In Vitro Modeling of Bradykinin-Mediated Angioedema States

 ,
,  ,
,
Abstract
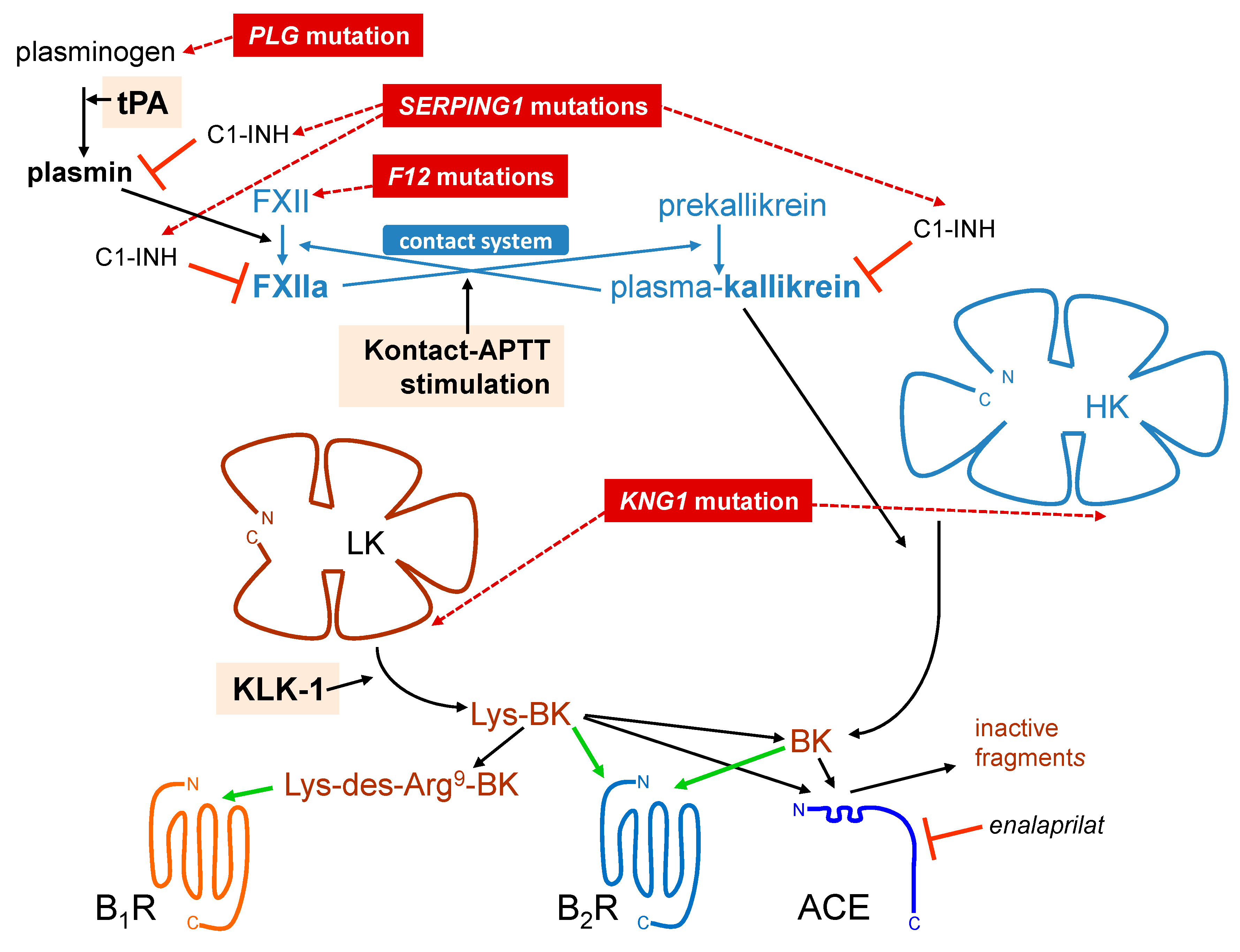
1. The Kallikrein–Kinin System
2. Bradykinin (BK)-Mediated Angioedema States
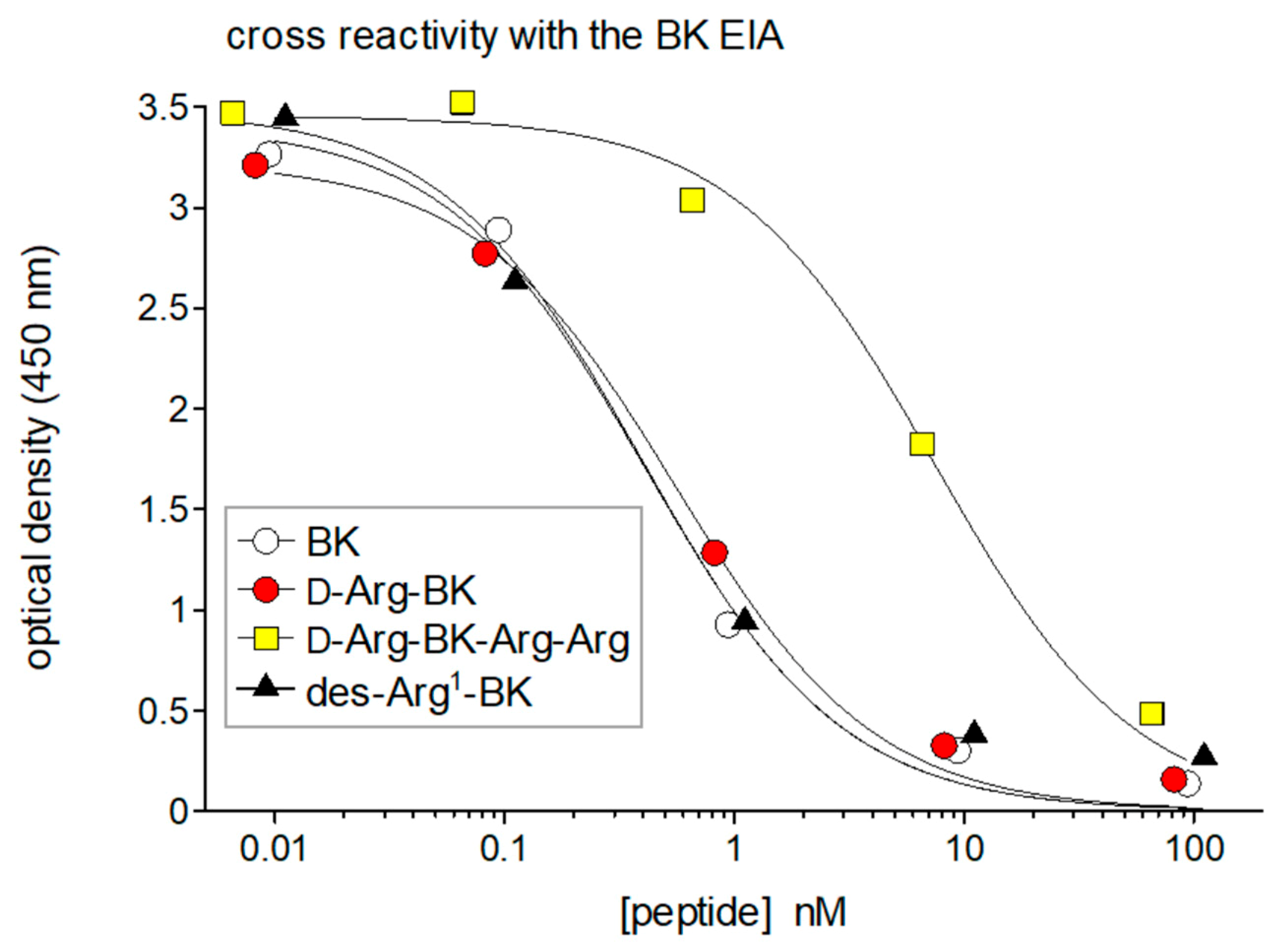
3. Antibody-Based Analytical Techniques for Kinins: Importance and Pitfalls
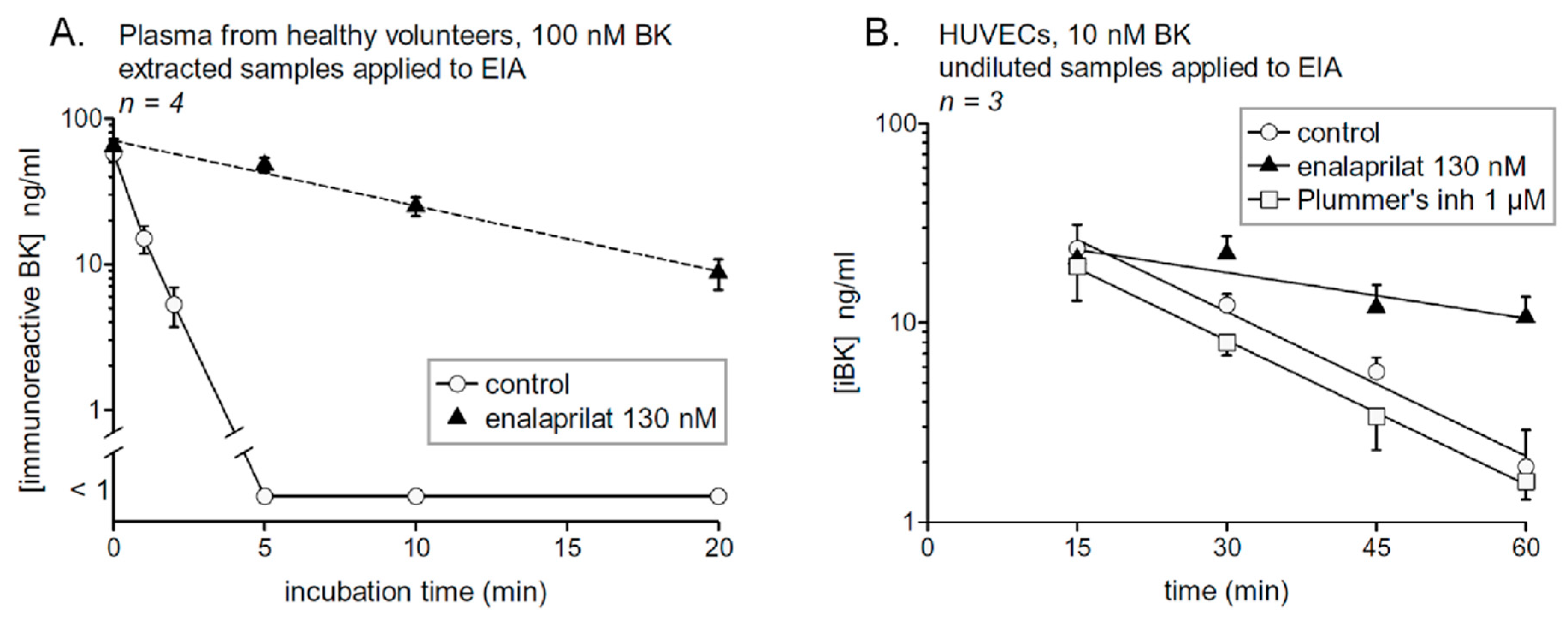
4. Probing Immunoreactive BK Degradation In Vitro
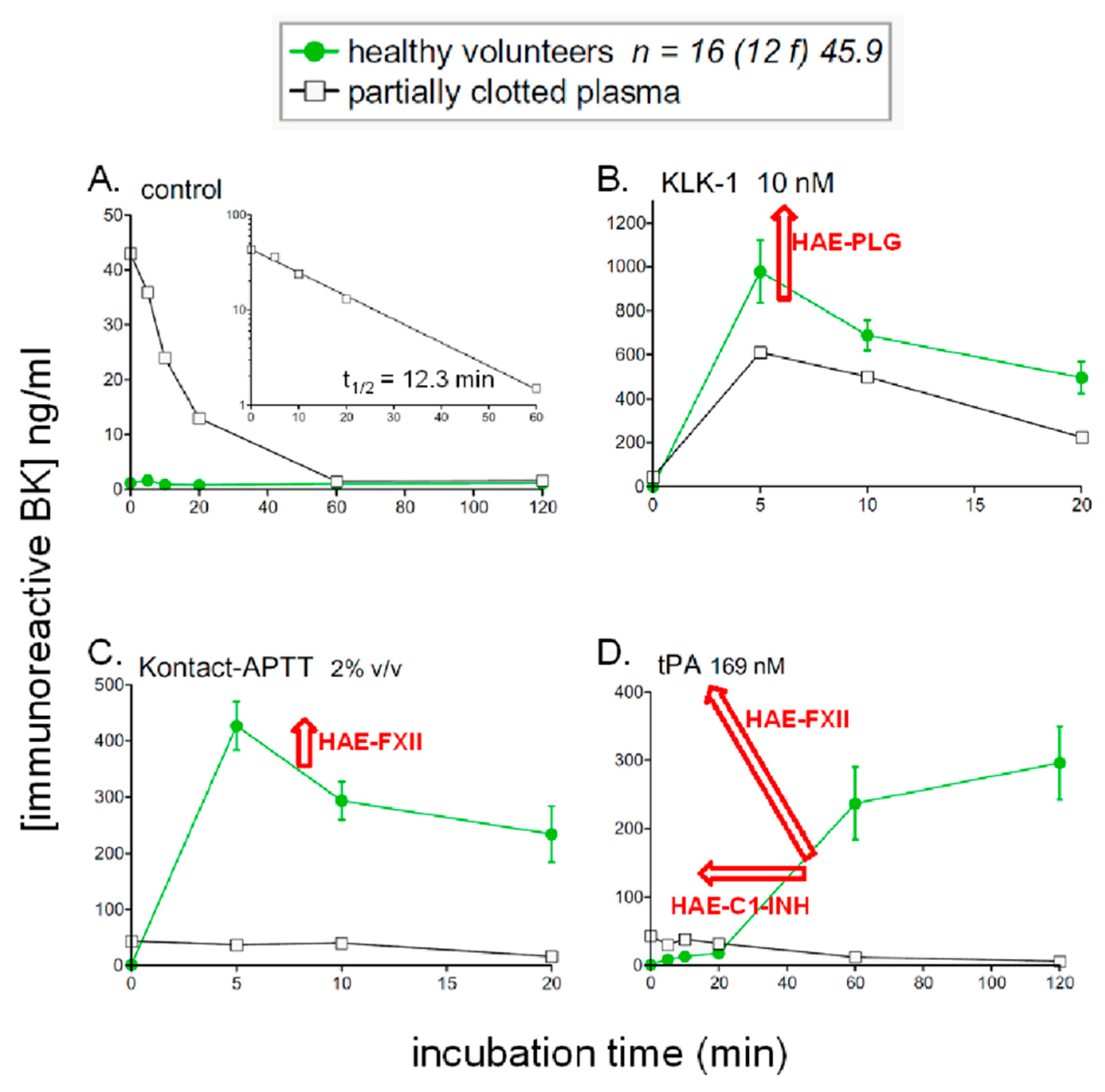
5. Profiling Immunoreactive BK Generation In Vitro: Modeling Hereditary Angioedema (HAE)
6. Areas of Uncertainty: Role of BK B1 Receptors and Tissue Kallikrein (KLK-1) in Angioedema States
7. Conclusions
8. Materials and Methods (for Original Results)
8.1. Degradation of BK by Human Endothelial Cells
8.2. Generation and Degradation of BK by Human Plasma in Health and Disease
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaplan, A.P. The bradykinin-forming cascade: A historical perspective. Chem. Immunol. Allergy 2014, 100, 205–213. [Google Scholar] [PubMed]
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar] [PubMed]
- Blais, C.; Marceau, F.; Rouleau, J.L.; Adam, A. The kallikrein-kininogen-kinin systems: Lessons from the quantification of endogenous kinins. Peptides 2000, 21, 1903–1940. [Google Scholar] [CrossRef]
- Alhenc-Gelas, F.; Bouby, N.; Girolami, J.P. Kallikrein/K1, Kinins, and ACE/Kininase II in homeostasis and in disease insight from human and experimental genetic studies, therapeutic implication. Front. Med. (Lausanne) 2019, 6, 136. [Google Scholar] [CrossRef]
- Leeb-Lundberg, L.M.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International Union of Pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef]
- Bergaya, S.; Matrougui, K.; Meneton, P.; Henrion, D.; Boulanger, C.M. Role of tissue kallikrein in response to flow in mouse resistance arteries. J. Hypertens. 2004, 22, 745–750. [Google Scholar] [CrossRef]
- Stadnicki, A.; Mazurek, U.; Plewka, D.; Wilczok, T. Intestinal tissue kallikrein-kallistatin profile in inflammatory bowel disease. Int. Immunopharmacol. 2003, 3, 939–944. [Google Scholar] [CrossRef]
- Sexton, D.J.; Chen, T.; Martik, D.; Kuzmic, P.; Kuang, G.; Chen, J.; Nixon, A.E.; Zuraw, B.L.; Forteza, R.M.; Abraham, W.M.; et al. Specific inhibition of tissue kallikrein 1 with a human monoclonal antibody reveals a potential role in airway diseases. Biochem. J. 2009, 422, 383–392. [Google Scholar] [CrossRef]
- Bouillet, L.; Mannic, T.; Arboleas, M.; Subileau, M.; Massot, C.; Drouet, C.; Huber, P.; Vilgrain, I. Hereditary angioedema: Key role for kallikrein and bradykinin in vascular endothelial-cadherin cleavage and edema formation. J. Allergy Clin. Immunol. 2011, 128, 232–234. [Google Scholar] [CrossRef]
- Kaplan, A.P.; Austen, K.F. A prealbumin activator of prekallikrein. II. Derivation of activators of prekallikrein from active Hageman factor by digestion with plasmin. J. Exp. Med. 1971, 133, 696–712. [Google Scholar] [CrossRef]
- Chao, J.; Chao, L. Biochemistry, regulation and potential function of kallistatin. Biol. Chem. Hoppe Seyler 1995, 376, 705–713. [Google Scholar] [PubMed]
- Charest-Morin, X.; Hébert, J.; Rivard, G.É.; Bonnefoy, A.; Wagner, E.; Marceau, F. Comparing pathways of bradykinin formation in whole blood from healthy volunteers and patients with hereditary angioedema due to C1 inhibitor deficiency. Front. Immunol. 2018, 9, 2183. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Magerl, M.; Metz, M.; Siebenhaar, F.; Weller, K.; Krause, K. Practical algorithm for diagnosing patients with recurrent wheals or angioedema. Allergy 2013, 68, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Bezalel, S.; Mahlab-Guri, K.; Asher, I.; Werner, B.; Sthoeger, Z.M. Angiotensin-converting enzyme inhibitor-induced angioedema. Am. J. Med. 2015, 128, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Aygören-Pürsün, E.; Magerl, M.; Maetzel, A.; Maurer, M. Epidemiology of Bradykinin-mediated angioedema: A systematic investigation of epidemiological studies. Orphanet J. Rare Dis. 2018, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Cyr, M.; Lepage, Y.; Blais, C.; Gervais, N.; Cugno, M.; Rouleau, J.L.; Adam, A. Bradykinin and des-Arg9-bradykinin metabolic pathways and kinetics of activation of human plasma. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H275–H283. [Google Scholar] [CrossRef]
- Fryer, R.M.; Segreti, J.; Banfor, P.N.; Widomski, D.L.; Backes, B.J.; Lin, C.W.; Ballaron, S.J.; Cox, B.F.; Trevillyan, J.M.; Reinhart, G.A.; et al. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: Rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br. J. Pharmacol. 2008, 153, 947–955. [Google Scholar] [CrossRef]
- Marceau, F.; Rivard, G.E.; Gauthier, J.M.; Binkley, K.; Bonnefoy, A.; Boccon-Gibod, I.; Bouillet, L.; Picard, M.; Levesque, G.; Elfassy, H.L.; et al. Measurement of bradykinin formation and degradation in blood plasma: Relevance for acquired angioedema associated with angiotensin converting enzyme inhibition and for hereditary angioedema due to factor XII or plasminogen gene variants. Front. Med. (Lausanne) 2020, 7, 358. [Google Scholar] [CrossRef]
- Hoover, T.; Lippmann, M.; Grouzmann, E.; Marceau, F.; Herscu, P. Angiotensin converting enzyme inhibitor induced angio-oedema: A review of the pathophysiology and risk factors. Clin. Exp. Allergy 2009, 40, 50–61. [Google Scholar] [CrossRef]
- Molinaro, G.; Gervais, N.; Adam, A. Biochemical basis of angioedema associated with recombinant tissue plasminogen activator treatment: An in vitro experimental approach. Stroke 2002, 33, 1712–1716. [Google Scholar] [CrossRef]
- Gauberti, M.; Potzeha, F.; Vivien, D.; Martinez de Lizarrondo, S. Impact of bradykinin generation during thrombolysis in ischemic stroke. Front. Med. (Lausanne) 2018, 5, 195. [Google Scholar] [CrossRef] [PubMed]
- Marcelino-Rodriguez, I.; Callero, A.; Mendoza-Alvarez, A.; Perez-Rodriguez, E.; Barrios-Recio, J.; Garcia-Robaina, J.C.; Flores, C. Bradykinin-mediated angioedema: An update of the genetic causes and the impact of genomics. Front. Genet. 2019, 10, 900. [Google Scholar] [CrossRef] [PubMed]
- de Maat, S.; Björkqvist, J.; Suffritti, C.; Wiesenekker, C.P.; Nagtegaal, W.; Koekman, A.; van Dooremalen, S.; Pasterkamp, G.; de Groot, P.G.; Cicardi, M.; et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J. Allergy Clin. Immunol. 2016, 138, 1414–1423. [Google Scholar] [CrossRef]
- Bafunno, V.; Firinu, D.; D’Apolito, M.; Cordisco, G.; Loffredo, S.; Leccese, A.; Bova, M.; Barca, M.P.; Santacroce, R.; Cicardi, M.; et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J. Allergy Clin. Immunol. 2018, 141, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Han, E.D.; MacFarlane, R.C.; Mulligan, A.N.; Scafidi, J.; Davis, A.E., 3rd. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J. Clin. Investig. 2002, 109, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Veronez, C.L.; Maghsodi, S.; Todiras, M.; Popova, E.; Rodrigues, A.F.; Qadri, F.; Pesquero, J.B.; Bader, M. Endothelial B2-receptor overexpression as an alternative animal model for hereditary angioedema. Allergy 2019, 74, 1998–2002. [Google Scholar] [CrossRef] [PubMed]
- Nicola, S.; Rolla, G.; Brussino, L. Breakthroughs in hereditary angioedema management: A systematic review of approved drugs and those under research. Drugs Context. 2019, 8, 212605. [Google Scholar] [PubMed]
- Lesage, A.; Gibson, C.; Marceau, F.; Ambrosi, H.-D.; Saupe, J.; Katzer, W.; Loenders, B.; Charest-Morin, X.; Knolle, J. In vitro pharmacological profile of new small molecule bradykinin B2 receptor antagonists. Front. Pharmacol. 2020, 11, 916. [Google Scholar] [CrossRef]
- Décarie, A.; Drapeau, G.; Closset, J.; Couture., R.; Adam, A. Development of digoxigenin-labeled peptide: Application to chemiluminoenzyme immunoassay of bradykinin in inflamed tissues. Peptides 1994, 5, 511–518. [Google Scholar]
- Raymond, P.; Drapeau, G.; Raut, R.; Audet, R.; Marceau, F.; Ong, H.; Adam, A. Quantification of des-Arg9-bradykinin using chemiluminescence enzyme immunoassay: Application to its kinetic profile during plasma activation. J. Immunol. Methods 1995, 180, 247–257. [Google Scholar] [CrossRef]
- Bachelard, H.; Charest-Morin, X.; Marceau, F. D-Arg0-bradykinin-Arg-Arg, a latent vasoactive bradykinin B2 receptor agonist metabolically activated by carboxypeptidases. Front. Pharmacol. 2018, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar] [PubMed]
- Adam, A.; Montpas, N.; Keire, D.; Désormeaux, A.; Brown, N.J.; Marceau, F.; Westenberger, B. Bradykinin forming capacity of oversulfated chondroitin sulfate contaminated heparin in vitro. Biomaterials 2010, 31, 5741–5748. [Google Scholar] [CrossRef]
- Ehlers, M.R.W.; Gordon, K.; Schwager, S.L.U.; Sturrock, E.D. Shedding the load of hypertension: The proteolytic processing of angiotensin-converting enzyme. S. Afr. Med. J. 2012, 102, 461–464. [Google Scholar] [CrossRef]
- Morissette, G.; Couture, J.P.; Désormeaux, A.; Adam, A.; Marceau, F. Lack of direct interaction between enalaprilat and the kinin B1 receptors. Peptides 2008, 29, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Koumbadinga, G.A.; Bawolak, M.T.; Marceau, E.; Adam, A.; Gera, L.; Marceau, F. A ligand-based approach to investigate the expression and function of angiotensin converting enzyme in intact human umbilical vein endothelial cells. Peptides 2010, 31, 1546–1554. [Google Scholar] [CrossRef]
- Graf, K.; Gräfe, M.; Auch-Schwelk, W.; Baumgarten, C.R.; Bossaler, C.; Fleck, E. Bradykinin degrading activity in cultured human endothelial cells. J. Cardiovasc. Pharmacol. 1992, 20 (Suppl. 9), S16–S20. [Google Scholar] [CrossRef]
- Sharma, U.; Cozier, G.E.; Sturrock, E.D.; Acharya, K.R. Molecular basis for omapatrilat and sampatrilat binding to neprilysin—Implications for dual inhibitor design with angiotensin converting enzyme. J. Med. Chem. 2020, 63, 5488–5500. [Google Scholar]
- Sheikh, I.A.; Kaplan, A.P. Studies of the digestion of bradykinin, Lys-bradykinin, and des-Arg9-bradykinin by angiotensin converting enzyme. Biochem. Pharmacol. 1986, 35, 1951–1956. [Google Scholar] [CrossRef]
- Pretorius, M.; McFarlane, J.A.; Vaughan, D.E.; Brown, N.J.; Murphey, L.J. Angiotensin-converting enzyme inhibition and smoking potentiate the kinin response to cardiopulmonary bypass. Clin. Pharmacol. Ther. 2004, 76, 379–387. [Google Scholar] [CrossRef]
- Suffritti, C.; Zanichelli, A.; Maggioni, L.; Bonanni, E.; Cugno, M.; Cicardi, M. High-molecular-weight kininogen cleavage correlates with disease states in the bradykinin-mediated angioedema due to hereditary C1-inhibitor deficiency. Clin. Exp. Allergy 2014, 44, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Baroso, R.; Sellier, P.; Defendi, F.; Charignon, D.; Ghannam, A.; Habib, M.; Drouet, C.; Favier, B. Kininogen cleavage assay: Diagnostic assistance for kinin-mediated angioedema conditions. PLoS ONE 2016, 11, e0163958. [Google Scholar] [CrossRef] [PubMed]
- Lara-Marquez, M.L.; Christiansen, S.C.; Riedl, M.A.; Herschbach, J.; Zuraw, B.L. Threshold-stimulated kallikrein activity distinguishes bradykinin- from histamine-mediated angioedema. Clin. Exp. Allergy 2018, 48, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Marceau, F.; Bachelard, H.; Rivard, G.É.; Hébert, J. Increased fibrinolysis-induced bradykinin formation in hereditary angioedema confirmed using stored plasma and biotechnological inhibitors. BMC Res. Notes 2019, 12, 291. [Google Scholar] [CrossRef]
- Zotter, Z.; Csuka, D.; Szabó, E.; Czaller, I.; Nébenführer, Z.; Temesszentandrási, G.; Fust, G.; Varga, L.; Farkas, H. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J. Rare Dis. 2014, 9, 44. [Google Scholar] [CrossRef]
- von Känel, R. Acute mental stress and hemostasis: When physiology becomes vascular harm. Thromb. Res. 2015, 135, S52–S55. [Google Scholar] [CrossRef]
- Duan, Q.L.; Binkley, K.; Rouleau, G.A. Genetic analysis of Factor XII and bradykinin catabolic enzymes in a family with estrogen-dependent inherited angioedema. J. Allergy Clin. Immunol. 2009, 123, 906–910. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Steinmüller-Magin, L.; Braenne, I.; Staubach-Renz, P.; Witzke, G.; Hardt, J. Hereditary angioedema with a mutation in the plasminogen gene. Allergy 2018, 73, 442–450. [Google Scholar] [CrossRef]
- Parsopoulou, F.; Charignon, D.; Tengo, M.; Psarros, F.; Maas, C.; Gonzalez-Quevedo, T.; Drouet, C.; Germenis, A.E.; Ghannam, A. Plasminogen glycoforms alteration and activation susceptibility associated with the missense variant p.Lys330Glu in HAE-PLG patients. Allergy 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Pantano, E.; Marchi, S.; Biagini, M.; Di Fede, M.; Nardi Dei, M.; Rossi Paccani, S.; Pizza, M.; Cartocci, E. NHBA is processed by kallikrein from human saliva. PLoS ONE 2019, 14, e02003234. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Witzke, G.; Machnig, T.; Hardt, J. Treatment of patients with hereditary angioedema with the c.988A>G (p.Lys330Glu) variant in the plasminogen gene. Orphanet J. Rare Dis. 2020, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Bossi, F.; Fischetti, F.; Regoli, D.; Durigutto, P.; Frossi, B.; Gobeil, F.; Ghebrehiwet, B.; Peerschke, E.I.; Cicardi, M.; Tedesco, F. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J. Allergy Clin. Immunol. 2009, 124, 1303–1310. [Google Scholar] [CrossRef]
- Koumbadinga, G.A.; Désormeaux, A.; Adam, A.; Marceau, F. Effect of interferon-γ on inflammatory cytokine-induced bradykinin B1 receptor expression in human vascular cells. Eur. J. Pharmacol. 2010, 647, 117–125. [Google Scholar] [CrossRef]
- Montinaro, V.; Cicardi, M. ACE inhibitor-mediated angioedema. Int. Immunopharmacol. 2020, 78, 106081. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.K.; Spencer, K.; Mascelli, M.; McCauley, T.G. Pharmacokinetics of single and repeat doses of icatibant. Clin. Pharmacol. Drug Dev. 2015, 4, 105–111. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Standing for |
|---|---|
| ACE | angiotensin-I converting enzyme |
| AE | angioedema |
| APTT | activated partial thromboplastin time |
| B1R | bradykinin B1 receptor |
| B2R | bradykinin B2 receptor |
| BK | bradykinin |
| C1-INH | C1-esterase inhibitor |
| c-Fos | a transcription factor |
| EIA | enzyme immunoassay |
| ERK1/2 | extracellular signal-regulated kinases 1 and 2 |
| F12 | gene encoding factor XII |
| FDA | U.S. Food and Drug Administration |
| FXI | coagulation factor XI |
| FXII | coagulation factor XII |
| HAE | hereditary angioedema |
| HAE-C1-INH | HAE caused by SERPING1 variants |
| HAE-FXII | HAE caused by F12 variants |
| HAE-PLG | HAE caused a PLG variant |
| HK | high molecular weight kininogen |
| HPLC | high-performance liquid chromatography |
| HUVEC | human umbilical vein endothelial cell |
| iBK | immunoreactive bradykinin |
| KD | binding dissociation constant |
| KLK-1 | tissue kallikrein |
| KNG1 | gene encoding kininogens |
| Kontact-APTT™ | a commcercial reagent that activates the contact system |
| LK | low molecular weight kininogen |
| Lys-BK | lysyl-bradykinin, kallidin |
| PI | propidium iodide |
| PLG | gene encoding plasminogen |
| s.e.m. | standard error of the mean |
| t1/2 | half-life |
| tPA | tissue plasminogen activator |
| VE-cadherin | vascular endothelial cadherin |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marceau, F.; Bachelard, H.; Charest-Morin, X.; Hébert, J.; Rivard, G.E. In Vitro Modeling of Bradykinin-Mediated Angioedema States. Pharmaceuticals 2020, 13, 201. https://doi.org/10.3390/ph13090201
Marceau F, Bachelard H, Charest-Morin X, Hébert J, Rivard GE. In Vitro Modeling of Bradykinin-Mediated Angioedema States. Pharmaceuticals. 2020; 13(9):201. https://doi.org/10.3390/ph13090201
Chicago/Turabian StyleMarceau, François, Hélène Bachelard, Xavier Charest-Morin, Jacques Hébert, and Georges E. Rivard. 2020. "In Vitro Modeling of Bradykinin-Mediated Angioedema States" Pharmaceuticals 13, no. 9: 201. https://doi.org/10.3390/ph13090201
APA StyleMarceau, F., Bachelard, H., Charest-Morin, X., Hébert, J., & Rivard, G. E. (2020). In Vitro Modeling of Bradykinin-Mediated Angioedema States. Pharmaceuticals, 13(9), 201. https://doi.org/10.3390/ph13090201