Adenosine Signaling in Autoimmune Disorders



Abstract
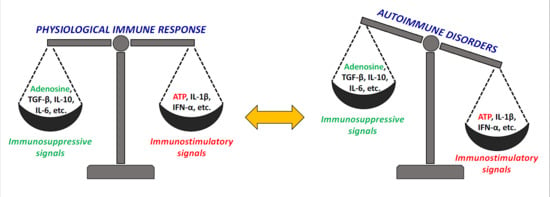
1. The Immunomodulatory Role of Adenosine
1.1. The Equilibrium between ATP and Adenosine Signaling in Inflammation and Traumatic Conditions
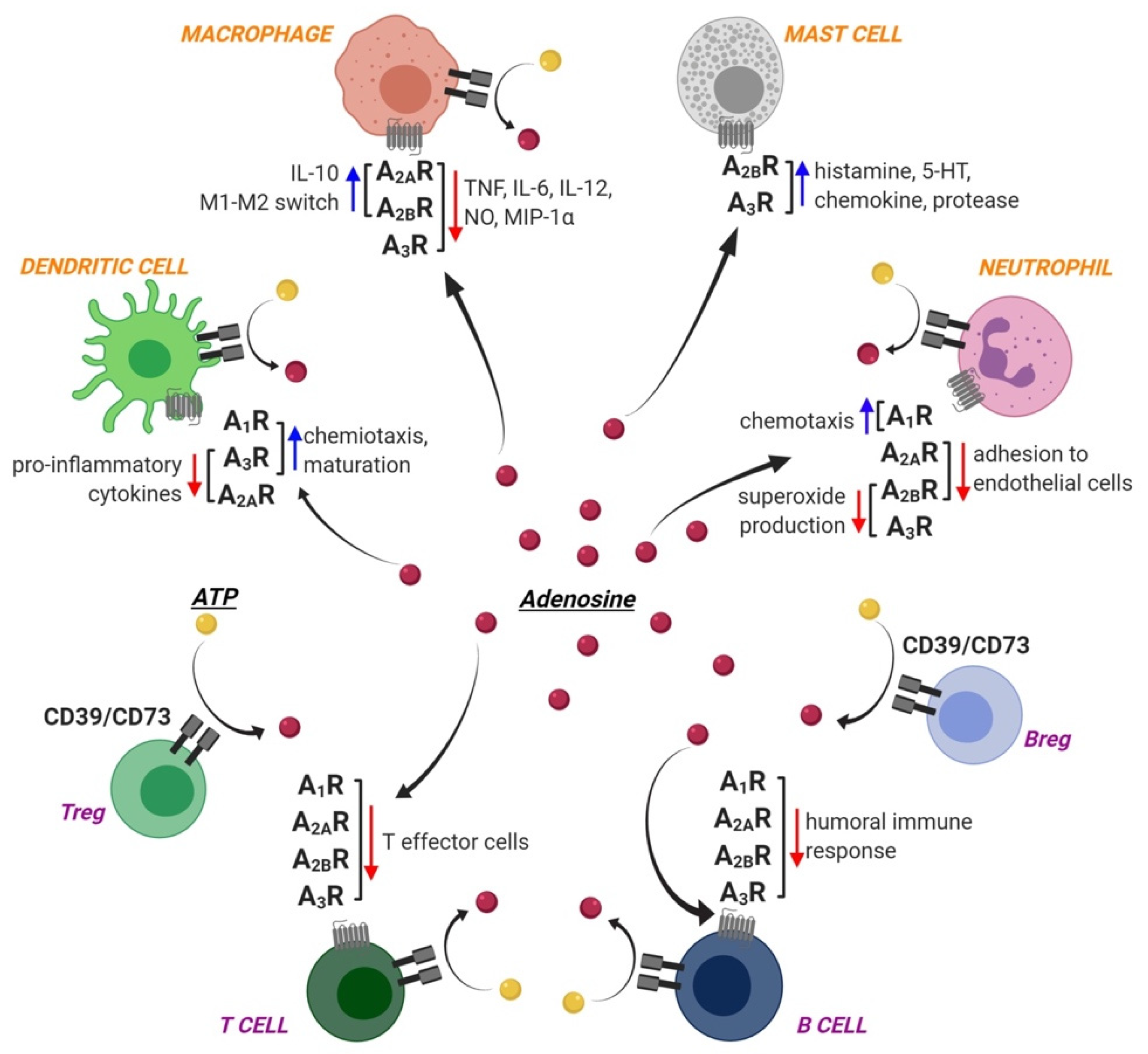
1.2. Adenosine Receptors and Immune Cells
1.2.1. Innate Immunity
1.2.2. Adaptive Immunity
1.3. Adenosine Signaling and Autoimmunity
2. Drugs Targeting the Adenosine System in Autoimmune Disorders: The Puzzling Cases of Methotrexate and NSAIDs
3. Towards New Ado-Based Immunomodulatory Approaches: Preclinical and Clinical Studies
3.1. The Beneficial Role of A1ARs in Autoimmunity
3.2. The Immunomodulatory Role of A2AARs: Can Its Derailment Play a Role in Autoimmune Disorders?
3.3. Interesting Hints from the Long Considered Elusive Member of AR Family: The A2BAR
3.4. A3AR Agonists: Clinical Trials Are already on Their Way to Find Novel Approaches to Autoimmune Disorders
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bouma, M.G.; van den Wildenberg, F.A.; Buurman, W.A. The anti-inflammatory potential of adenosine in ischemia-reperfusion injury: Established and putative beneficial actions of a retaliatory metabolite. Shock 1997, 8, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine signaling and the immune system: When a lot could be too much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef]
- Hoebe, K.; Janssen, E.; Beutler, B. The interface between innate and adaptive immunity. Nat. Immunol. 2004, 5, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Green, H.; Chan, T. Pyrimidine starvation induced by adenosine in fibroblasts and lymphoid cells: Role of adenosine deaminase. Science 1973, 182, 836–837. [Google Scholar] [CrossRef] [PubMed]
- Giblett, E.R.; Anderson, J.E.; Cohen, F.; Pollara, B.; Meuwissen, H.J. Adenosine deaminase deficiency in two patients with severely impaired cellular immunity. Lancet 1972, 2, 1067–1069. [Google Scholar] [CrossRef]
- Schnurr, M.; Toy, T.; Shin, A.; Hartmann, G.; Rothenfusser, S.; Soellner, J.; Davis, I.D.; Cebon, J.; Maraskovsky, E. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood 2004, 103, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Pacheco, R.; Martinez-Navio, J.M.; Lejeune, M.; Climent, N.; Oliva, H.; Gatell, J.M.; Gallart, T.; Mallol, J.; Lluis, C.; Franco, R. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 9583–9588. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast cell: A multi-functional master cell. Front. Immunol. 2015, 6, 620. [Google Scholar] [CrossRef]
- Gao, Z.G.; Jacobson, K.A. Purinergic signaling in mast cell degranulation and asthma. Front. Pharmacol. 2017, 8, 947. [Google Scholar] [CrossRef]
- Rudich, N.; Ravid, K.; Sagi-Eisenberg, R. Mast cell adenosine receptors function: A focus on the A3 adenosine receptor and inflammation. Front. Immunol. 2012, 3, 134. [Google Scholar] [CrossRef]
- Cohen, H.B.; Ward, A.; Hamidzadeh, K.; Ravid, K.; Mosser, D.M. IFN-γ prevents adenosine receptor (A2bR) upregulation to sustain the macrophage activation response. J. Immunol. 2015, 195, 3828–3837. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Zanin, R.F.; Braganhol, E.; Bergamin, L.S.; Campesato, L.F.; Filho, A.Z.; Moreira, J.C.; Morrone, F.B.; Sévigny, J.; Schetinger, M.R.; de Souza Wyse, A.T.; et al. Differential macrophage activation alters the expression profile of NTPDase and ecto-5’-nucleotidase. PLoS ONE 2012, 7, e31205. [Google Scholar] [CrossRef] [PubMed]
- Barletta, K.E.; Ley, K.; Mehrad, B. Regulation of neutrophil function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 856–864. [Google Scholar] [CrossRef]
- Wang, X.; Chen, D. Purinergic regulation of neutrophil function. Front. Immunol. 2018, 9, 399. [Google Scholar] [CrossRef]
- Frasson, A.P.; Menezes, C.B.; Goelzer, G.K.; Gnoatto, S.C.B.; Garcia, S.C.; Tasca, T. Adenosine reduces reactive oxygen species and interleukin-8 production by Trichomonas vaginalis-stimulated neutrophils. Purinergic Signal. 2017, 13, 569–577. [Google Scholar] [CrossRef]
- Corriden, R.; Self, T.; Akong-Moore, K.; Nizet, V.; Kellam, B.; Briddon, S.J.; Hill, S.J. Adenosine-A3 receptors in neutrophil microdomains promote the formation of bacteria-tethering cytonemes. EMBO Rep. 2013, 14, 726–732. [Google Scholar] [CrossRef]
- Xu, K.; Cooney, K.A.; Shin, E.Y.; Wang, L.; Deppen, J.N.; Ginn, S.C.; Levit, R.D. Adenosine from a biologic source regulates neutrophil extracellular traps (NETs). J. Leukoc. Biol. 2019, 105, 1225–1234. [Google Scholar] [CrossRef]
- Przybyla, T.; Sakowicz-Burkiewicz, M.; Pawelczyk, T. Purinergic signaling in B cells. Acta Biochim. Pol. 2018, 65, 1–7. [Google Scholar] [CrossRef]
- Figueiró, F.; Muller, L.; Funk, S.; Jackson, E.K.; Battastini, A.M.; Whiteside, T.L. Phenotypic and functional characteristics of CD39(high) human regulatory B cells (Breg). Oncoimmunology 2016, 5, e1082703. [Google Scholar] [CrossRef] [PubMed]
- Saze, Z.; Schuler, P.J.; Hong, C.S.; Cheng, D.; Jackson, E.K.; Whiteside, T.L. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood 2013, 122, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Cekic, C. Regulation of lymphocyte function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.K.; Silva, M.; Labuda, J.; Thayer, M.; Cain, D.W.; Philbrook, P.; Sethumadhavan, S.; Hatfield, S.; Ohta, A.; Sitkovsky, M. The GS protein-coupled A2a adenosine receptor controls T cell help in the germinal center. J. Biol. Chem. 2017, 292, 1211–1217. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Cavanaugh, C.; Pereira, M.; Babu, U.; Williams, K. Susceptibility of aging mice to listeriosis: Role of anti-inflammatory responses with enhanced Treg-cell expression of CD39/CD73 and Th-17 cells. Int. J. Med. Microbiol. 2020, 310, 151397. [Google Scholar] [CrossRef]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Hüls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am. J. Physiol. Cell Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef]
- Liang, D.; Woo, J.I.; Shao, H.; Born, W.K.; O’Brien, R.L.; Kaplan, H.J.; Sun, D. Ability of γδ T cells to modulate the Foxp3 T cell response is dependent on adenosine. PLoS ONE 2018, 13, e0197189. [Google Scholar] [CrossRef]
- Ehrentraut, H.; Westrich, J.A.; Eltzschig, H.K.; Clambey, E.T. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS ONE 2012, 7, e32416. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Feng, M.; Du, S.; Wei, X.; Song, H.; Yixin, X.; Song, J.; Wenxian, G. Adenosine generated by regulatory T cells induces CD8+ T cell exhaustion in gastric cancer through A2aR pathway. Biomed. Res. Int. 2019, 2019, 4093214. [Google Scholar] [CrossRef] [PubMed]
- Allard, D.; Allard, B.; Stagg, J. On the mechanism of anti-CD39 immune checkpoint therapy. J. Immunother. Cancer 2020, 8, e000186. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.; Kuchroo, V.K. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand. J. Immunol. 2010, 72, 173–184. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J.J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Rizzo, R.; Malavasi, F. The role of extracellular adenosine generation in the development of autoimmune diseases. Mediat. Inflamm. 2018, 2018, 7019398. [Google Scholar] [CrossRef]
- Vuerich, M.; Harshe, R.P.; Robson, S.C.; Longhi, M.S. Dysregulation of adenosinergic signaling in systemic and organ-specific autoimmunity. Int. J. Mol. Sci. 2019, 20, 528. [Google Scholar] [CrossRef]
- Han, L.; Sugiyama, H.; Zhang, Q.; Yan, K.; Fang, X.; McCormick, T.S.; Cooper, K.D.; Huang, Q. Phenotypical analysis of ectoenzymes CD39/CD73 and adenosine receptor 2A in CD4 + CD25 high Foxp3 + regulatory T-cells in psoriasis. Australas J. Dermatol. 2018, 59, e31–e38. [Google Scholar] [CrossRef]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef]
- Cross, M.; Smith, E.; Hoy, D.; Carmona, L.; Wolfe, F.; Vos, T.; Williams, B.; Gabriel, S.; Lassere, M.; Johns, N.; et al. The global burden of rheumatoid arthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1316–1322. [Google Scholar] [CrossRef]
- Pal, Y.; Bandyopadhyay, N.; Pal, R.S.; Ahmed, S.; Bandopadhyay, S. Perspective and potential of A2A and A3 adenosine receptors as therapeutic targets for the treatment of rheumatoid arthritis. Curr. Pharm. Des. 2019, 25, 2859–2874. [Google Scholar] [CrossRef] [PubMed]
- Volkov, M.; van Schie, K.A.; van der Woude, D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunol. Rev. 2020, 294, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, V.; Pavlovic-Popovic, Z.; Vukmirovic, S.; Cvejic, J.; Mooranian, A.; Al-Salami, H.; Mikov, M.; Golocorbin-Kon, S. Molecular mechanism of action and pharmacokinetic properties of methotrexate. Mol. Biol. Rep. 2020, 47, 4699–4708. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Angelis-Stoforidis, P.; Vajda, F.J.; Christophidis, N. Methotrexate polyglutamate levels in circulating erythrocytes and polymorphs correlate with clinical efficacy in rheumatoid arthritis. Clin. Exp. Rheumatol. 1999, 17, 313–320. [Google Scholar] [PubMed]
- Cronstein, B.N.; Sitkovsky, M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 41–51. [Google Scholar] [CrossRef]
- Montesinos, M.C.; Yap, J.S.; Desai, A.; Posadas, I.; McCrary, C.T.; Cronstein, B.N. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: Evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000, 43, 656–663. [Google Scholar] [CrossRef]
- Kobold, N.; Jenko, B.; Tomšič, M.; Dolžan, V.; Praprotnik, S. ADORA2A Polymorphisms Influence Methotrexate Adverse Events in Rheumatoid Arthritis. Isr. Med. Assoc. J. 2019, 5, 333–338. [Google Scholar]
- Burnstock, G. Purinergic signalling: Therapeutic developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef]
- Singh, A.; Misra, R.; Aggarwal, A. Baseline adenosine receptor mRNA expression in blood as predictor of response to methotrexate therapy in patients with rheumatoid arthritis. Rheumatol. Int. 2019, 39, 1431–1438. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C. Association of the ATIC 347 C/G polymorphism with responsiveness to and toxicity of methotrexate in rheumatoid arthritis: A meta-analysis. Rheumatol. Int. 2016, 36, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Peres, R.S.; Liew, F.Y.; Talbot, J.; Carregaro, V.; Oliveira, R.D.; Almeida, S.L.; França, R.F.; Donate, P.B.; Pinto, L.G.; Ferreira, F.I.; et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Montesinos, M.C.; Weissmann, G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB. Proc. Natl. Acad. Sci. USA 1999, 96, 6377–6381. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Reitman, M.L. Adenosine-Related Mechanisms in Non-Adenosine Receptor Drugs. Cells 2020, 9, 956. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Safarzadeh, E.; Jadidi-Niaragh, F.; Motallebnezhad, M.; Yousefi, M. The role of adenosine and adenosine receptors in the immunopathogenesis of multiple sclerosis. Inflamm. Res. 2016, 65, 511–520. [Google Scholar] [CrossRef]
- Mayne, M.; Shepel, P.N.; Jiang, Y.; Geiger, J.D.; Power, C. Dysregulation of adenosine A1 receptor-mediated cytokine expression in peripheral blood mononuclear cells from multiple sclerosis patients. Ann. Neurol. 1999, 45, 633–639. [Google Scholar] [CrossRef]
- Johnston, J.B.; Silva, C.; Gonzalez, G.; Holden, J.; Warren, K.G.; Metz, L.M.; Power, C. Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann. Neurol. 2001, 49, 650–658. [Google Scholar] [CrossRef]
- Tsutsui, S.; Schnermann, J.; Noorbakhsh, F.; Henry, S.; Yong, V.W.; Winston, B.W.; Warren, K.; Power, C. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J. Neurosci. 2004, 24, 1521–1529. [Google Scholar] [CrossRef]
- Chen, G.Q.; Chen, Y.Y.; Wang, X.S.; Wu, S.Z.; Yang, H.M.; Xu, H.Q.; He, J.C.; Wang, X.T.; Chen, J.F.; Zheng, R.Y. Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in Wistar rats. Brain Res. 2010, 1309, 116–125. [Google Scholar] [CrossRef]
- Wang, T.; Xi, N.N.; Chen, Y.; Shang, X.F.; Hu, Q.; Chen, J.F.; Zheng, R.Y. Chronic caffeine treatment protects against experimental autoimmune encephalomyelitis in mice: Therapeutic window and receptor subtype mechanism. Neuropharmacology 2014, 86, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Vergote, D.; Shariat, N.; Warren, K.; Ferguson, S.S.; Power, C. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: Reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells. FASEB J. 2008, 22, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, W.; Guo, J.; Kong, F.; Zhou, S.; Chen, S.; Wang, Z.; Zang, D. Adenosine binds predominantly to adenosine receptor A1 subtype in astrocytes and mediates an immunosuppressive effect. Brain Res. 2018, 1700, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, G.; Guo, J.; Kong, F.; Chen, S.; Wang, Z. Pro-inflammatory effect of downregulated CD73 expression in EAE astrocytes. Front. Cell. Neurosci. 2019, 13, 233. [Google Scholar] [CrossRef]
- Bru-Tari, E.; Cobo-Vuilleumier, N.; Alonso-Magdalena, P.; Dos Santos, R.S.; Marroqui, L.; Nadal, A.; Gauthier, B.R.; Quesada, I. Pancreatic alpha-cell mass in the early-onset and advanced stage of a mouse model of experimental autoimmune diabetes. Sci. Rep. 2019, 9, 9515. [Google Scholar] [CrossRef]
- Yip, L.; Taylor, C.; Whiting, C.C.; Fathman, G. Diminished Adenosine A1 Receptor Expression in Pancreatic α-cells May Contribute to the Pathology of Type 1 Diabetes. Diabetes 2013, 62, 4208–4219. [Google Scholar] [CrossRef]
- Yang, P.; Chen, P.; Wang, T.; Zhan, Y.; Zhou, M.; Xia, L.; Cheng, R.; Guo, Y.; Zhu, L.; Zhang, J. Loss of A(1) adenosine receptor attenuates alpha-naphthylisothiocyanate-induced cholestatic liver injury in mice. Toxicol. Sci. 2013, 131, 128–138. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The belated US FDA approval of the adenosine A(2A) receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Ohta, A. The ‘danger’ sensors that STOP the immune response: The A2 adenosine receptors? Trends Immunol. 2005, 26, 299–304. [Google Scholar] [CrossRef]
- Varani, K.; Vincenzi, F.; Tosi, A.; Targa, M.; Masieri, F.F.; Ongaro, A.; De Mattei, M.; Massari, L.; Borea, P.A. Expression and functional role of adenosine receptors in regulating inflammatory responses in human synoviocytes. Br. J. Pharmacol. 2010, 160, 101–115. [Google Scholar] [CrossRef]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and Function of A(2A) and A₃ Adenosine Receptors in Patients with Ankylosing Spondylitis, Psoriatic Arthritis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef]
- Montes, G.C.; Hammes, N.; da Rocha, M.D.; Montagnoli, T.L.; Fraga, C.A.; Barreiro, E.J.; Sudo, R.T.; Zapata-Sudo, G. Treatment with adenosine receptor agonist ameliorates pain induced by acute and chronic inflammation. J. Pharmacol. Exp. Ther. 2016, 358, 315–323. [Google Scholar] [CrossRef]
- Varani, K.; Padovan, M.; Vincenzi, F.; Targa, M.; Trotta, F.; Govoni, M.; Borea, P.A. A2A and A3 adenosine receptor expression in rheumatoid arthritis: Upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res. Ther. 2011, 13, R197. [Google Scholar] [CrossRef]
- Odashima, M.; Bamias, G.; Rivera-Nieves, J.; Linden, J.; Nast, C.C.; Moskaluk, C.A.; Marini, M.; Sugawara, K.; Kozaiwa, K.; Otaka, M.; et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology 2005, 129, 26–33. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacol. Rev. 2019, 71, 345–382. [Google Scholar] [CrossRef]
- Bortoluzzi, A.; Vincenzi, F.; Govoni, M.; Padovan, M.; Ravani, A.; Borea, P.A.; Varani, K. A2A adenosine receptor upregulation correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 192. [Google Scholar] [CrossRef]
- Yao, S.-Q.; Li, Z.-Z.; Huang, Q.-Y.; Li, F.; Wang, Z.-W.; Augusto, E.; He, J.-C.; Wang, X.-T.; Chen, J.-F.; Zheng, R.-Y. Genetic inactivation of the adenosine A(2A) receptor exacerbates brain damage in mice with experimental autoimmune encephalomyelitis. J. Neurochem. 2012, 123, 100–112. [Google Scholar] [CrossRef]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Merighi, S.; Gessi, S.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Varani, K. Multiple sclerosis lymphocytes upregulate A2A adenosine receptors that are anti-inflammatory when stimulated. Eur. J. Immunol. 2013, 43, 2206–2216. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Z.X.; Zheng, L.P.; Wang, L.; Liu, Y.F.; Yin, W.Y.; Chen, Y.Y.; Wang, X.S.; Hou, S.T.; Chen, J.F.; et al. The adenosine A2A receptor antagonist SCH58261 reduces macrophage/microglia activation and protects against experimental autoimmune encephalomyelitis in mice. Neurochem. Int. 2019, 129, 104490. [Google Scholar] [CrossRef]
- Rajasundaram, S. Adenosine A2A Receptor Signaling in the Immunopathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Liang, D.; Shao, H.; Born, W.K.; O’Brien, R.L.; Kaplan, H.J.; Sun, D. High level expression of A2ARs is required for the enhancing function, but not for the inhibiting function, of γδ T cells in the autoimmune responses of EAU. PLoS ONE 2018, 13, e0199601. [Google Scholar] [CrossRef]
- Muhammad, F.; Wang, D.; Montieth, A.; Lee, S.; Preble, J.; Foster, C.S.; Larson, T.A.; Ding, K.; Dvorak, J.D.; Lee, D.J. PD-1+ melanocortin receptor dependent-Treg cells prevent autoimmune disease. Sci. Rep. 2019, 9, 16941. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, E.A.; White, P.J.; May, L.T. The adenosine A(2B) G protein-coupled receptor: Recent advances and therapeutic implications. Pharmacol. Ther. 2019, 198, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Du, C.; Lv, J.; Zhao, G.; Li, Z.; Wu, Z.; Haskó, G.; Xie, X. Blocking A2B adenosine receptor alleviates pathogenesis of experimental autoimmune encephalomyelitis via inhibition of IL-6 production and Th17 differentiation. J. Immunol. 2013, 190, 138–146. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Y.; Du, B.; Wang, Y.; Yang, G.Y.; Bi, X. Mesenchymal stem cells attenuated blood-brain barrier disruption via downregulation of aquaporin-4 expression in EAE mice. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Chen, M.; Liang, D.; Zuo, A.; Shao, H.; Kaplan, H.J.; Sun, D. An A2B adenosine receptor agonist promotes Th17 autoimmune responses in Experimental Autoimmune Uveitis (EAU) via dendritic cell activation. PLoS ONE 2015, 10, e0132348. [Google Scholar] [CrossRef]
- Liang, D.; Zuo, A.; Shao, H.; Chen, M.; Kaplan, H.J.; Sun, D. A2B adenosine receptor activation switches differentiation of bone marrow cells to a CD11c+Gr-1+ dendritic cell subset that promotes the Th17 response. Immunol. Inflamm. Dis. 2015, 3, 360–373. [Google Scholar] [CrossRef]
- Bazzichi, L.; Trincavelli, L.; Rossi, A.; De Feo, F.; Lucacchini, A.; Bombardieri, S.; Martini, C. A2B adenosine receptor activity is reduced in neutrophils from patients with systemic sclerosis. Arthritis Res. Ther. 2005, 7, R189–R195. [Google Scholar] [CrossRef]
- Karmouty-Quintana, H.; Molina, J.G.; Philip, K.; Bellocchi, C.; Gudenkauf, B.; Wu, M.; Chen, N.Y.; Collum, S.D.; Ko, J.; Agarwal, S.K.; et al. The antifibrotic effect of A2b adenosine receptor antagonism in a mouse model of dermal fibrosis. Arthritis Rheumatol. 2018, 70, 1673–1684. [Google Scholar] [CrossRef]
- Németh, Z.H.; Bleich, D.; Csóka, B.; Pacher, P.; Mabley, J.G.; Himer, L.; Vizi, E.S.; Deitch, E.A.; Szabó, C.; Cronstein, B.N.; et al. Adenosine receptor activation ameliorates type 1 diabetes. FASEB J. 2007, 21, 2379–2388. [Google Scholar] [CrossRef]
- Chia, J.S.; McRae, J.L.; Thomas, H.E.; Fynch, S.; Elkerbout, L.; Hill, P.; Murray-Segal, L.; Robson, S.C.; Chen, J.F.; d’Apice, A.J.; et al. The protective effects of CD39 overexpression in multiple low-dose streptozotocin-induced diabetes in mice. Diabetes 2013, 62, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Shakya, A.K.; Naik, R.R.; Almasri, I.M.; Kaur, A. Role and function of adenosine and its receptors in inflammation, neuroinflammation, IBS, autoimmune inflammatory disorders, rheumatoid arthritis and psoriasis. Curr. Pharm. Des. 2019, 25, 2875–2891. [Google Scholar] [CrossRef]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Kolachala, V.L.; Bajaj, R.; Chalasani, M.; Sitaraman, S.V. Purinergic receptors in gastrointestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G401–G410. [Google Scholar] [CrossRef] [PubMed]
- Kolachala, V.L.; Vijay-Kumar, M.; Dalmasso, G.; Yang, D.; Linden, J.; Wang, L.; Gewirtz, A.; Ravid, K.; Merlin, D.; Sitaraman, S.V. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology 2008, 135, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Fornai, M.; Awwad, O.; Giustarini, G.; Pellegrini, C.; Tuccori, M.; Caputi, V.; Qesari, M.; Castagliuolo, I.; Brun, P.; et al. Role of the A(2B) receptor-adenosine deaminase complex in colonic dysmotility associated with bowel inflammation in rats. Br. J. Pharmacol. 2014, 171, 1314–1329. [Google Scholar] [CrossRef]
- Aherne, C.M.; Collins, C.B.; Rapp, C.R.; Olli, K.E.; Perrenoud, L.; Jedlicka, P.; Bowser, J.L.; Mills, T.W.; Karmouty-Quintana, H.; Blackburn, M.R.; et al. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight 2018, 3, e121521. [Google Scholar] [CrossRef]
- Bernuzzi, F.; Fenoglio, D.; Battaglia, F.; Fravega, M.; Gershwin, M.E.; Indiveri, F.; Ansari, A.A.; Podda, M.; Invernizzi, P.; Filaci, G. Phenotypical and functional alterations of CD8 regulatory T cells in primary biliary cirrhosis. J. Autoimmun. 2010, 35, 176–180. [Google Scholar] [CrossRef]
- Lavoie, E.G.; Fausther, M.; Goree, J.R.; Dranoff, J.A. The cholangiocyte adenosine-IL-6 axis regulates survival during biliary cirrhosis. Gene Exp. 2017, 17, 327–340. [Google Scholar] [CrossRef]
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 200, 151–199. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A(3) Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Cohen, S. The A3 adenosine receptor (A3AR): Therapeutic target and predictive biological marker in rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. [Google Scholar] [CrossRef]
- Bar-Yehuda, S.; Silverman, M.H.; Kerns, W.D.; Ochaion, A.; Cohen, S.; Fishman, P. The anti-inflammatory effect of A3 adenosine receptor agonists: A novel targeted therapy for rheumatoid arthritis. Expert Opin. Investig. Drugs 2007, 16, 1601–1613. [Google Scholar] [CrossRef]
- Balogh, E.A.; Bashyam, A.M.; Ghamrawi, R.I.; Feldman, S.R. Emerging systemic drugs in the treatment of plaque psoriasis. Expert Opin. Emerg. Drugs 2020, 25, 89–100. [Google Scholar] [CrossRef]
- Costa, L.; Del Puente, A.; Peluso, R.; Tasso, M.; Caso, P.; Chimenti, M.S.; Sabbatino, V.; Girolimetto, N.; Benigno, C.; Bertolini, N.; et al. Small molecule therapy for managing moderate to severe psoriatic arthritis. Expert Opin. Pharmacother. 2017, 18, 1557–1567. [Google Scholar] [CrossRef]
- Magni, G.; Pedretti, S.; Audano, M.; Caruso, D.; Mitro, N.; Ceruti, S. Glial cell activation and altered metabolic profile in the spinal-trigeminal axis in a rat model of multiple sclerosis associated with the development of trigeminal sensitization. Brain Behav. Immun. 2020. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Giancotti, L.A.; Lauro, F.; Mufti, F.; Salvemini, D. Treatment of chronic neuropathic pain: Purine receptor modulation. Pain 2020, 161, 1425–1441. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Receptor | Autoimmune Disease | Hypothesized Role | References |
|---|---|---|---|
| A1 | Multiple sclerosis | Protective role | [56] |
| Type 1 diabetes | Reduced expression in pancreatic α-cells contributes to the pathology | [66] | |
| Autoimmune liver disorders | Lack of receptors limits the efflux of toxic biliary constituents | [67] | |
| A2A | Reumathoid arthritis | Anti-inflammatory activity | [46] |
| Inflammatory bowel disease | Anti-inflammatory activity | [74] | |
| Multiple sclerosis/EAE | Opposite conflicting results | [77,79] | |
| Experimental autoimmune uveitis | Activation promotes disease progression | [81] | |
| Systemic Lupus Erythematosus | Anti-inflammatory activity | [75,76] | |
| A2B | Multiple sclerosis | Pro-inflammatory actions | [84] |
| Experimental autoimmune uveitis | Pro-inflammatory actions | [86] | |
| Systemic sclerosis | Pro-fibrotic regulator in vivo | [89] | |
| Type 1 diabetes | Inhibition of pro-inflammatory cytokine release | [90] | |
| Inflammatory bowel disease | Pro-inflammatory actions | [92] | |
| Autoimmune liver disorders | Promotion of IL-6 release | [99] | |
| A3 | Reumathoid arthritis | Dampening of immune response (Phase II clinical trials with selective agonists) | [103] |
| Psoriasis | Dampening of immune response (Phase III clinical trials with selective agonists) | [105] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magni, G.; Ceruti, S. Adenosine Signaling in Autoimmune Disorders. Pharmaceuticals 2020, 13, 260. https://doi.org/10.3390/ph13090260
Magni G, Ceruti S. Adenosine Signaling in Autoimmune Disorders. Pharmaceuticals. 2020; 13(9):260. https://doi.org/10.3390/ph13090260
Chicago/Turabian StyleMagni, Giulia, and Stefania Ceruti. 2020. "Adenosine Signaling in Autoimmune Disorders" Pharmaceuticals 13, no. 9: 260. https://doi.org/10.3390/ph13090260
APA StyleMagni, G., & Ceruti, S. (2020). Adenosine Signaling in Autoimmune Disorders. Pharmaceuticals, 13(9), 260. https://doi.org/10.3390/ph13090260