick, J. Development of unified statistical potentials describing protein–protein interactions. Biophys. J. 2003, 84, 1895–1901. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-Y.; Zou, X. An iterative knowledge-based scoring function for protein–protein recognition. Proteins 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Muyldermans, S. Single domain camel antibodies: Current status. J. Biotechnol. 2001, 74, 277–302. [Google Scholar] [CrossRef]
- Hassanzadeh-Ghassabeh, G.; Devoogdt, N.; De Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, L.S.; Colwell, L.J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. 2018, 31, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, L.S.; Colwell, L.J. Comparative analysis of nanobody sequence and structure data. Proteins 2018, 86, 697–706. [Google Scholar] [CrossRef]
- Olson, M.A.; Legler, P.M.; Zabetakis, D.; Turner, K.B.; Anderson, G.P.; Goldman, E.R. Sequence Tolerance of a Single-Domain Antibody with a High Thermal Stability: Comparison of Computational and Experimental Fitness Profiles. ACS Omega 2019, 4, 10444–10454. [Google Scholar] [CrossRef] [PubMed]
- Zavrtanik, U.; Lukan, J.; Loris, R.; Lah, J.; Hadži, S. Structural Basis of Epitope Recognition by Heavy-Chain Camelid Antibodies. J. Mol. Biol. 2018, 430, 4369–4386. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Nadzirin, N.; Velankar, S.; Wodak, S.J. Modeling protein–protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins Struct. Funct. Bioinf. 2020, 88, 916–938. [Google Scholar] [CrossRef] [PubMed]
- Akiba, H.; Tamura, H.; Kiyoshi, M.; Yanaka, S.; Sugase, K.; Caaveiro, J.M.M.; Tsumoto, K. Structural and thermodynamic basis for the recognition of the substrate-binding cleft on hen egg lysozyme by a single-domain antibody. Sci. Rep. 2019, 9, 15481. [Google Scholar] [CrossRef]
- Keskin, O.; Ma, B.; Nussinov, R. Hot regions in protein--protein interactions: The organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 2005, 345, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Lafont, V.; Schaefer, M.; Stote, R.H.; Altschuh, D.; Dejaegere, A. Protein–protein recognition and interaction hot spots in an antigen-antibody complex: Free energy decomposition identifies “efficient amino acids”. Proteins 2007, 67, 418–434. [Google Scholar] [CrossRef]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—a review of the protein–protein interface determinant amino-acid residues. Proteins 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Kidera, A.; Konishi, Y.; Oka, M.; Ooi, T.; Scheraga, H.A. Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J. Protein Chem. 1985, 4, 23–55. [Google Scholar] [CrossRef]
- Yang, L.; Shu, M.; Ma, K.; Mei, H.; Jiang, Y.; Li, Z. ST-scale as a novel amino acid descriptor and its application in QSAM of peptides and analogues. Amino Acids 2010, 38, 805–816. [Google Scholar] [CrossRef]
- Georgiev, A.G. Interpretable numerical descriptors of amino acid space. J. Comput. Biol. 2009, 16, 703–723. [Google Scholar] [CrossRef]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Baker, T.; Fuchs, A.; Georges, G.; Shi, J.; Deane, C.M. SAbDab: The structural antibody database. Nucleic Acids Res. 2014, 42, D1140–D1146. [Google Scholar] [CrossRef]
- Adolf-Bryfogle, J.; Xu, Q.; North, B.; Lehmann, A.; Dunbrack, R.L., Jr. PyIgClassify: A database of antibody CDR structural classifications. Nucleic Acids Res. 2015, 43, D432–D438. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Davis, I.W.; Arendall, W.B., 3rd; Richardson, D.C.; Richardson, J.S. The backrub motion: How protein backbone shrugs when a sidechain dances. Structure 2006, 14, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Wallner, B. DockQ: A Quality Measure for Protein–protein Docking Models. PLoS ONE 2016, 11, e0161879. [Google Scholar] [CrossRef] [Green Version]
- Stranges, P.B.; Kuhlman, B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013, 22, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruciani, G.; Baroni, M.; Carosati, E.; Clementi, M.; Valigi, R.; Clementi, S. Peptide studies by means of principal properties of amino acids derived from MIF descriptors. J. Chemom. 2004, 18, 146–155. [Google Scholar] [CrossRef]
- Liang, G.; Li, Z. Factor Analysis Scale of Generalized Amino Acid Information as the Source of a New Set of Descriptors for Elucidating the Structure and Activity Relationships of Cationic Antimicrobial Peptides. QSAR Comb. Sci. 2007, 26, 754–763. [Google Scholar] [CrossRef]
- Mei, H.; Liao, Z.H.; Zhou, Y.; Li, S.Z. A new set of amino acid descriptors and its application in peptide QSARs. Biopolymers 2005, 80, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Osorio, D.; Rondón-Villarreal, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. Small 2015, 12, 44–444. [Google Scholar] [CrossRef]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef]
- Tian, F.; Zhou, P.; Li, Z. T-scale as a novel vector of topological descriptors for amino acids and its application in QSARs of peptides. J. Mol. Struct. 2007, 830, 106–115. [Google Scholar] [CrossRef]
- Van Westen, G.J.; Swier, R.F.; Wegner, J.K.; Ijzerman, A.P.; van Vlijmen, H.W.; Bender, A. Benchmarking of protein descriptor sets in proteochemometric modeling (part 1): Comparative study of 13 amino acid descriptor sets. J. Cheminform. 2013, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Zaliani, A.; Gancia, E. MS-WHIM Scores for Amino Acids: A New 3D-Description for Peptide QSAR and QSPR Studies. J. Chem. Inf. Comput. Sci. 1999, 39, 525–533. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Zhang, D.; Gong, Y. The Comparison of LightGBM and XGBoost Coupling Factor Analysis and Prediagnosis of Acute Liver Failure. IEEE Access 2020, 8, 220990–221003. [Google Scholar] [CrossRef]
- Sharma, A.; Verbeke, W.J.M.I. Improving Diagnosis of Depression With XGBOOST Machine Learning Model and a Large Biomarkers Dutch Dataset (n = 11,081). Front. Big Data 2020, 3, 15. [Google Scholar] [CrossRef]
- Dhaliwal, S.S.; Nahid, A.-A.; Abbas, R. Effective Intrusion Detection System Using XGBoost. Information 2018, 9, 149. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, S.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAPRI | DockQ | Labeling |
|---|---|---|
| Incorrect | 0.00–0.23 | Non-native-like (0) |
| Acceptable | 0.23–0.49 | Native-like (1) |
| Medium | 0.49–0.80 | |
| High | 0.80–1.00 |
| Profile | Feature Group | Number of Features | Description |
|---|---|---|---|
| Contact | Interface residue count | 2 | Count of residues in paratope and epitope |
| Interacting CDR count | 3 | Count of interacting residues from each CDR loop | |
| CDR full length | 3 | Full length of each CDR loop | |
| Interacting CDR residues in paratope | 1 | Proportion of total CDR residue in paratope | |
| Interacting CDR residues versus full length | 3 | Proportion of CDR residue versus full length of each CDR loop | |
| Amino acid count | 40 | Count of individual amino acid in paratope and epitope | |
| Energy | Rosetta InterfaceAnalyzer energy terms | 20 | Rosetta energy descriptors of the interface |
| FoldX AnalyseComplex energy Terms | 44 | FoldX energy descriptors of paratope and epitope | |
| Interface Properties | Total properties of paratope and epitope | 132 | Summation of each of 66 aaDescriptors of paratope and epitope |
| Feature Set | Labeling | Partition Sets | Number of PDBs | Model | Classification Type | Validation Method | Ranking Method |
|---|---|---|---|---|---|---|---|
| Energy, Contact and Interface Properties Profiles | Native-like OR Non-native-like | Training | 80% (96 PDBs) | XGBoost | Binary | K-fold Validation (k = 5) | Descending Order of Average Classification Probability of Refined Poses |
| Test | 20% (25 PDBs) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, C.; Kumar, A.; Zhang, K.Y.J. NbX: Machine Learning-Guided Re-Ranking of Nanobody–Antigen Binding Poses. Pharmaceuticals 2021, 14, 968. https://doi.org/10.3390/ph14100968
Tam C, Kumar A, Zhang KYJ. NbX: Machine Learning-Guided Re-Ranking of Nanobody–Antigen Binding Poses. Pharmaceuticals. 2021; 14(10):968. https://doi.org/10.3390/ph14100968
Chicago/Turabian StyleTam, Chunlai, Ashutosh Kumar, and Kam Y. J. Zhang. 2021. "NbX: Machine Learning-Guided Re-Ranking of Nanobody–Antigen Binding Poses" Pharmaceuticals 14, no. 10: 968. https://doi.org/10.3390/ph14100968
APA StyleTam, C., Kumar, A., & Zhang, K. Y. J. (2021). NbX: Machine Learning-Guided Re-Ranking of Nanobody–Antigen Binding Poses. Pharmaceuticals, 14(10), 968. https://doi.org/10.3390/ph14100968