Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
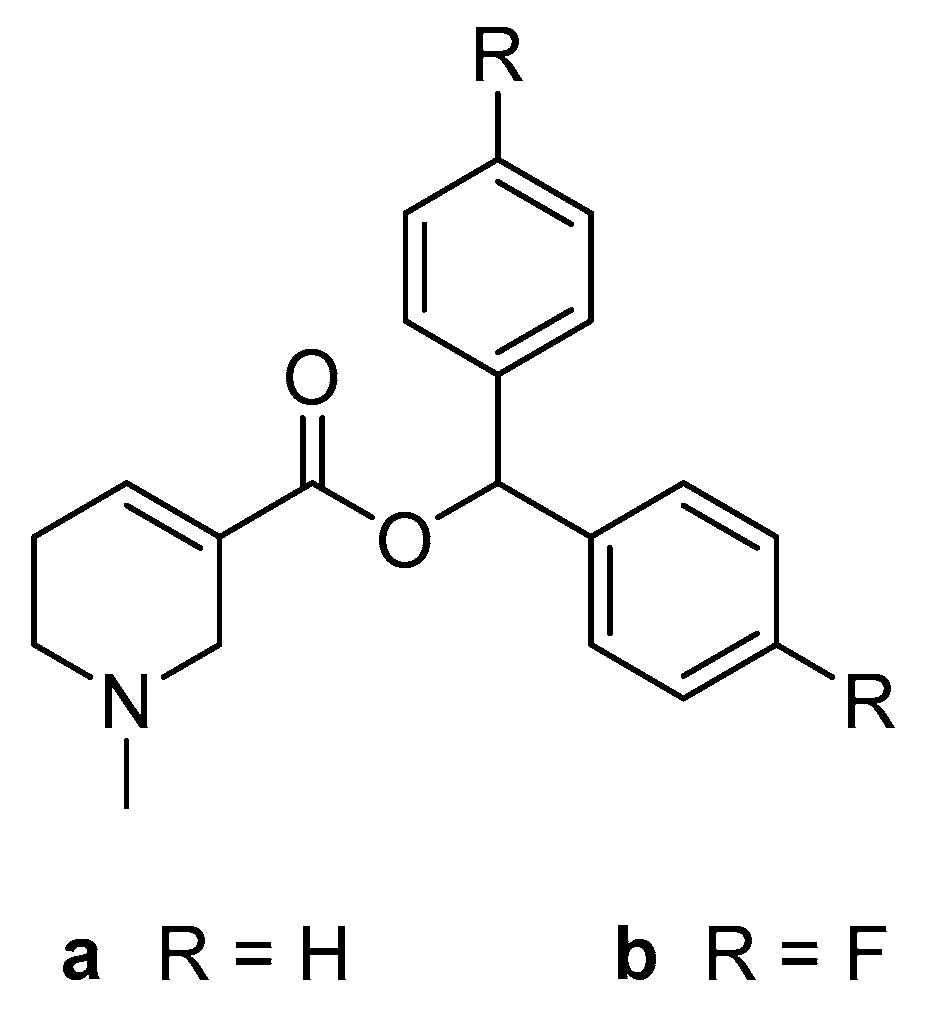
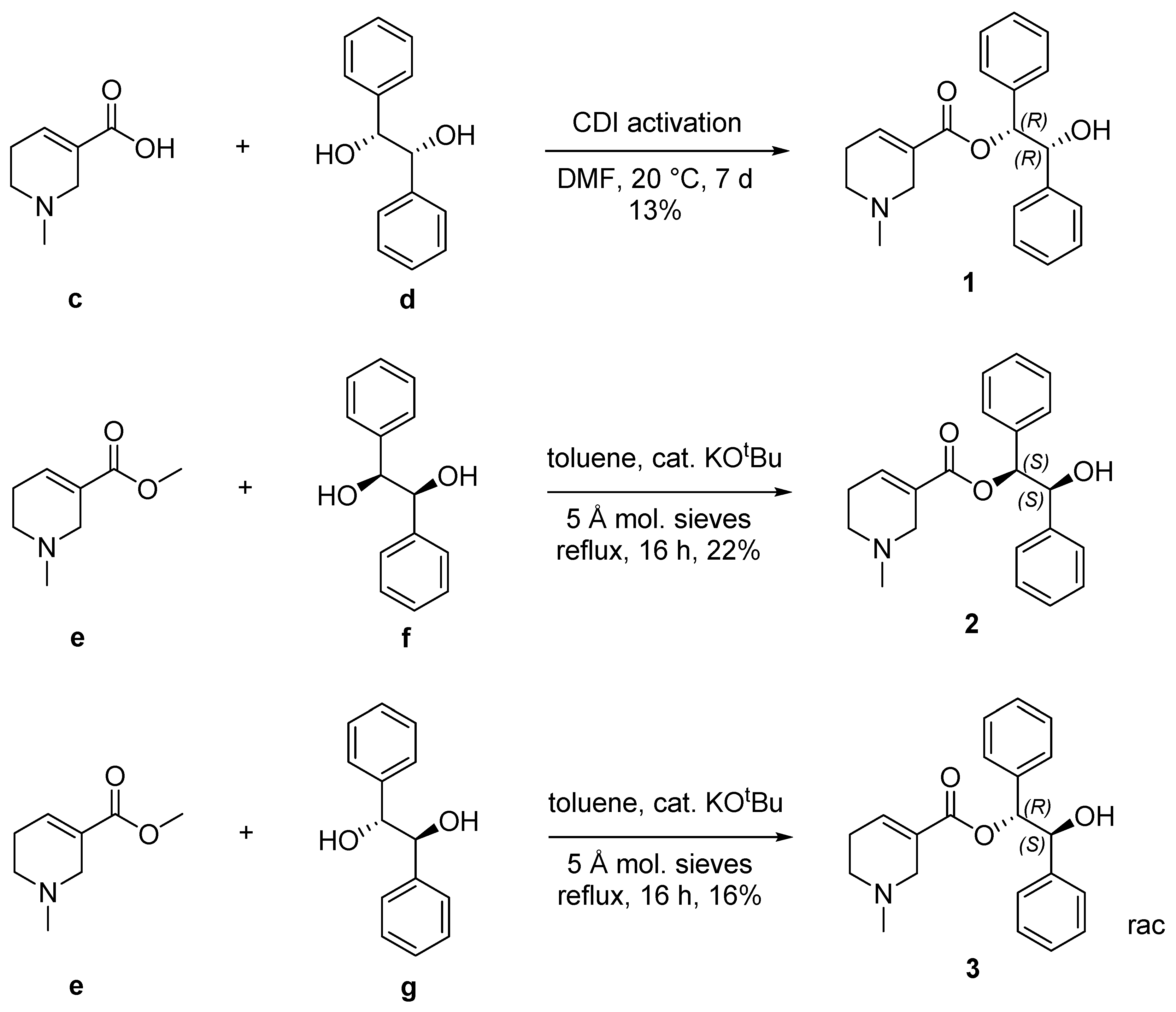
2.1. Chemistry
2.2. Evaluation of Physico-Chemical Parameters and Stability
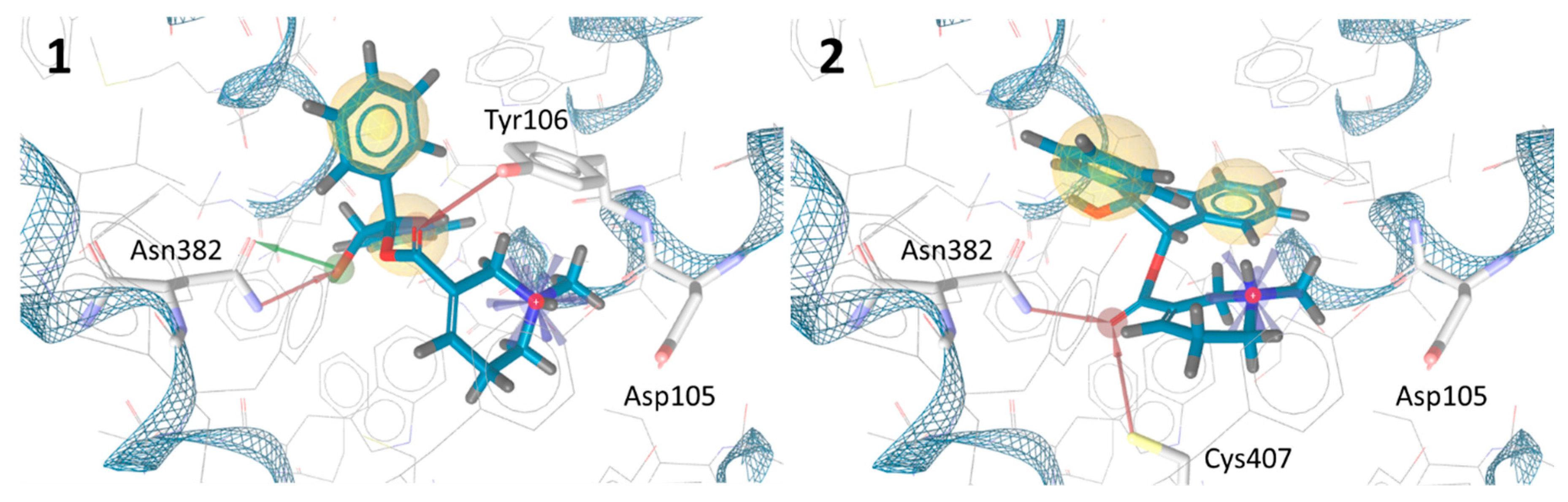
2.3. In Silico Docking Experiments
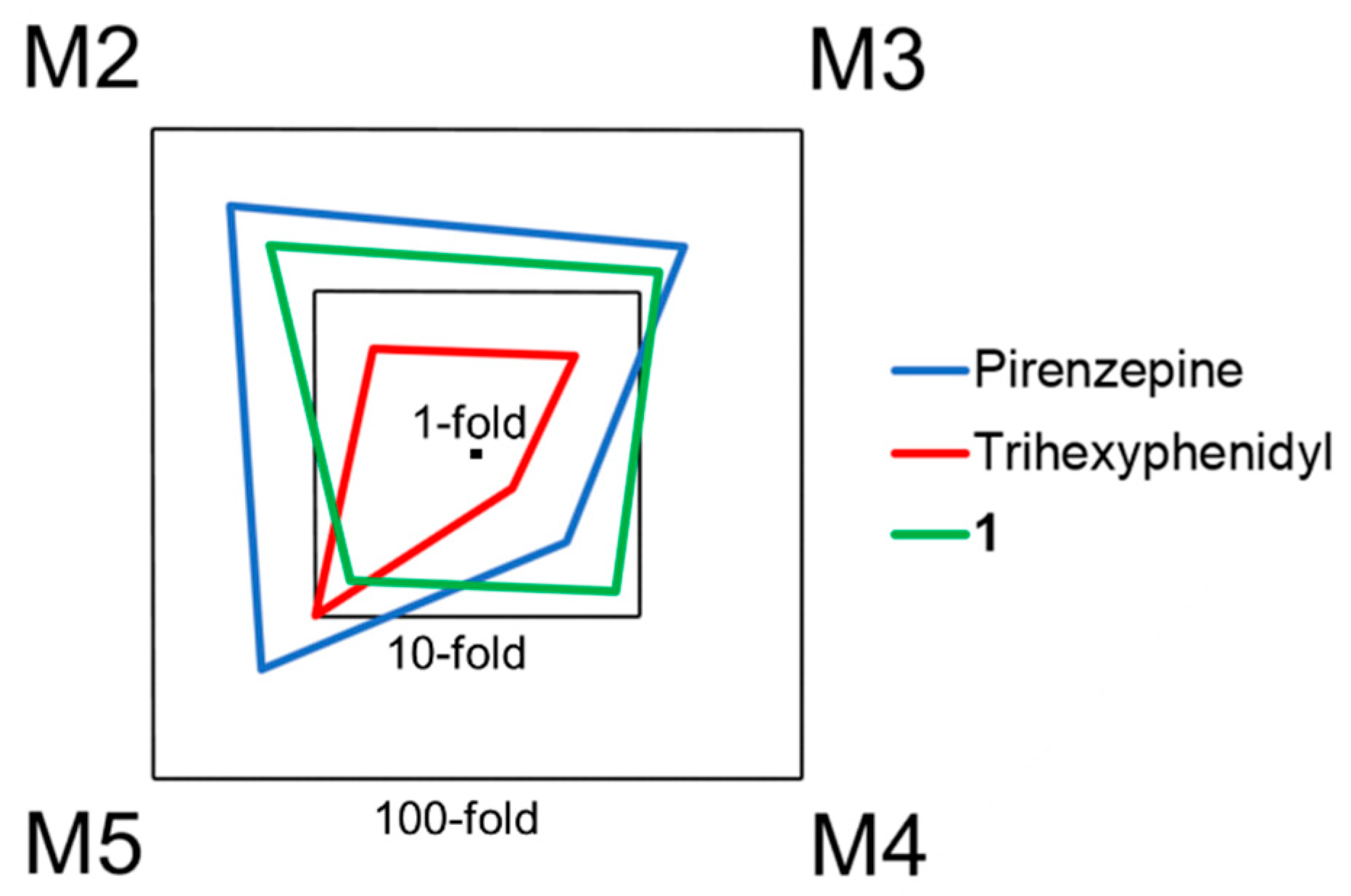
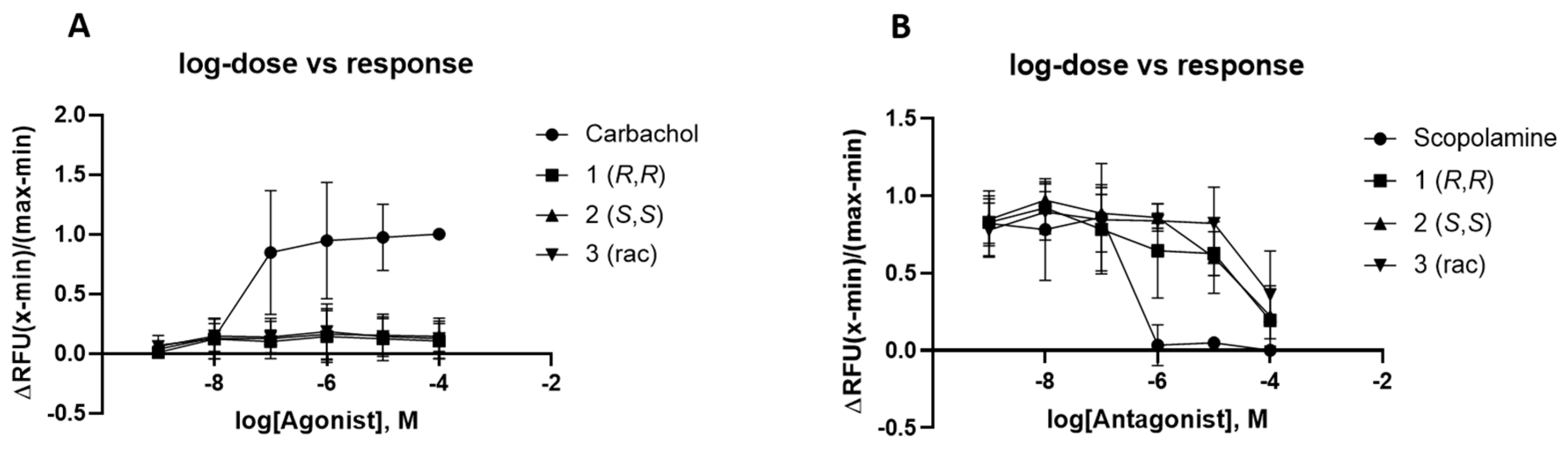
2.4. Biological Evaluation
3. Conclusions
4. Experimental
4.1. Materials
4.2. Instrumentation
4.3. Stability Measurements in DPBS and Cell Culture Media
4.4. Molecular Docking Studies
4.5. Cell Culture
4.6. Affinity Assay Toward mAChRs M1-M5 on CHO Membranes
4.7. Calcium Efflux Assay for Agonist/Antagonists Discrimination
4.8. Calculations and Statistics
4.9. Synthesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haga, T. Molecular properties of muscarinic acetylcholine receptors. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, P.; Kiechle, T.; Bakker, R.A. Differential Inverse Agonism at the Human Muscarinic M3 Receptor. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 81–101. [Google Scholar]
- Cheng, C.-Q.; Xu, H.; Liu, L.; Wang, R.-N.; Liu, Y.-T.; Li, J.; Zhou, X.-K. Efficacy and Safety of Pilocarpine for Radiation-Induced Xerostomia in Patients with Head and Neck Cancer. J. Am. Dent. Assoc. 2016, 147, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Gaitonde, S.; Malik, R.D.; Christie, A.L.; Zimmern, P.E. Bethanechol: Is It Still Being Prescribed for Bladder Dysfunction in Women? Int. J. Clin. Pract. 2019, 73, e13248. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, A.; Tripathi, T.; Kumar, A. Muscarinic and Nicotinic Acetylcholine Receptor Agonists: Current Scenario in Alzheimer’s Disease Therapy. J. Pharm. Pharmacol. 2018, 70, 985–993. [Google Scholar] [CrossRef]
- Xiang, Z.; Thompson, A.D.; Jones, C.K.; Lindsley, C.W.; Conn, P.J. Roles of the M1 Muscarinic Acetylcholine Receptor Subtype in the Regulation of Basal Ganglia Function and Implications for the Treatment of Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2012, 340, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Holle, G.E. Pathophysiology and Modern Treatment of Ulcer Disease. Int. J. Mol. Med. 2010, 25, 483–491. [Google Scholar] [CrossRef]
- Gribkoff, V.K.; Kaczmarek, L.K. The Need for New Approaches in CNS Drug Discovery: Why Drugs Have Failed, and What Can Be Done to Improve Outcomes. Neuropharmacology 2017, 120, 11–19. [Google Scholar] [CrossRef]
- Toyohara, J.; Sakata, M.; Ishiwata, K. Human Brain Imaging of Acetylcholine Receptors. In Imaging of the Human Brain in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2014; pp. 113–160. [Google Scholar]
- Eckelman, W.C.; Kilbourn, M.R.; Mathis, C.A. Discussion of Targeting Proteins in Vivo: In Vitro Guidelines. Nucl. Med. Biol. 2006, 33, 449–451. [Google Scholar] [CrossRef]
- Moulton, B.C.; Fryer, A.D. Muscarinic Receptor Antagonists, from Folklore to Pharmacology; Finding Drugs That Actually Work in Asthma and COPD: Anticholinergics in Treatment of Airway Disease. Br. J. Pharmacol. 2011, 163, 44–52. [Google Scholar] [CrossRef]
- Ozenil, M.; Pacher, K.; Balber, T.; Vraka, C.; Roller, A.; Holzer, W.; Spreitzer, H.; Mitterhauser, M.; Wadsak, W.; Hacker, M.; et al. Enhanced Arecoline Derivatives as Muscarinic Acetylcholine Receptor M1 Ligands for Potential Application as PET Radiotracers. Eur. J. Med. Chem. 2020, 204, 112623. [Google Scholar] [CrossRef] [PubMed]
- Nagar, S.; Korzekwa, K. Commentary: Nonspecific Protein Binding versus Membrane Partitioning: It Is Not Just Semantics. Drug Metab. Dispos. 2012, 40, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Vraka, C.; Nics, L.; Wagner, K.-H.; Hacker, M.; Wadsak, W.; Mitterhauser, M. Log P, a Yesterday’s Value? Nucl. Med. Biol. 2017, 50, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Nagamine, T.; Inomata, K.; Endo, Y.; Paquette, L.A. Amino Acid Mediated Intramolecular Asymmetric Aldol Reaction to Construct a New Chiral Bicyclic Enedione Containing a Seven-Membered Ring: Remarkable Inversion of Enantioselectivity Compared to the Six-Membered Ring Example. J. Org. Chem. 2007, 72, 123–131. [Google Scholar] [CrossRef]
- Ward, S.D.C.; Curtis, C.A.M.; Hulme, E.C. Alanine-Scanning Mutagenesis of Transmembrane Domain 6 of the M1 Muscarinic Acetylcholine Receptor Suggests That Tyr381 Plays Key Roles in Receptor Function. Mol. Pharm. 1999, 56, 1031–1041. [Google Scholar] [CrossRef]
- Motulsky, H.J.; Neubig, R.R. Analyzing Binding Data. Curr. Protoc. Neurosci. 2010, 52, 7.5.1–7.5.65. [Google Scholar] [CrossRef]
- Bolden, C.; Cusack, B.; Richelson, E. Antagonism by Antimuscarinic and Neuroleptic Compounds at the Five Cloned Human Muscarinic Cholinergic Receptors Expressed in Chinese Hamster Ovary Cells. J. Pharmacol. Exp. Ther. 1992, 260, 576–580. [Google Scholar] [PubMed]
- del Bello, F.; Bonifazi, A.; Giorgioni, G.; Piergentili, A.; Sabbieti, M.G.; Agas, D.; Dell’Aera, M.; Matucci, R.; Górecki, M.; Pescitelli, G.; et al. Novel Potent Muscarinic Receptor Antagonists: Investigation on the Nature of Lipophilic Substituents in the 5- and/or 6-Positions of the 1,4-Dioxane Nucleus. Med. Chem. 2020, 63, 5763–5782. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.J.; Kuc, R.E.; Davenport, A.P. Radioligand Binding Assays and Their Analysis. In Receptor Binding Techniques; Davenport, A.P., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 31–77. [Google Scholar]
- Ma, Q.; Ye, L.; Liu, H.; Shi, Y.; Zhou, N. An Overview of Ca2+ Mobilization Assays in GPCR Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, C.; Dolzer, J.; Huan, B.; Yu, M.; Lydford, S.; Huggins, C. Cell Based Assays on the FLIPRTETRA® System: Comparison of the FLIPR® Calcium 5 Assay Kit to Other Fluorescence Based Calcium Flux Assays. In Molecular Devices Scientific Poster; Molecular Devices, Inc.: San Jose, CA, USA, 2017. [Google Scholar]
- Zeeberg, B.R. Pharmacokinetic computer simulations of the relationship between in vivo and in vitro neuroreceptor subtype selectivity of radioligands. Nucl. Med. Biol. 1999, 26, 803–809. [Google Scholar] [CrossRef]
- Colom, M.; Vidal, B.; Zimmer, L. Is There a Role for GPCR Agonist Radiotracers in PET Neuroimaging? Front Mol. Neurosci. 2019, 12, 255. [Google Scholar] [CrossRef]
- Song, S.; Zhu, S.-F.; Pu, L.-Y.; Zhou, Q.-L. Iridium-Catalyzed Enantioselective Hydrogenation of Unsaturated Heterocyclic Acids. Angew. Chem. Int. Ed. 2013, 52, 6072–6075. [Google Scholar] [CrossRef]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal Structures of the M1 and M4 Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef]
- Murthy, K.S.K.; Rey, A.W.; Tjepkema, M. Enantioselective Synthesis of 3-Substituted-4-Aryl Piperidines Useful for the Preparation of Paroxetine. Tetrahedron Lett. 2003, 44, 5355–5358. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M1 | M2 | M3 | M4 | M5 | Source | |
|---|---|---|---|---|---|---|
| 1 (R,R) | 99 ± 19 | 1900 ± 300 | 1300 ± 600 | 700 ± 300 | 600 ± 75 | this work |
| 2 (S,S) | 800 ± 200 | 8000 ± 2000 | 1600 ± 300 | 2700 ± 600 | 1300 ± 300 | this work |
| 3 (rac) | 380 ± 90 | 3700 ± 1000 | 3200 ± 500 | 1600 ± 200 | 970 ± 90 | this work |
| arecoline | 20,300 ± 2400 | 3800 ± 890 | 16,700 ± 5900 | 4700 ± 410 | 7000 ± 570 | [13] |
| pirenzepine | 8 ± 1 | 270 ± 10 | 150 ± 10 | 28 ± 1 | 170 ± 10 | [23] |
| trihexyphenidyl | 1.6 ± 0.2 | 7 ± 1 | 6.4 ± 0.4 | 2.6 ± 0.4 | 15.9 ± 0.1 | [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozenil, M.; Aronow, J.; Piljak, D.; Vraka, C.; Holzer, W.; Spreitzer, H.; Wadsak, W.; Hacker, M.; Pichler, V. Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1. Pharmaceuticals 2020, 13, 437. https://doi.org/10.3390/ph13120437
Ozenil M, Aronow J, Piljak D, Vraka C, Holzer W, Spreitzer H, Wadsak W, Hacker M, Pichler V. Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1. Pharmaceuticals. 2020; 13(12):437. https://doi.org/10.3390/ph13120437
Chicago/Turabian StyleOzenil, Marius, Jonas Aronow, Daniela Piljak, Chrysoula Vraka, Wolfgang Holzer, Helmut Spreitzer, Wolfgang Wadsak, Marcus Hacker, and Verena Pichler. 2020. "Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1" Pharmaceuticals 13, no. 12: 437. https://doi.org/10.3390/ph13120437
APA StyleOzenil, M., Aronow, J., Piljak, D., Vraka, C., Holzer, W., Spreitzer, H., Wadsak, W., Hacker, M., & Pichler, V. (2020). Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1. Pharmaceuticals, 13(12), 437. https://doi.org/10.3390/ph13120437