Development and Validation of Liquid Chromatography-Tandem Mass Spectrometry Method for Simple Analysis of Sumatriptan and its Application in Bioequivalence Study


Abstract
1. Introduction
2. Results and Discussion
2.1. Method Development
2.2. Method Validation
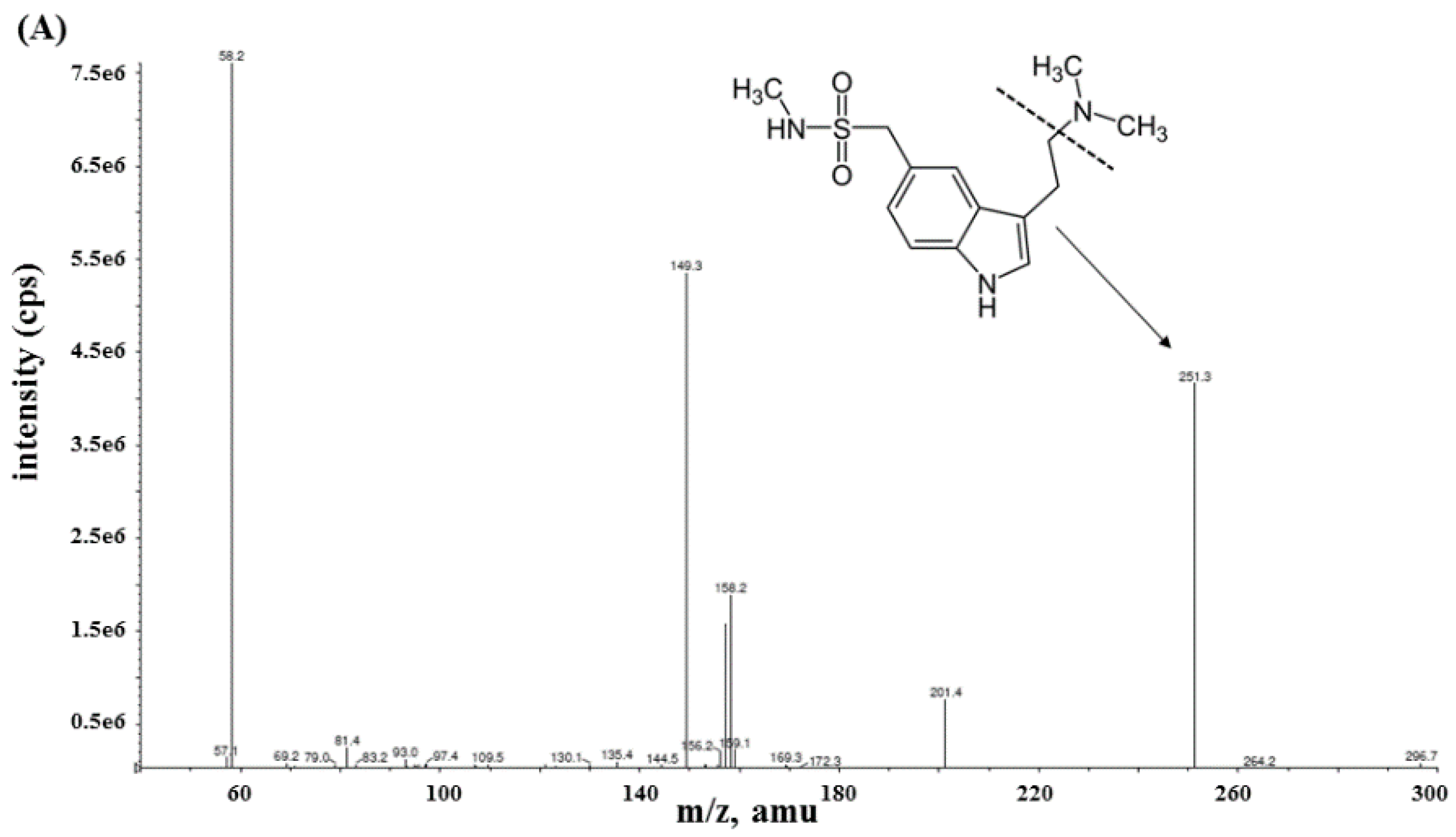
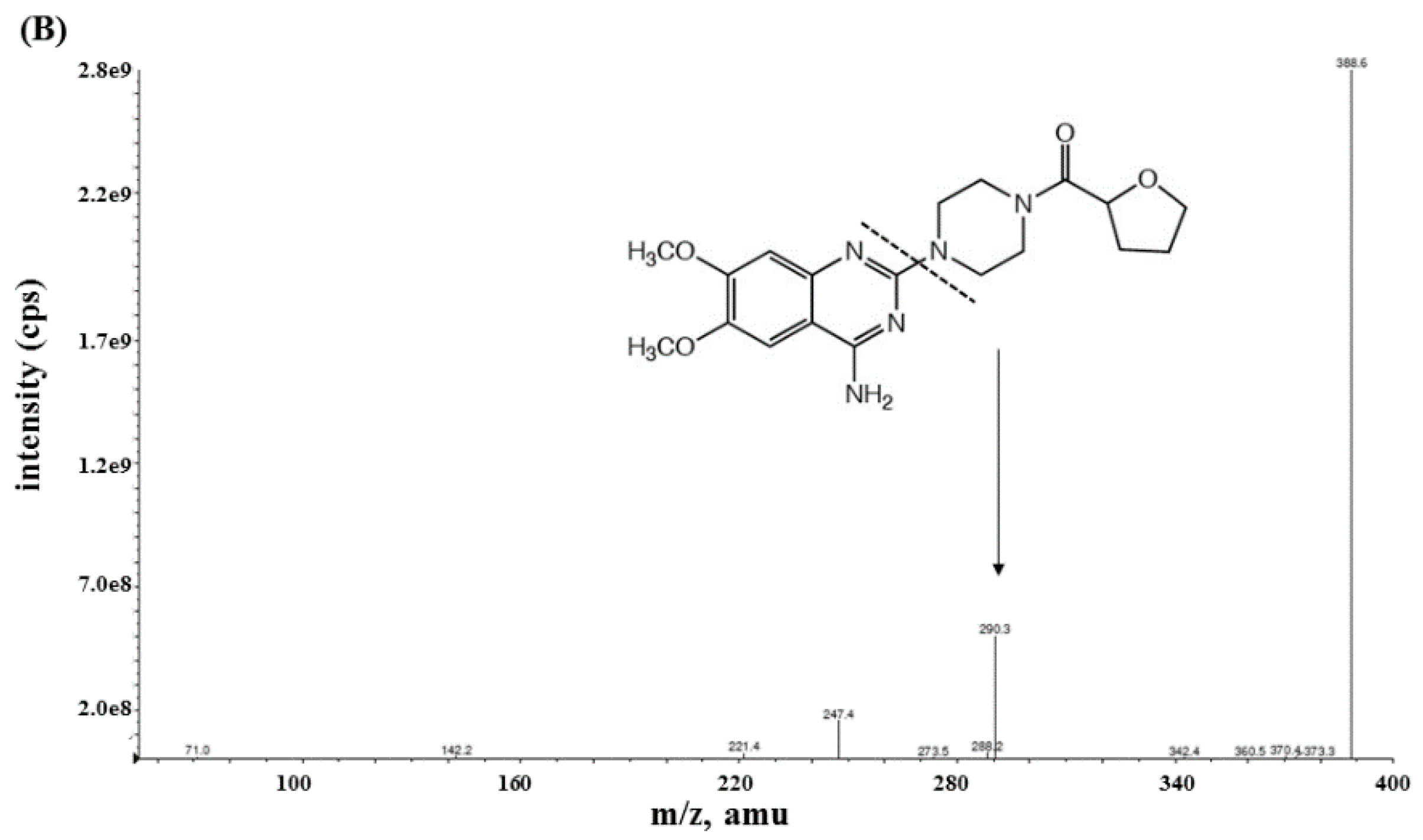
2.2.1. Specificity and Sensitivity
2.2.2. Calibration Curves and Linearity
2.2.3. Accuracy and Precision
2.2.4. Extraction Recovery
2.2.5. Matrix, Hemolytic, and Lipemic Effects
2.2.6. Dilution Integrity
2.2.7. Stability
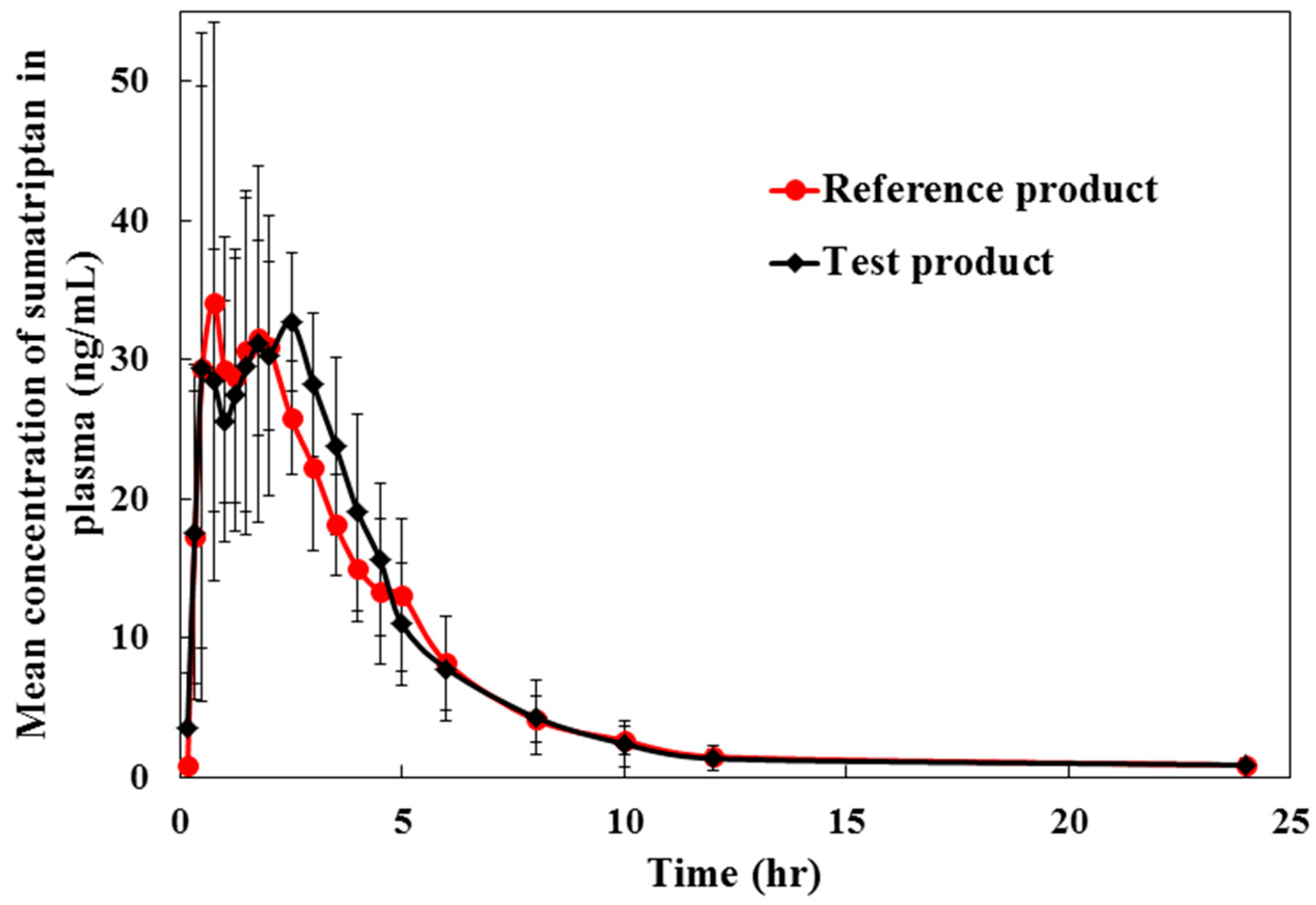
2.3. Method Application for Pharmacokinetic Study
3. Materials and Methods
3.1. Drugs and Chemicals
3.2. Instrument Conditions
3.3. Preparation of Stock Standard Solutions, Calibration Standards, and QC Samples
3.4. Sample Preparation
3.5. Method Validation
3.5.1. Specificity and Sensitivity
3.5.2. Calibration Curve and Linearity
3.5.3. Accuracy and Precision
3.5.4. Recovery
3.5.5. Dilution Integrity
3.5.6. Matrix Effect
3.5.7. Hemolytic and Lipemic Effects
3.5.8. Stability
3.6. Pharmacokinetic and Bioequivalence Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Solomon, S. Major Therapeutic Advances in the Past 25 Years. Headache 2007, 47, S20–S22. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.C.; Cady, R.K.; Landy, S.; O’Carroll, P.; Kwong, W.J.; Burch, S.P. Treatment satisfaction and efficacy of the rapid release formulation of sumatriptan 100 mg tablets utilising an early intervention paradigm in patients previously unsatisfied with sumatriptan. Int. J. Clin. Pract. 2008, 62, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.J.; Park, J.; Bae, M.H.; Lim, M.S.; Seong, S.J.; Lee, J. Rapid determination of sumatriptan in human plasma by ultra-performance liquid chromatography–tandem mass spectrometry and its application to clinical pharmacokinetic study. J. Chromatogr. B 2013, 919–920, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Sanghavi, B.J.; Kalambate, P.K.; Karna, S.P.; Srivastava, A.K. Voltammetric determination of sumatriptan based on a graphene/gold nanoparticles/Nafion composite modified glassy carbon electrode. Talanta 2014, 120, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Pakdel, Z.; Bezaatpour, A.; Shahrokhian, S. Electrocatalytic determination of sumatriptan on the surface of carbon-paste electrode modified with a composite of cobalt/Schiff-base complex and carbon nanotube. Bioelectrochemistry 2011, 81, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, K.N.; Basavaiah, K.; Xavier, C.M. Development and validation of UV-spectrophotometric methods for the determination of sumatriptan succinate in bulk and pharmaceutical dosage form and its degradation behavior under varied stress conditions. J. Assoc. Arab Univ. Basic Appl. Sci. 2014, 15, 43–52. [Google Scholar] [CrossRef]
- Femenía-Font, A.; Merino, V.; Rodilla, V.; López-Castellano, A. High-performance liquid chromatographic determination of sumatriptan after in vitro transdermal diffusion studies. J. Pharm. Biomed. Anal. 2005, 37, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Cózar-Bernal, M.J.; Rabasco, A.M.; González-Rodríguez, M.L. Development and validation of a high performance chromatographic method for determining sumatriptan in niosomes. J. Pharm. Biomed. Anal. 2013, 72, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Nozal, M.J.; Bernal, J.L.; Toribio, L.; Martı́n, M.T.; Diez, F.J. Development and validation of an LC assay for sumatriptan succinate residues on surfaces in the manufacture of pharmaceuticals. J. Pharm. Biomed. Anal. 2002, 30, 285–291. [Google Scholar] [CrossRef]
- Ge, Z.; Tessier, E.; Neirinck, L.; Zhu, Z. High performance liquid chromatographic method for the determination of sumatriptan with fluorescence detection in human plasma. J. Chromatogr. B 2004, 806, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Andrew, P. Fully automated assay for the determination of sumatriptan in human serum using solid-phase extraction and high-performance liquid chromatography with electrochemical detection. J. Pharm. Biomed. Anal. 1996, 14, 721–726. [Google Scholar] [CrossRef]
- Franklin, M.; Odontiadis, J.; Clement, E.M. Determination of sumatriptan succinate in human plasma by high-performance liquid chromatography with coulometric detection and utilization of solid-phase extraction. J. Chromatogr. B 1996, 681, 416–420. [Google Scholar] [CrossRef]
- Tan, A.; Hang, P.; Couture, J.; Hussain, S.; Vallée, F. An evaporation-free solid-phase extraction method for rapid and accurate analysis of sumatriptan in human plasma by LC–MS/MS. J. Chromatogr. B 2007, 856, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.P.; Sharma, P.; Sanyal, M.; Singhal, P.; Shrivastav, P.S. Challenges in the simultaneous quantitation of sumatriptan and naproxen in human plasma: Application to a bioequivalence study. J. Chromatogr. B 2012, 902, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.N.; Redrup, M.J.; Barrow, A.; Williams, P.N. Validation of a liquid chromatographic tandem mass spectrometric method for the determination of sumatriptan in human biological fluids. J. Pharm. Biomed. Anal. 1998, 17, 399–408. [Google Scholar] [CrossRef]
- Vishwanathan, K.; Bartlett, M.G.; Stewart, J.T. Determination of antimigraine compounds rizatriptan, zolmitriptan, naratriptan and sumatriptan in human serum by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 168–172. [Google Scholar] [CrossRef]
- McLoughlin, D.A.; Olah, T.V.; Ellis, J.D.; Gilbert, J.D.; Halpin, R.A. Quantitation of the 5HT1D agonists MK-462 and sumatriptan in plasma by liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 1996, 726, 115–124. [Google Scholar] [CrossRef]
- Food and Drug Administration of the United States (US-FDA); U.S. Department of Health and Human Services (DHHS); Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Bioanalytical Method Validation. In Guidance for Industry; US-FDA: Rockville, MD, USA, 2018. [Google Scholar]
- European Medicines Agency (EMA); Committee for Medical Products for Human Use (CHMP). Guidance on Bioanalytical Method Validation; EMA: London, United Kingdom, 2011. [Google Scholar]
- Ghosh, C.; Gaur, S.; Shinde, C.P.; Chakraborty, B. A systematic approach to overcome the matrix effect during LC-ESI-MS/MS analysis by different sample extraction techniques. J. Bioequivalence. Bioavailab. 2011, 3, 122–127. [Google Scholar] [CrossRef]
- Wells, D.A.; Wells, D. Liquid Liquid Extraction: Strategies for method development and optimization. In High Throughput Bioanalytical Sample Preparation-Methods and Automation Strategies; Elsevier: Amsterdam, The Netherlands, 2003; pp. 307–326. [Google Scholar]
- Center for Drug Evaluation and Research (CDER); Food and Drug Administration of the United States (US-FDA). Food-Effect Bioavailability and Fed Bioequivalence Studies. In Guidance for Industry; US-FDA: Rockville, MD, USA, 2002. [Google Scholar]
- Center for Drug Evaluation and Research (CDER); Food and Drug Administration of the United States (US-FDA). Bioavailability and Bioequivalence Studies for Orally Administered Drug Products−General Considerations. In Guidance for Industry; US-FDA: Rockville, MD, USA, 2002. [Google Scholar]
- Manish, Y.; Pranav, S.S. Incurred sample reanalysis (ISR): A decisive tool in bioanalytical research. Bioanalysis 2011, 3, 1007–1024. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration (ng/mL) | Back-Calculated Concentration (ng/mL) | Mean Back-Calculated Concentration (ng/mL) | %Deviation | %CV | ||
|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | ||||
| 0.5 | 0.51 | 0.50 | 0.50 | 0.50 | 0.53 | 0.61 |
| 1.0 | 0.97 | 1.00 | 1.01 | 0.99 | −0.97 | 2.06 |
| 2.5 | 2.57 | 2.50 | 2.48 | 2.52 | 0.61 | 1.92 |
| 10.0 | 9.61 | 9.39 | 9.19 | 9.40 | −6.01 | 2.25 |
| 20.0 | 19.52 | 20.95 | 20.89 | 20.45 | 2.27 | 3.94 |
| 35.0 | 36.93 | 35.87 | 35.70 | 36.17 | 3.33 | 1.84 |
| 50.0 | 50.04 | 49.58 | 50.95 | 50.19 | 0.38 | 1.39 |
| Nominal Concentration (ng/mL) | Within-Run (n = 6) | Between-Run (n = 18) | ||||
|---|---|---|---|---|---|---|
| Mean Back-Calculated Concentration (ng/mL) | Accuracy (%Deviation) | Precision (%CV) | Mean Back-Calculated Concentration (ng/mL) | Accuracy (%Deviation) | Precision (%CV) | |
| 0.5 | 0.52 | 3.23 | 5.40 | 0.54 | 8.30 | 7.62 |
| 1.5 | 1.42 | −5.48 | 3.91 | 1.45 | −3.60 | 5.05 |
| 25.0 | 24.85 | −0.58 | 3.93 | 23.24 | −7.05 | 7.33 |
| 45.0 | 40.33 | −10.38 | 4.43 | 41.98 | −6.71 | 9.51 |
| 50.0 | 49.94 | −0.13 | 2.15 | 46.37 | −7.27 | 7.39 |
| Concentration Added (ng/mL) | Plasma Lot | Matrix Factor (%) | IS-Normalized Matrix Effect | %CV | |
|---|---|---|---|---|---|
| Sumatriptan | Terazosin (IS) | ||||
| 1.5 | No.1 | 67.5 | 88.3 | 0.765 | 5.99 |
| No.2 | 65.2 | 86.9 | 0.751 | ||
| No.3 | 63.7 | 85.1 | 0.748 | ||
| No.4 | 66.2 | 86.9 | 0.761 | ||
| No.5 | 64.5 | 85.2 | 0.756 | ||
| No.6 | 65.4 | 83.3 | 0.785 | ||
| No.7 | 80.8 | 90.0 | 0.898 | ||
| No.8 | 67.6 | 86.1 | 0.785 | ||
| No.9 | 67.1 | 88.2 | 0.760 | ||
| 45 | No.1 | 79.7 | 91.4 | 0.871 | 1.89 |
| No.2 | 78.7 | 91.6 | 0.859 | ||
| No.3 | 77.8 | 89.0 | 0.875 | ||
| No.4 | 77.0 | 89.2 | 0.863 | ||
| No.5 | 74.9 | 88.1 | 0.850 | ||
| No.6 | 80.5 | 89.6 | 0.898 | ||
| No.7 | 84.0 | 94.2 | 0.892 | ||
| No.8 | 80.6 | 90.3 | 0.892 | ||
| No.9 | 79.3 | 89.9 | 0.882 | ||
| Nominal Concentration (ng/mL) | Mean Back-Calculated Concentration (ng/mL) | Accuracy (%Deviation) | Precision (%CV) | |
|---|---|---|---|---|
| Hemolytic effect | 1.5 | 1.28 | −14.62 | 6.33 |
| 45.0 | 43.60 | −3.11 | 2.34 | |
| Lipemic effect | 1.5 | 1.28 | −14.67 | 4.26 |
| 45.0 | 43.07 | −4.29 | 3.65 |
| Stability: Storage Condition | Nominal Concentration (ng/mL) | Back-Calculated Concentration (ng/mL) | %Deviation | %CV |
|---|---|---|---|---|
| Freeze and thaw stability | ||||
| After 4th cycle at −70 °C | 1.5 | 1.32 | −12.11 | 6.67 |
| 45.0 | 39.01 | −13.32 | 4.39 | |
| Short-term stability | ||||
| Bench-top at 25 °C for 4 h | 1.5 | 1.48 | −1.02 | 9.91 |
| 45.0 | 41.76 | −7.19 | 1.70 | |
| Long-term stability | ||||
| 77 days at −70 °C | 1.5 | 1.38 | −8.27 | 3.67 |
| 45.0 | 42.47 | −5.62 | 2.79 | |
| Post-preparative stability | ||||
| Dry state after extraction at 2–8 °C for 2 days | 1.5 | 1.33 | −11.44 | 3.27 |
| 45.0 | 38.67 | −14.07 | 2.46 | |
| Autosampler at 4 °C for 48 h | 1.5 | 1.64 | 9.09 | 6.97 |
| 45.0 | 49.67 | 10.37 | 4.15 | |
| Reinjection at 4 °C for 48 h | 1.5 | 1.53 | 1.84 | 5.47 |
| 45.0 | 49.67 | 6.23 | 6.43 |
| Pharmacokinetic Parameters | Mean (SD) | Ratio of Least Square Mean T/R (%) | 90% Confidence Intervals (T/R) | Intra-Subject CV (%) | |
|---|---|---|---|---|---|
| Test Product | Reference Product | ||||
| Cmax (ng/mL) | 44.08 (14.18) | 43.07 (16.40) | 103.88 | 87.18–123.78 | 9.69 |
| AUC0→t (ng·/mL) | 153.07 (45.54) | 146.90 (30.80) | 102.91 | 92.68–114.26 | 14.31 |
| AUC0→∞ (ng·h/mL) | 156.95 (46.14) | 152.26 (31.74) | 101.91 | 90.47–114.78 | 8.52 |
| Tmax (h) | 1.83 (0.86) | 1.46 (0.68) | - | - | - |
| t1/2 (h) | 2.62 (0.79) | 3.42 (2.03) | - | - | - |
| ke (h−1) | 0.28 (0.08) | 0.249 (0.10) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wichitnithad, W.; Nantaphol, S.; Vicheantawatchai, P.; Kiatkumjorn, T.; Wangkangwan, W.; Rojsitthisak, P. Development and Validation of Liquid Chromatography-Tandem Mass Spectrometry Method for Simple Analysis of Sumatriptan and its Application in Bioequivalence Study. Pharmaceuticals 2020, 13, 21. https://doi.org/10.3390/ph13020021
Wichitnithad W, Nantaphol S, Vicheantawatchai P, Kiatkumjorn T, Wangkangwan W, Rojsitthisak P. Development and Validation of Liquid Chromatography-Tandem Mass Spectrometry Method for Simple Analysis of Sumatriptan and its Application in Bioequivalence Study. Pharmaceuticals. 2020; 13(2):21. https://doi.org/10.3390/ph13020021
Chicago/Turabian StyleWichitnithad, Wisut, Siriwan Nantaphol, Petploy Vicheantawatchai, Thanyaporn Kiatkumjorn, Wachirasak Wangkangwan, and Pornchai Rojsitthisak. 2020. "Development and Validation of Liquid Chromatography-Tandem Mass Spectrometry Method for Simple Analysis of Sumatriptan and its Application in Bioequivalence Study" Pharmaceuticals 13, no. 2: 21. https://doi.org/10.3390/ph13020021
APA StyleWichitnithad, W., Nantaphol, S., Vicheantawatchai, P., Kiatkumjorn, T., Wangkangwan, W., & Rojsitthisak, P. (2020). Development and Validation of Liquid Chromatography-Tandem Mass Spectrometry Method for Simple Analysis of Sumatriptan and its Application in Bioequivalence Study. Pharmaceuticals, 13(2), 21. https://doi.org/10.3390/ph13020021