Multidirectional Efficacy of Biologically Active Nitro Compounds Included in Medicines



Abstract
:1. Introduction
2. Drugs Used in Cardiovascular Diseases
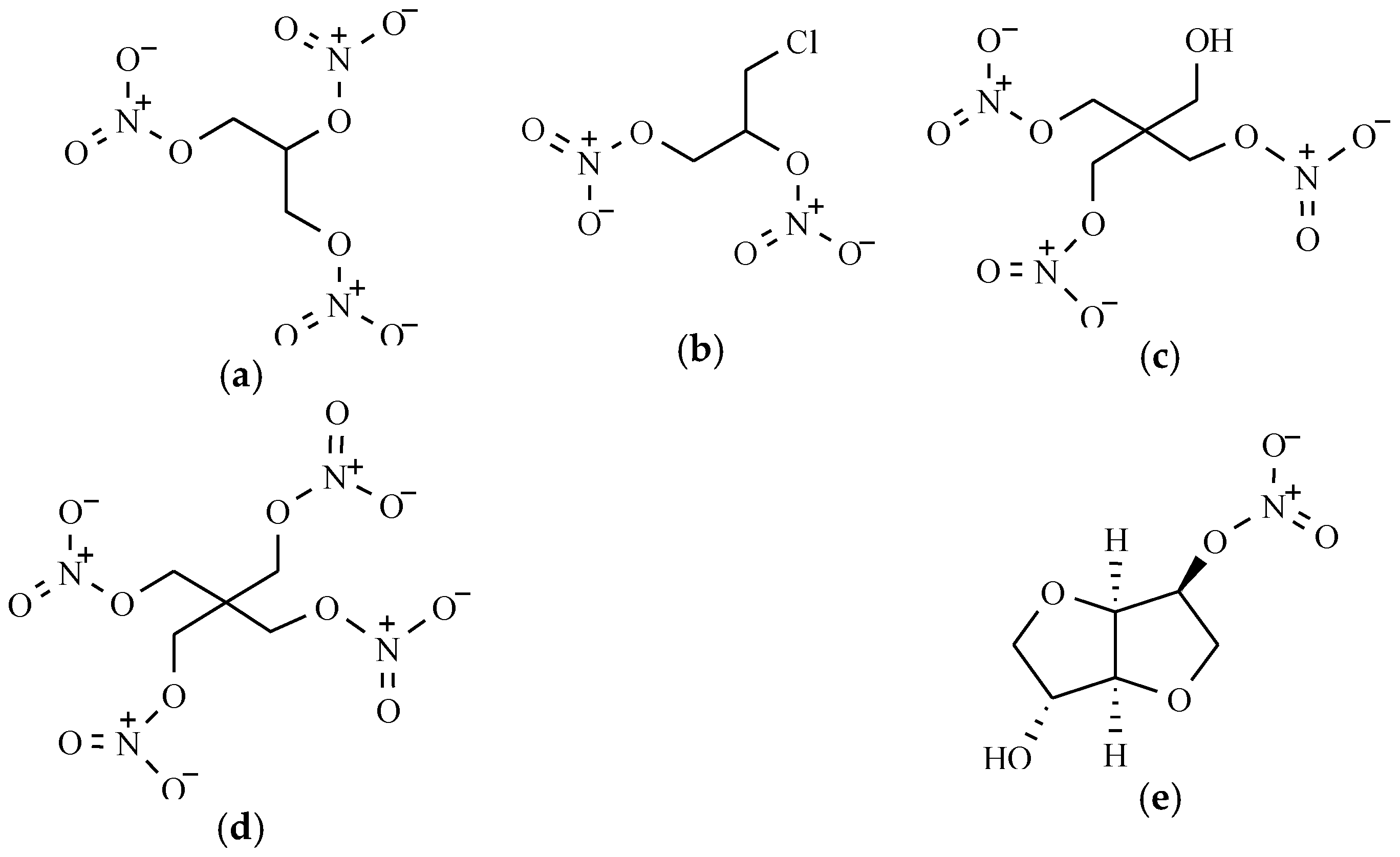
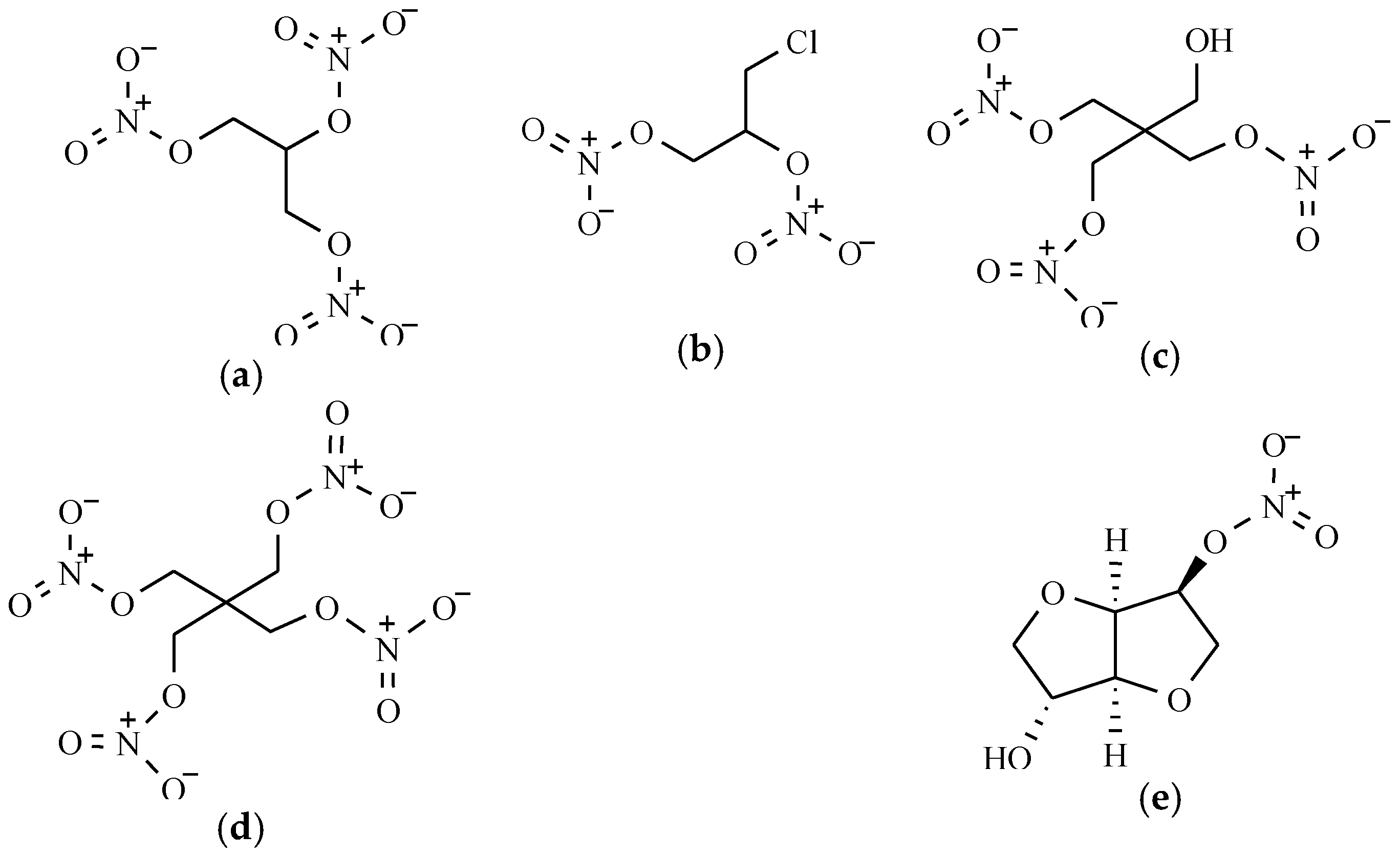
2.1. Organic Nitrates
Organic Nitrates’ Mechanism of Action
2.2. β-Blockers Others Cardiovascular Drugs
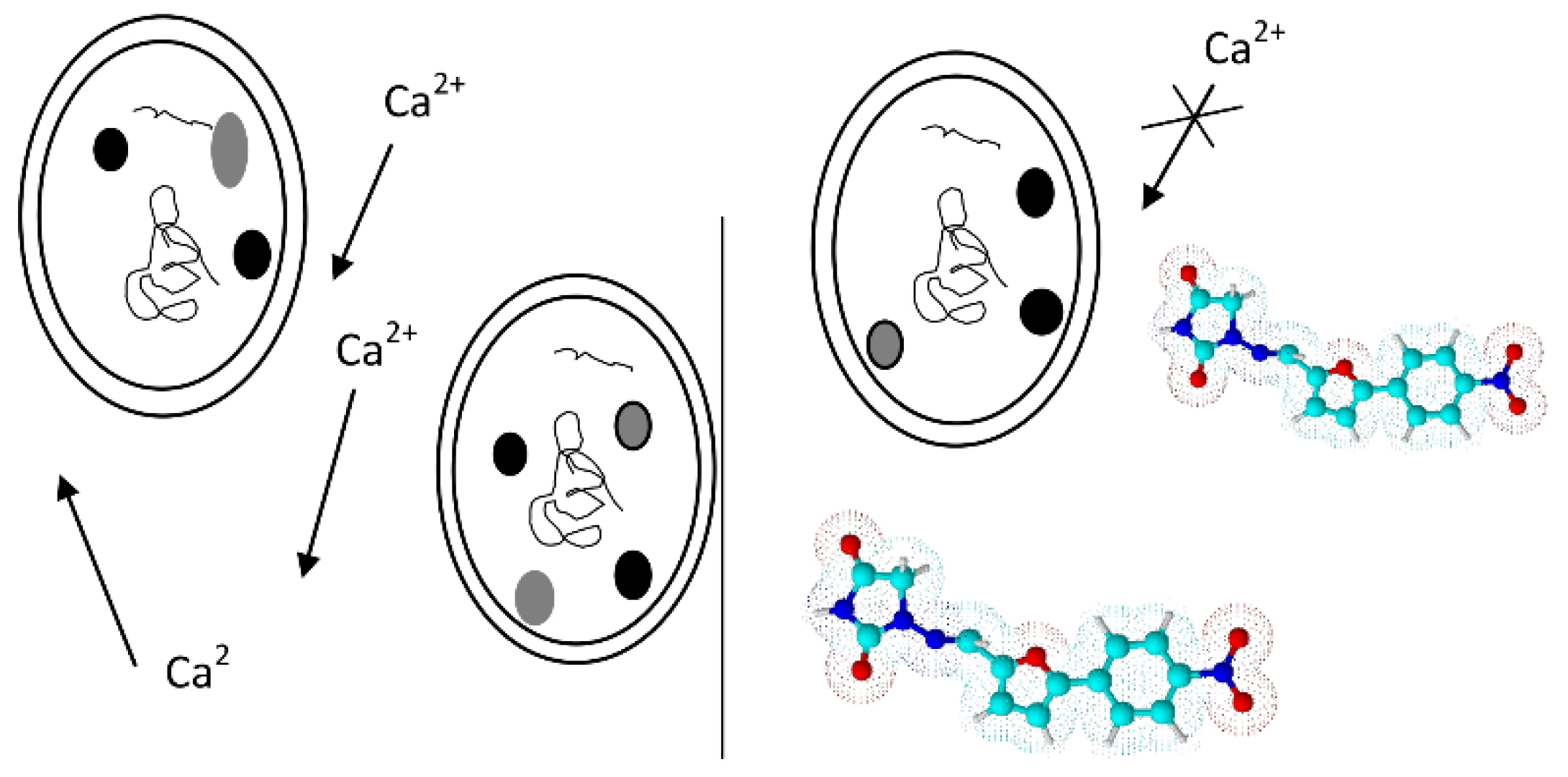
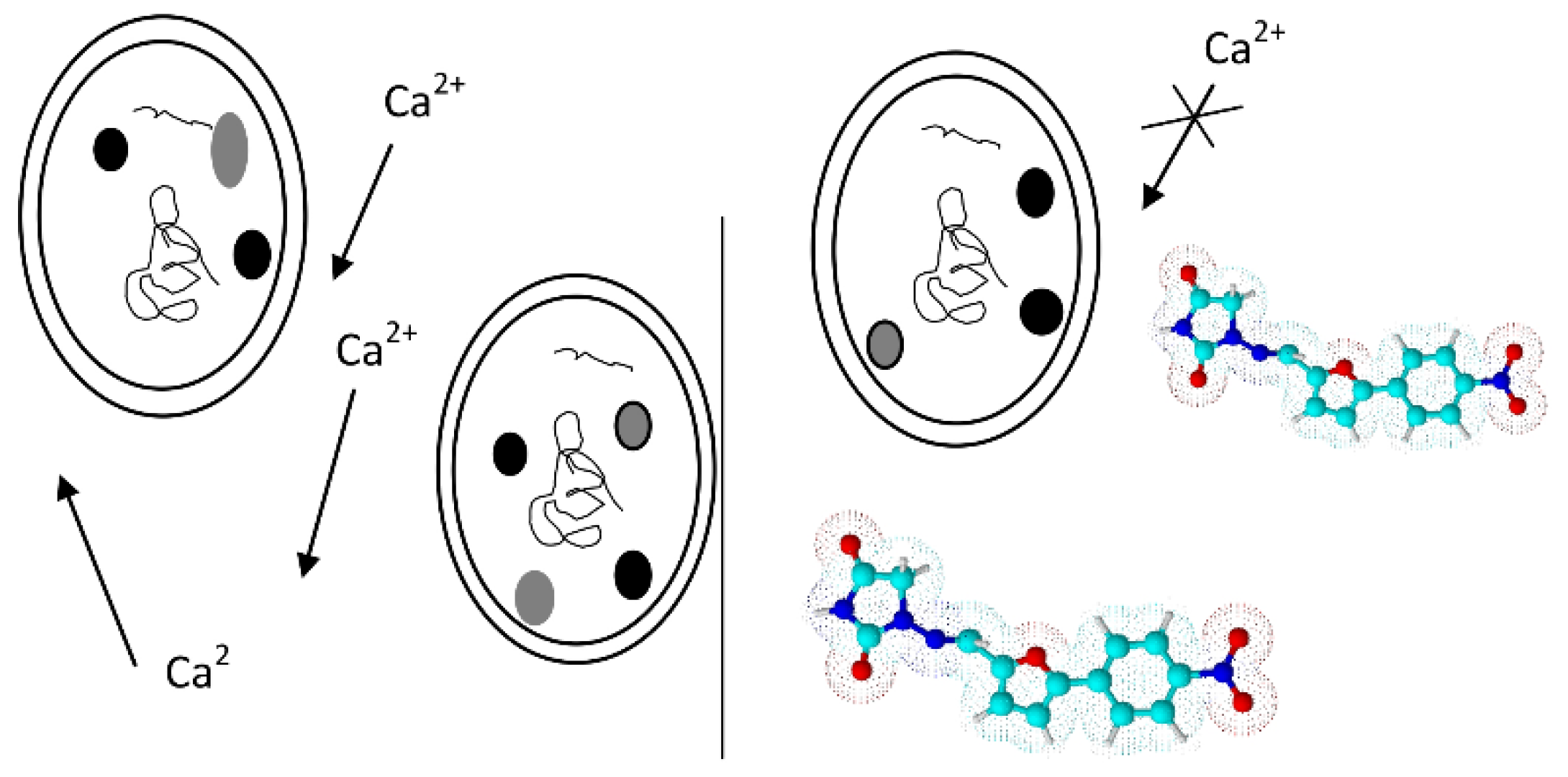
2.3. Calcium Channel Blockers
Nifedipine Derivatives’ Mechanism of Action
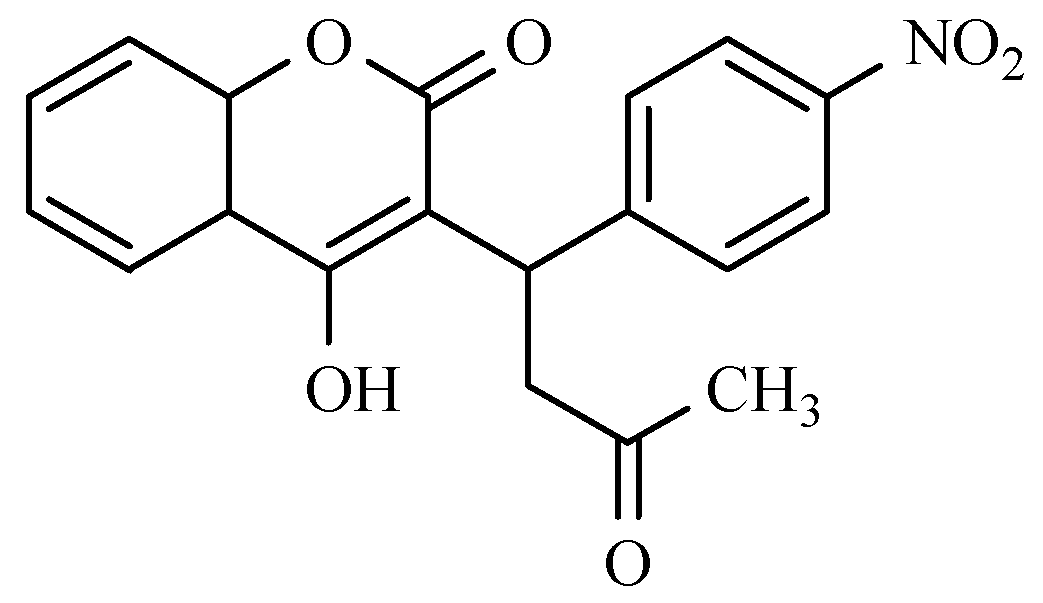
2.4. Treatment of Thromboembolic Disease
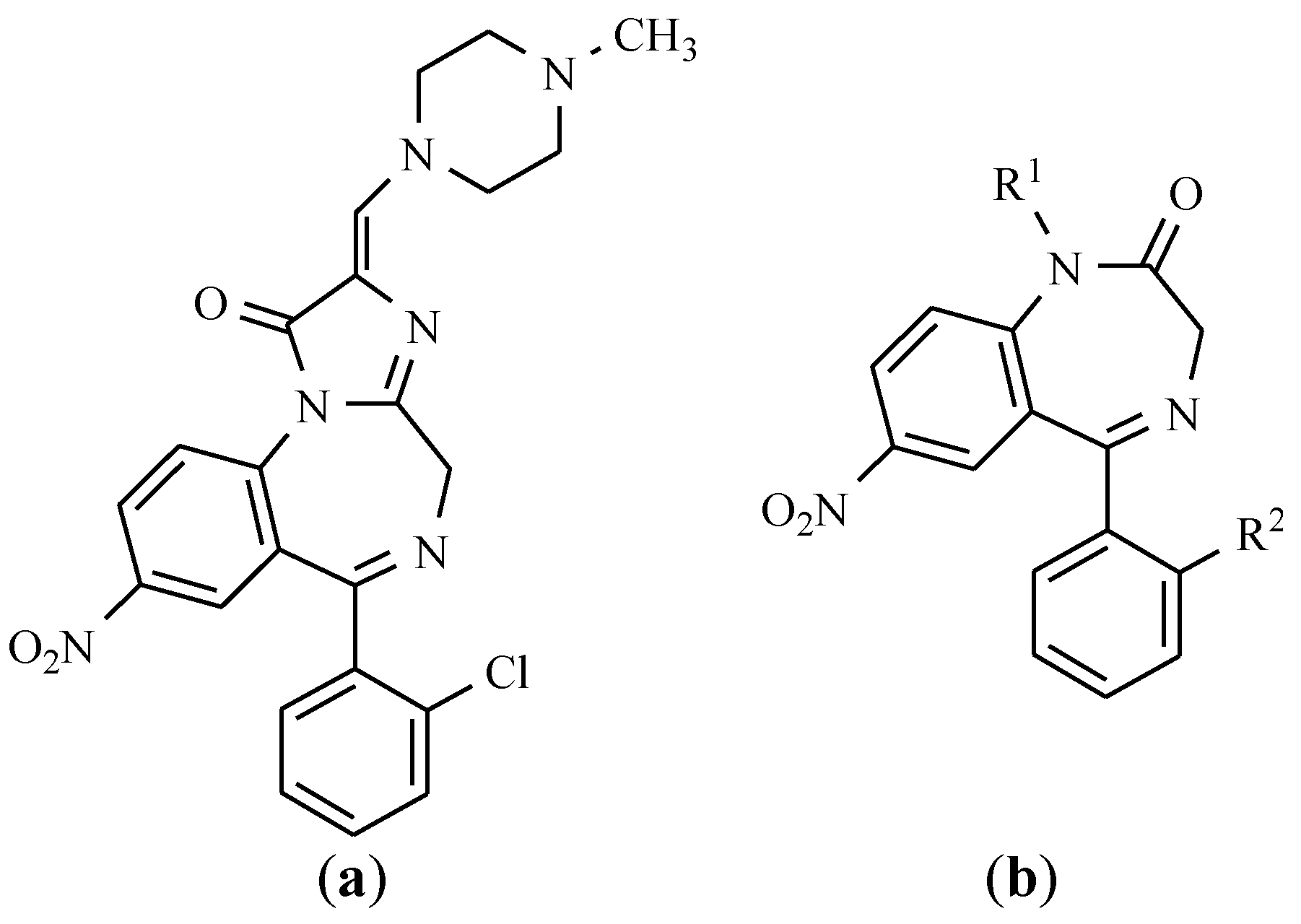
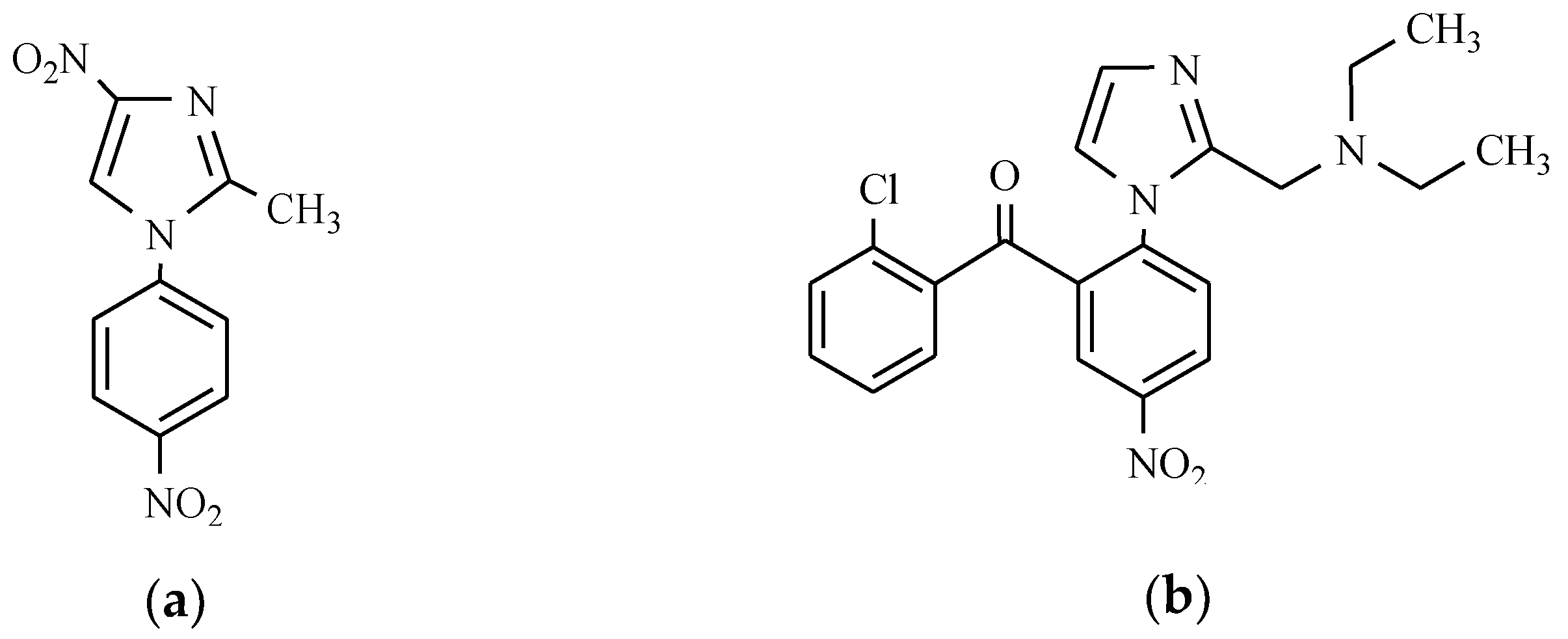
3. Anxiolytics
- Nitrazepam is used in short-term insomnia, and as adjunctive therapy in the treatment of epilepsy and preparation for surgery (a day before surgery overnight) [10].
- Flunitrazepam is a fluorine-benzodiazepine derivative with a strong sedative and hypnotic activity being applied for the treatment of sleep disorders, including premedication as an agent in anesthesia and intensive care [11].
- Clonazepam is a chloro derivative of nitrazepam, which is characterized by anti-convulsant activity. Clonazepam, more than other benzodiazepines, is of benefit in the treatment of some types of myoclonus. Its mechanism of action is to facilitate GABAergic transmission in the brain directly on benzodiazepine receptors [12]. It is one of the most effective antiepileptic drugs.
- Nimetazepam is the N-methyl derivative of nitrazepam with a sedative and hypnotic effect. This drug is not registered in Poland.
- Loprazolam is a tricyclic derivative of imidazo-1,4-benzodiazepine, and it mainly exhibits hypnotics, anxiolytics, sedatives, anticonvulsants, and muscle relaxants effects [13].
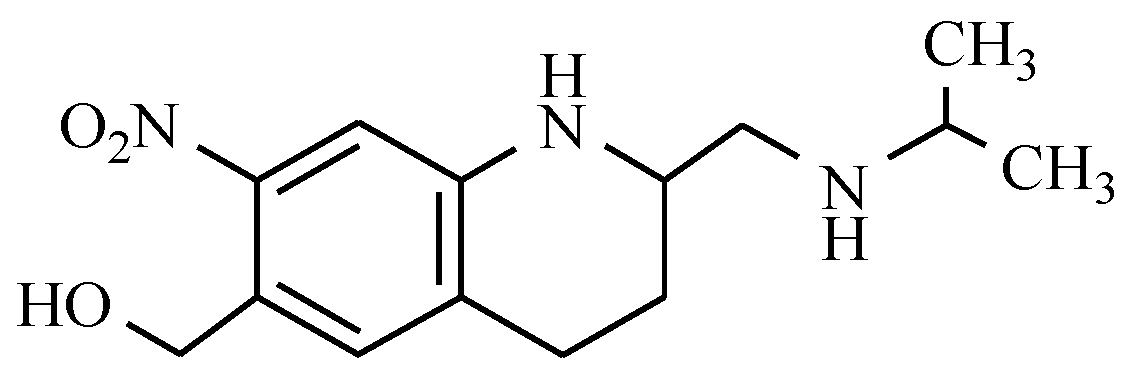
4. Drugs Used in Parkinson’s Disease
5. Drugs Used in Peptic Ulcer
6. Anticancer Agents
6.1. Flutamide and Nilutamide
6.2. Azathioprine
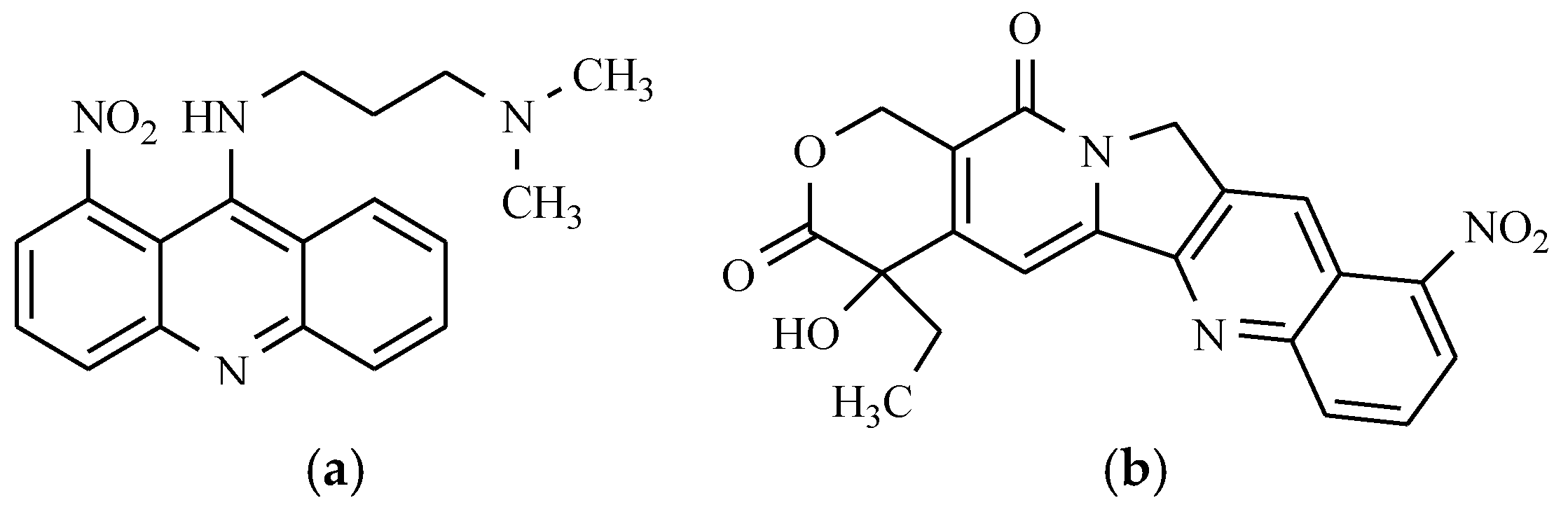
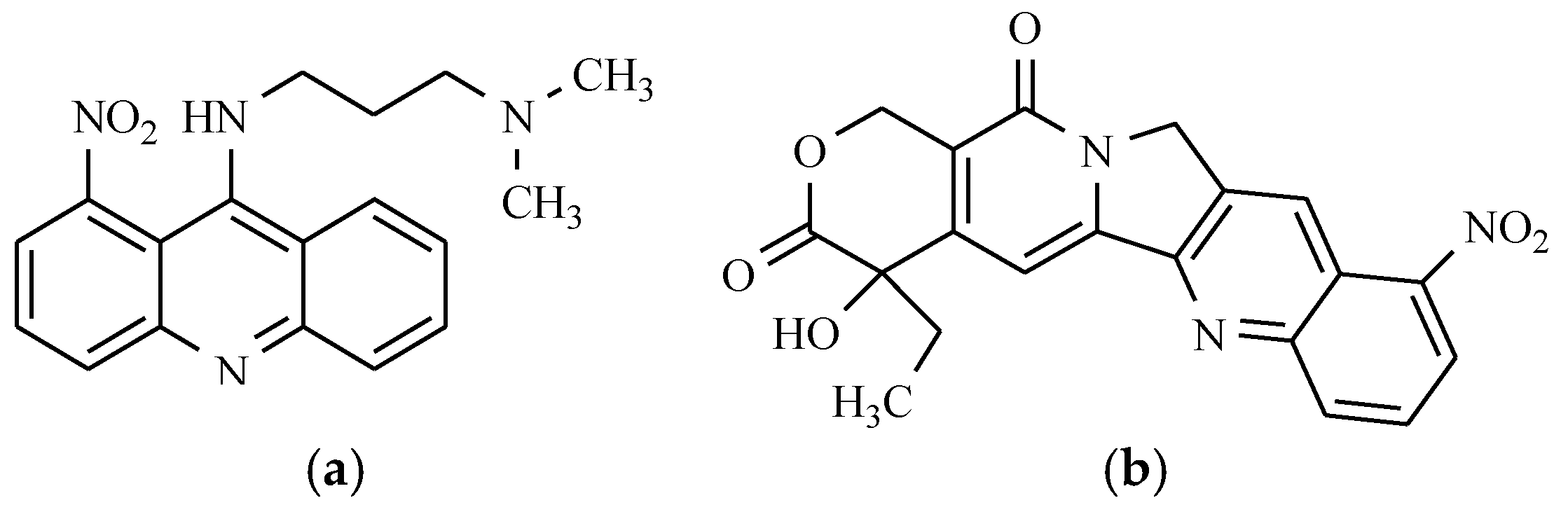
6.3. Nitracrine and Rubitecan
7. Antibacterial Drugs
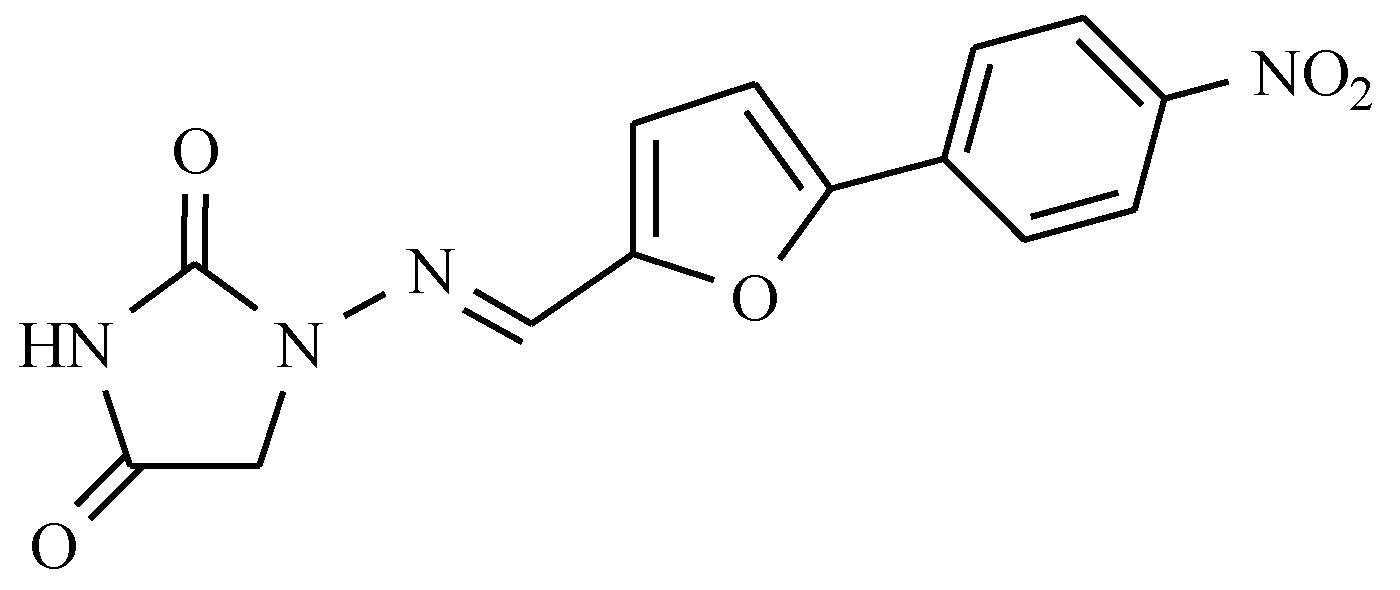
7.1. Derivatives of 5-Nitrofuran
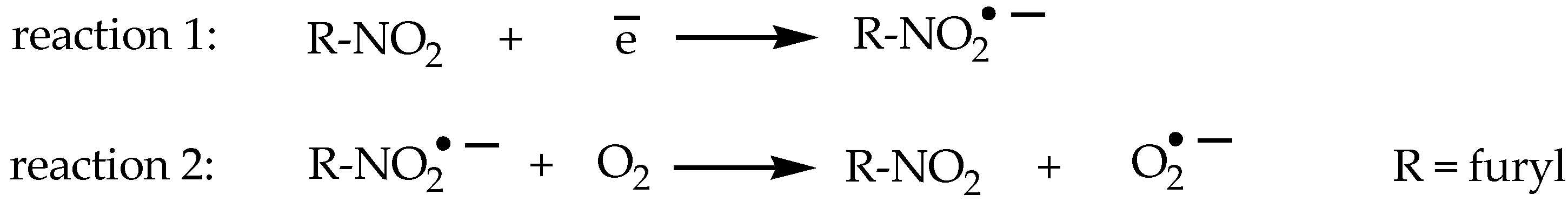
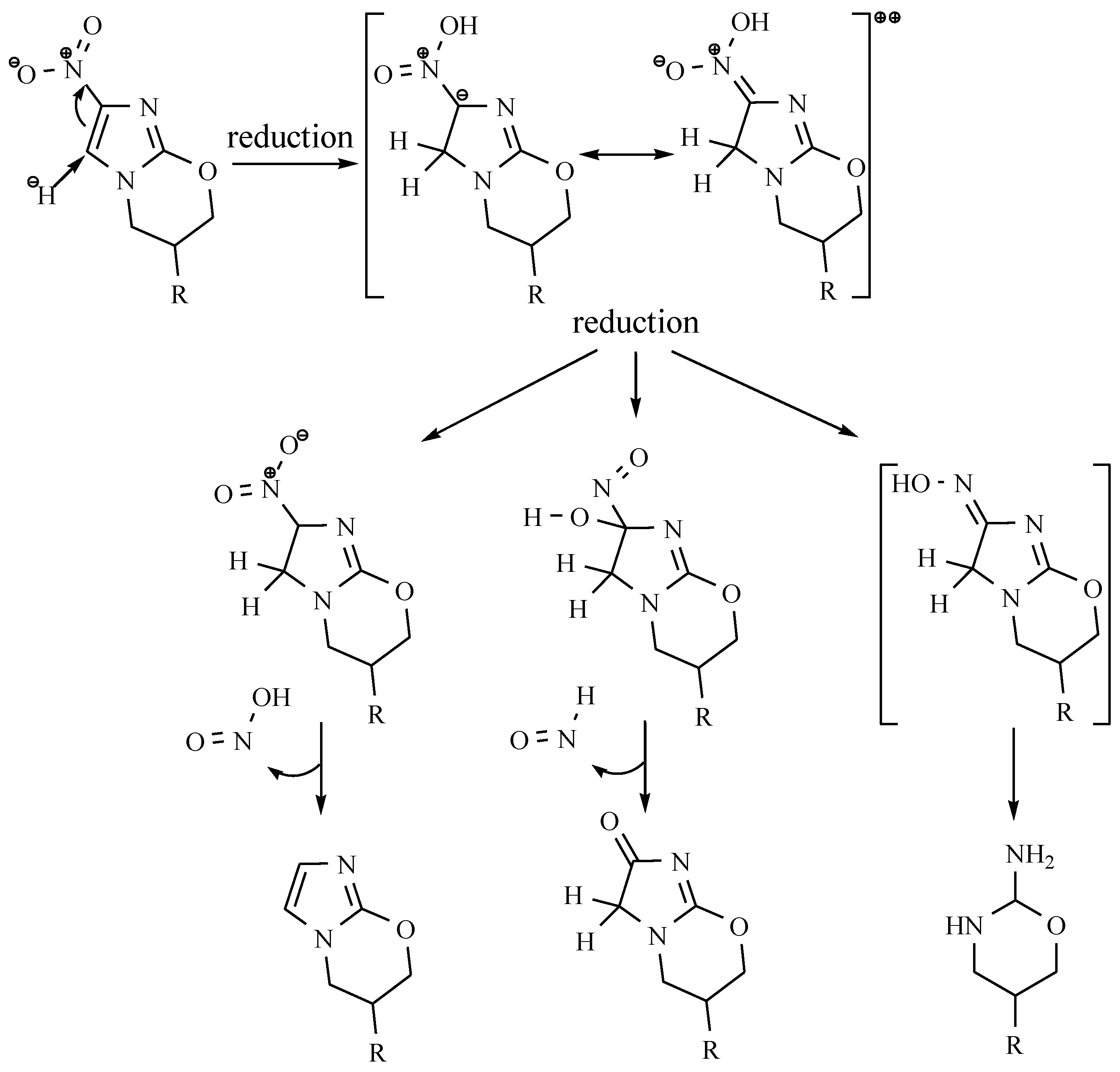
5-Nitrofuran Derivatives’ Mechanism of Action
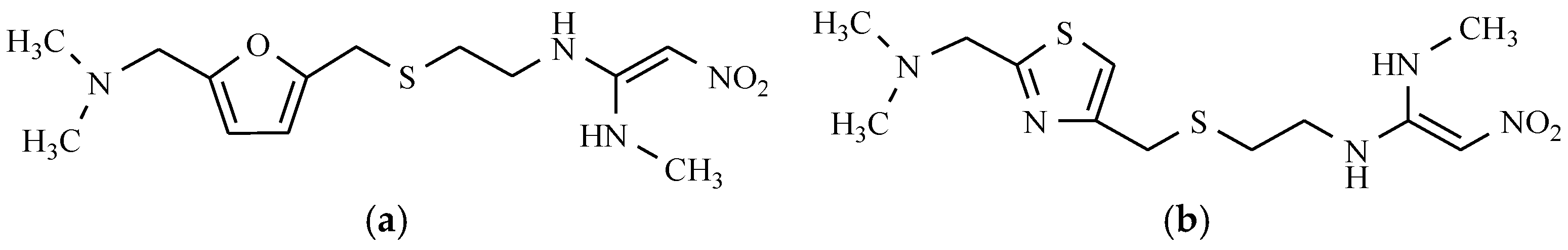
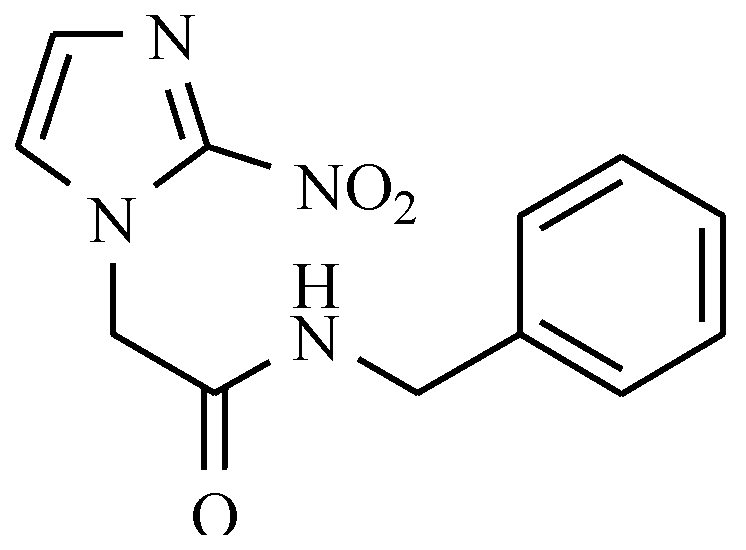
7.2. Derivatives of 2-Nitro-, 5-Nitroimidazole and 5-Nitrothiazole
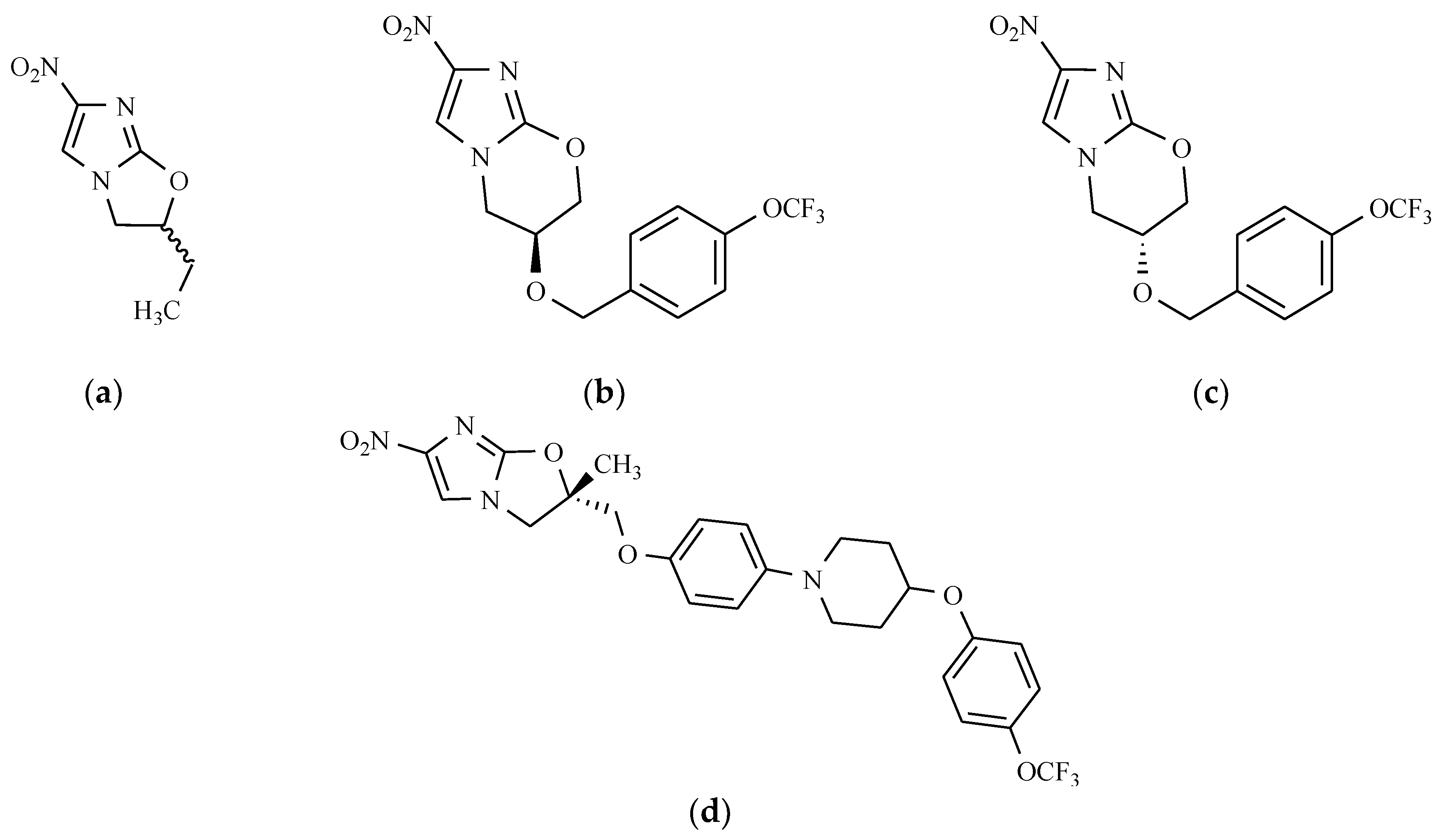
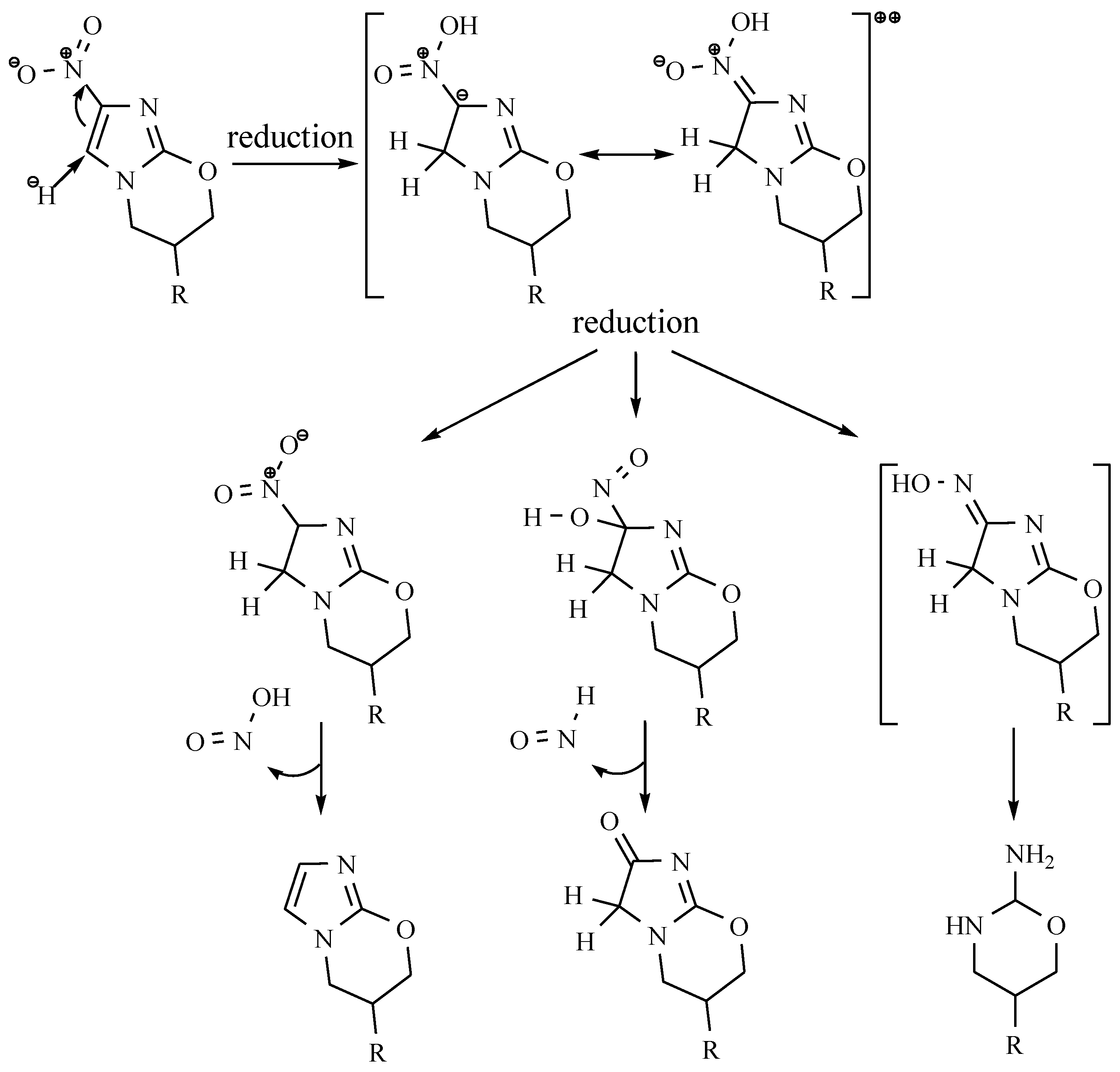
7.3. Antituberculotic Activity of Nitroimidazole Derivatives
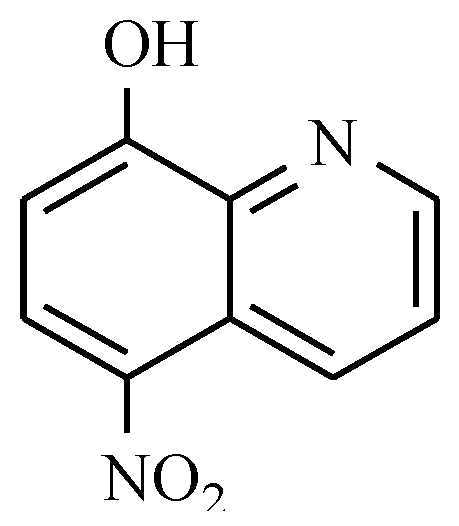
7.4. Quinoline Derivatives
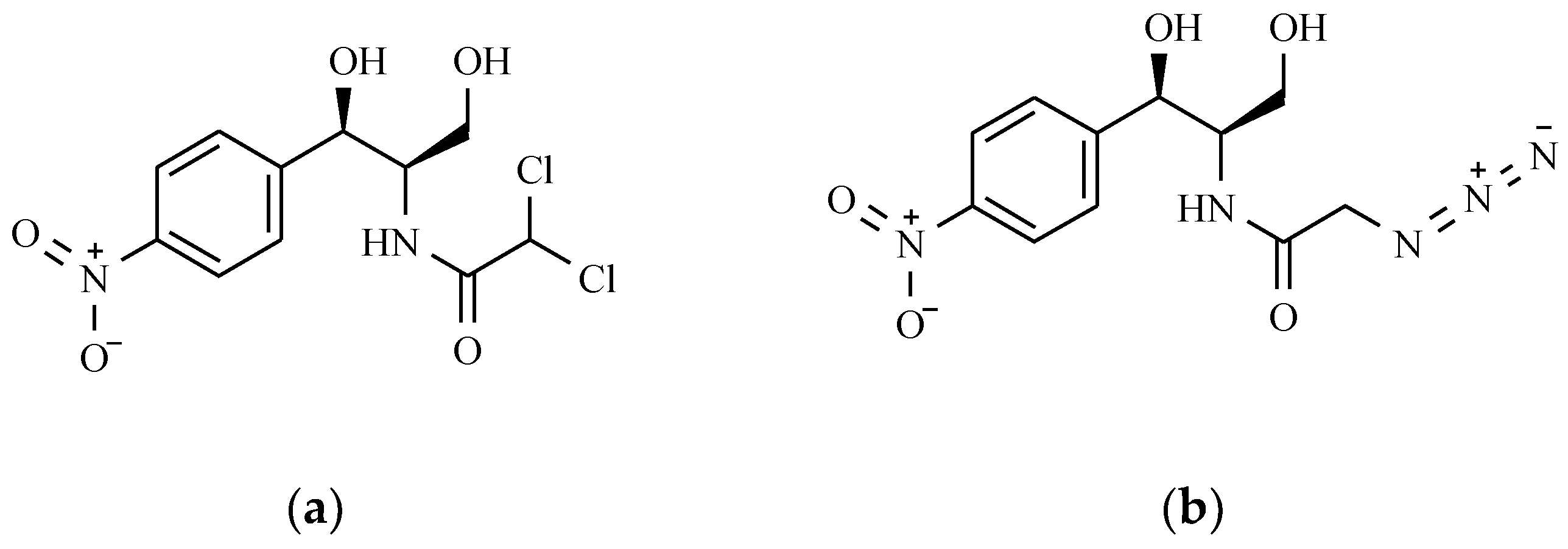
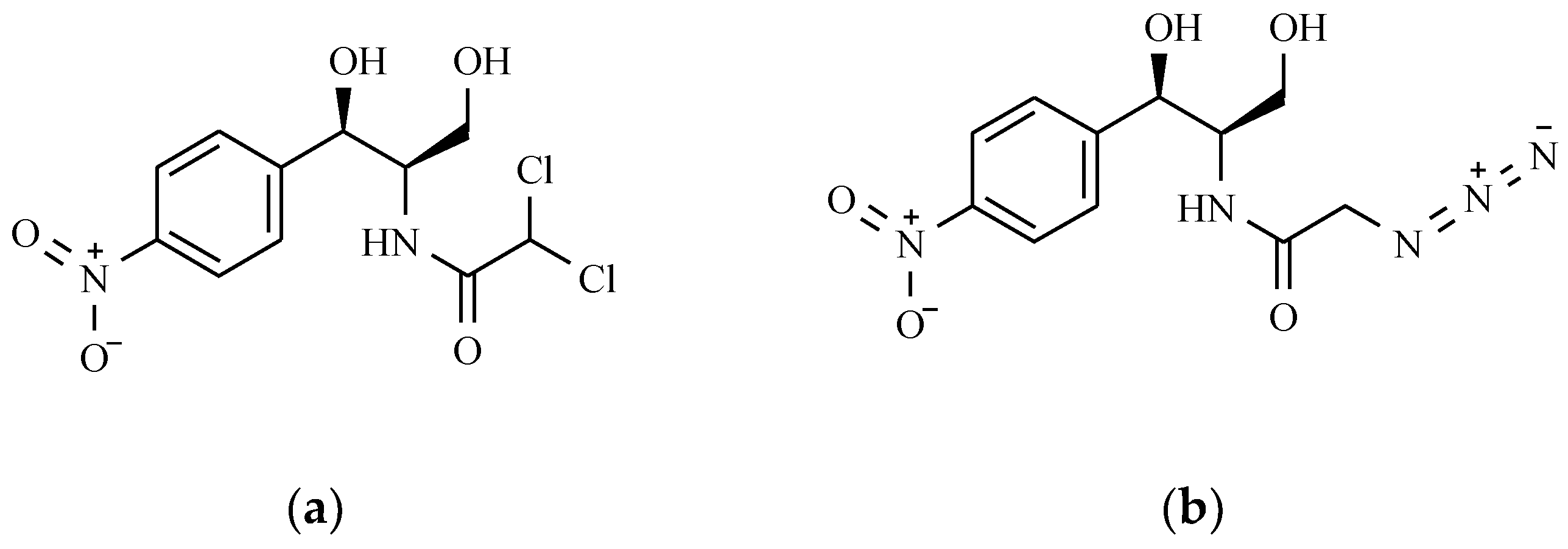
7.5. Chloramphenicol and Its Derivatives
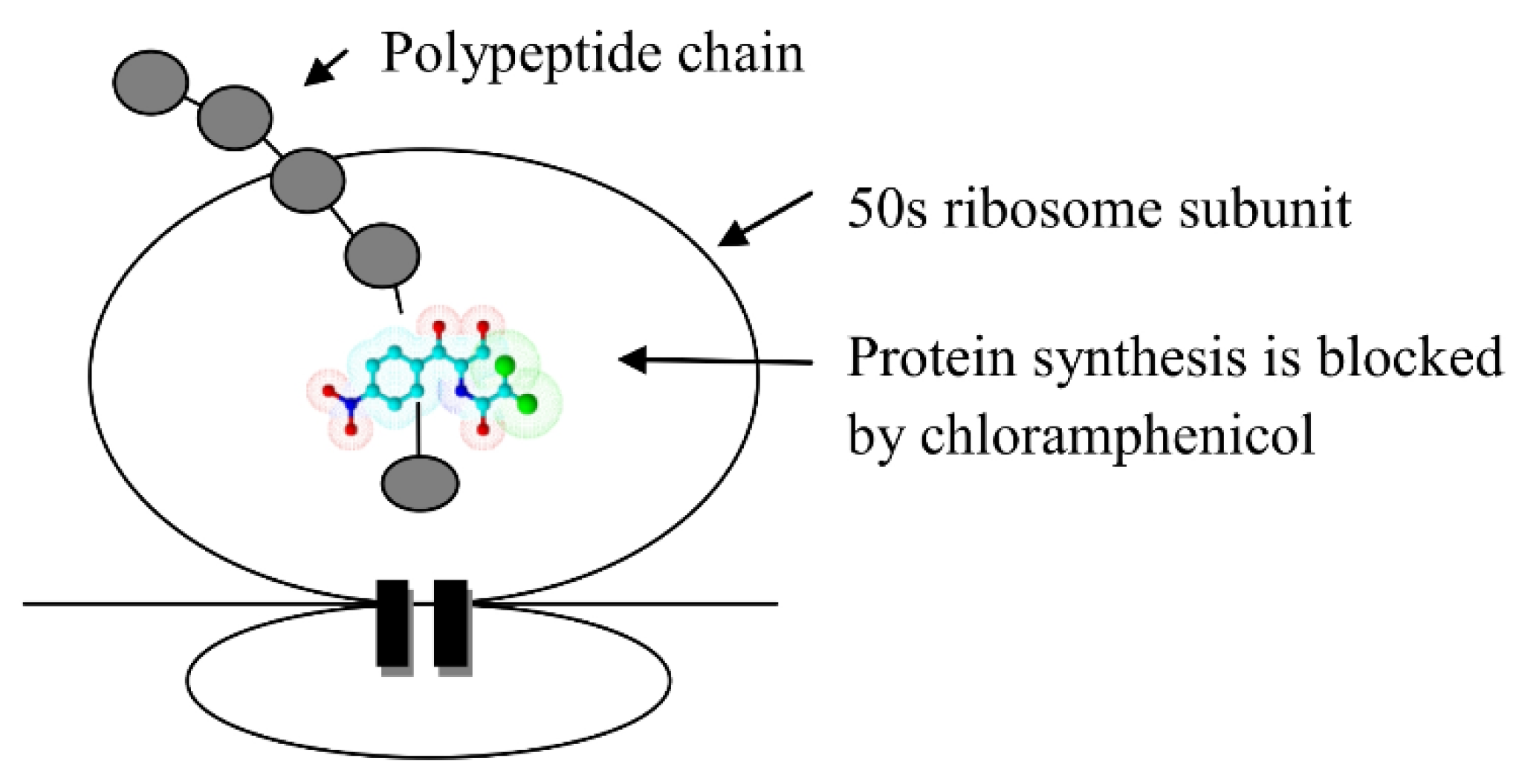
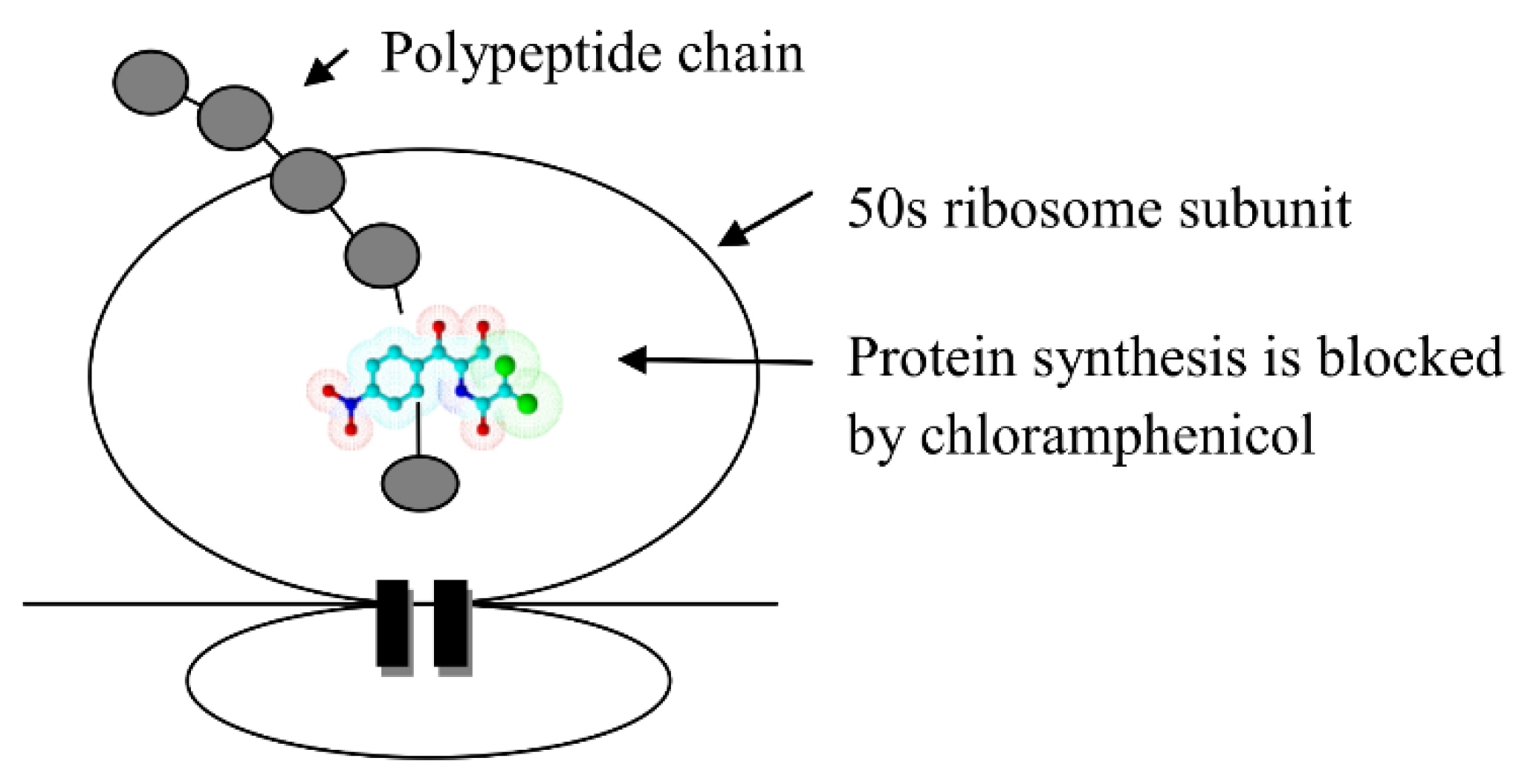
The Mechanism of the Antibacterial Action of Chloramphenicol
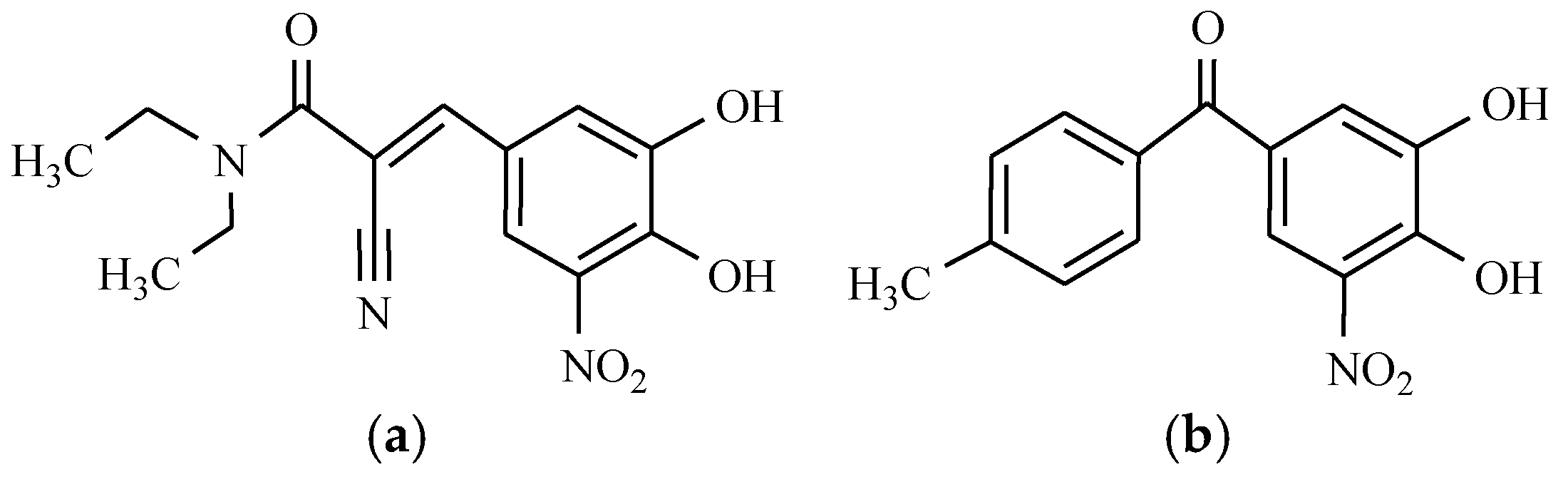
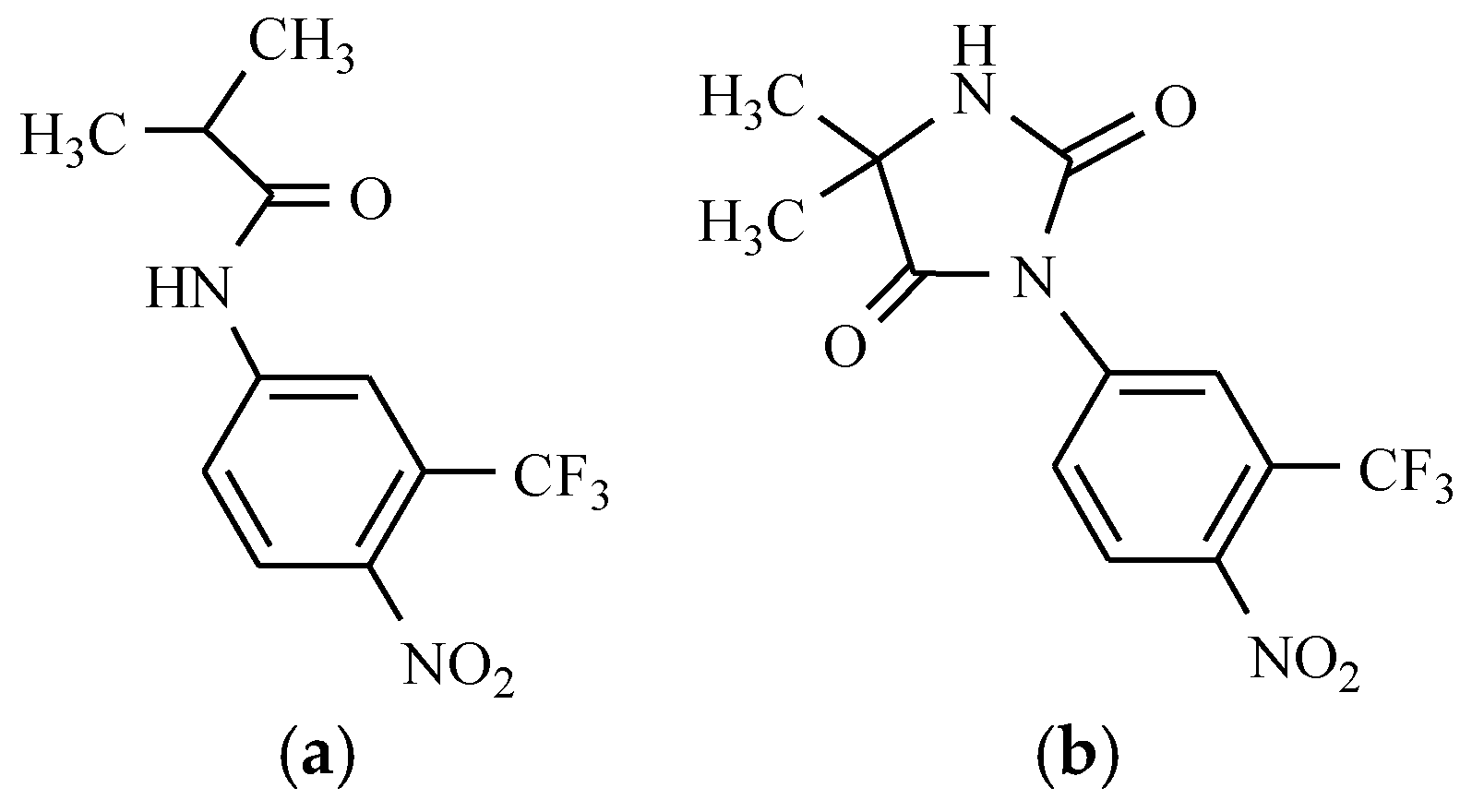
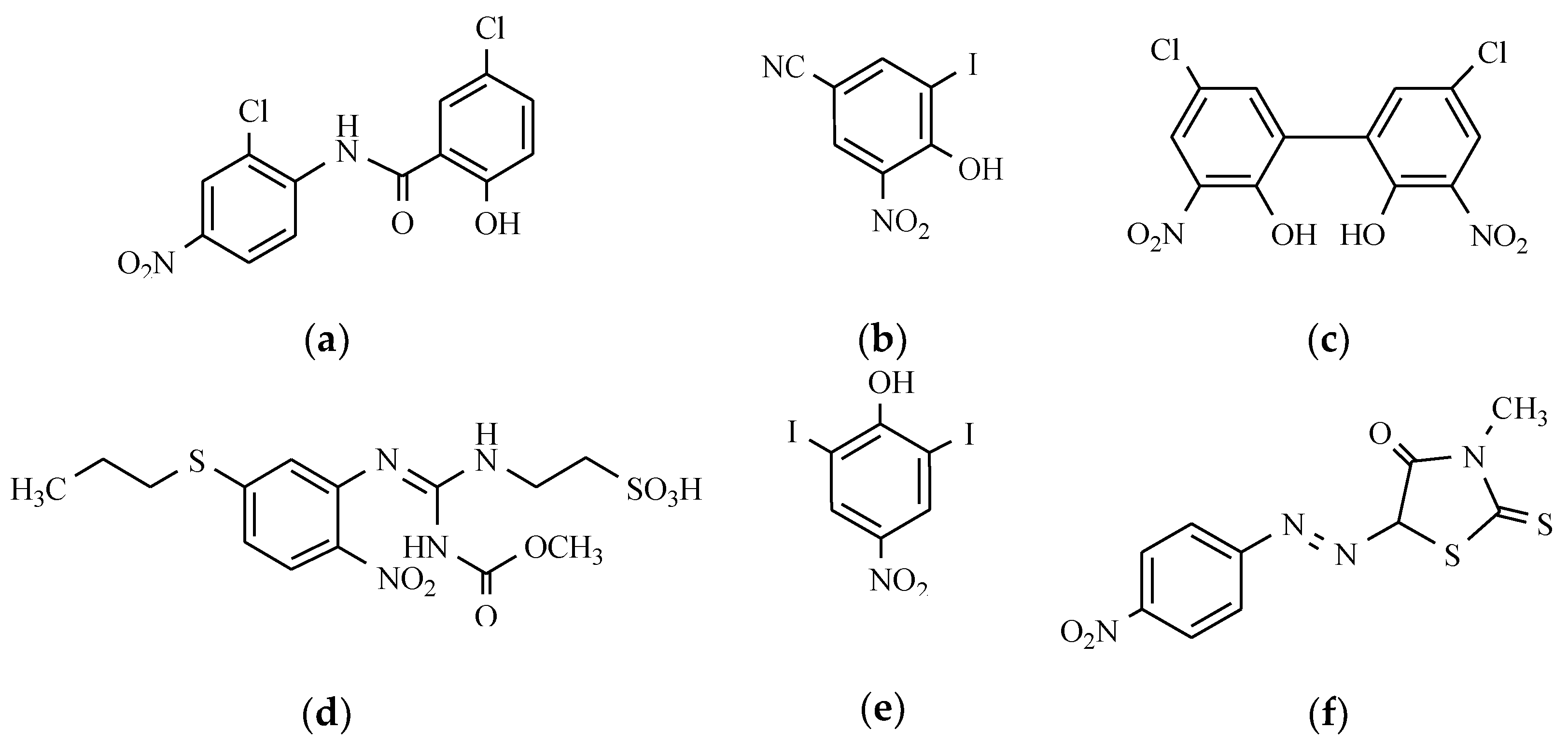
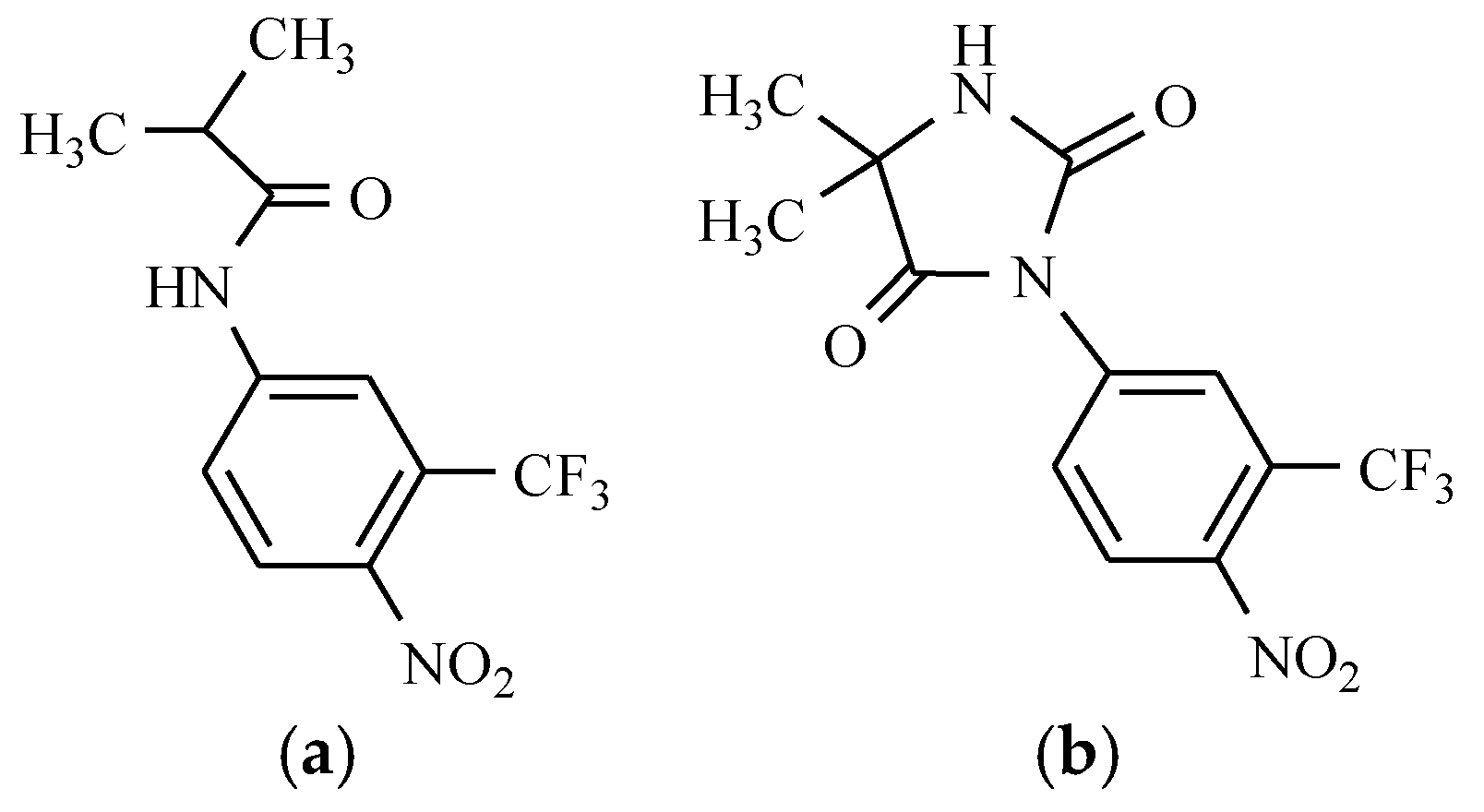
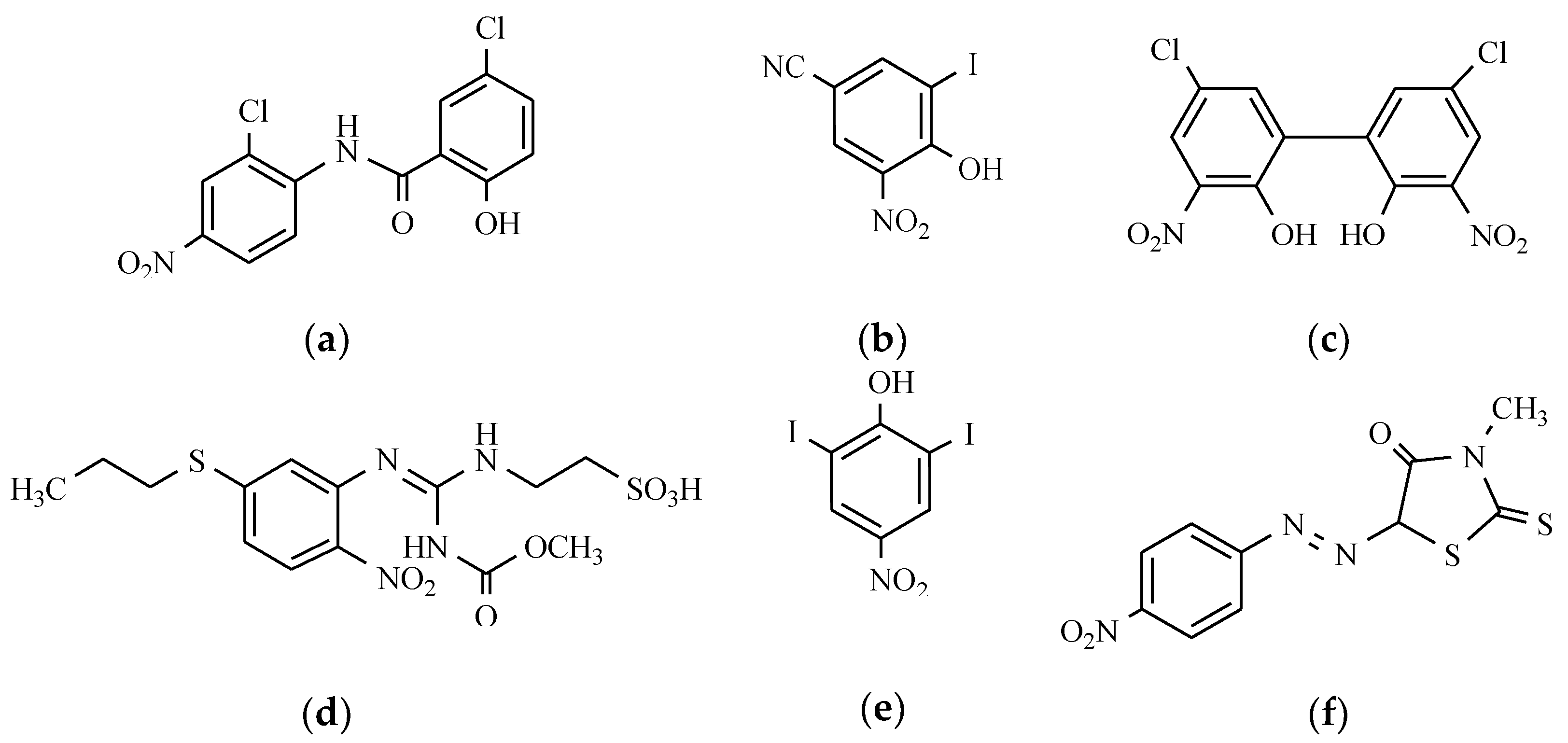
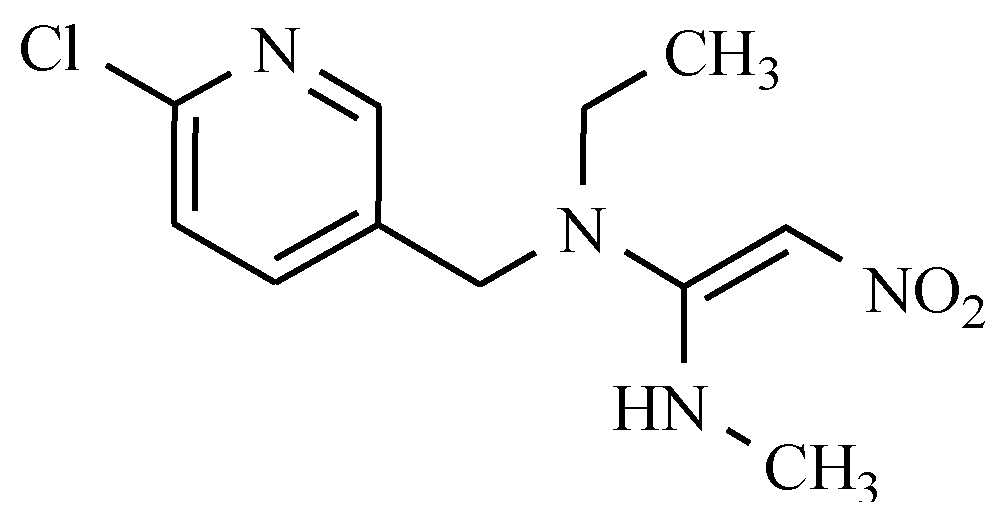
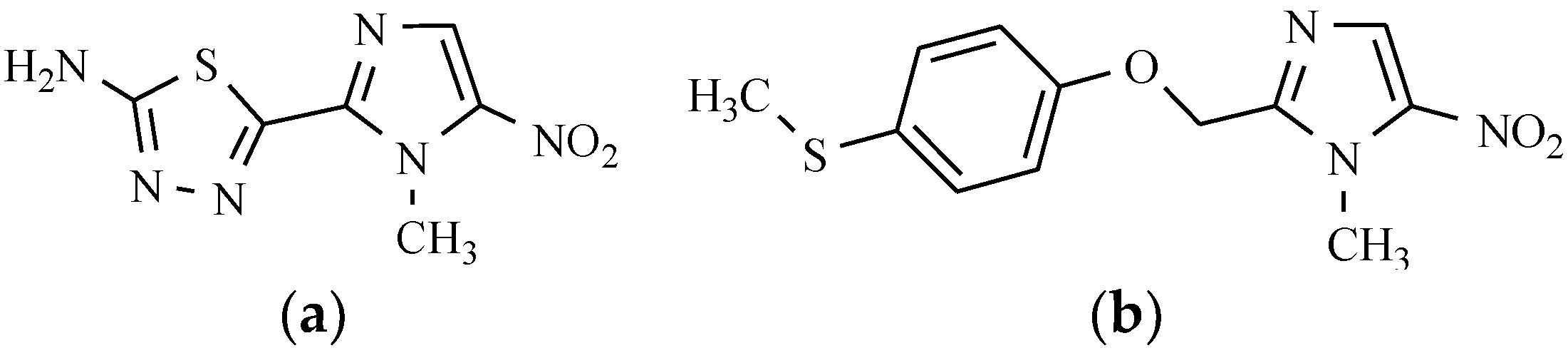
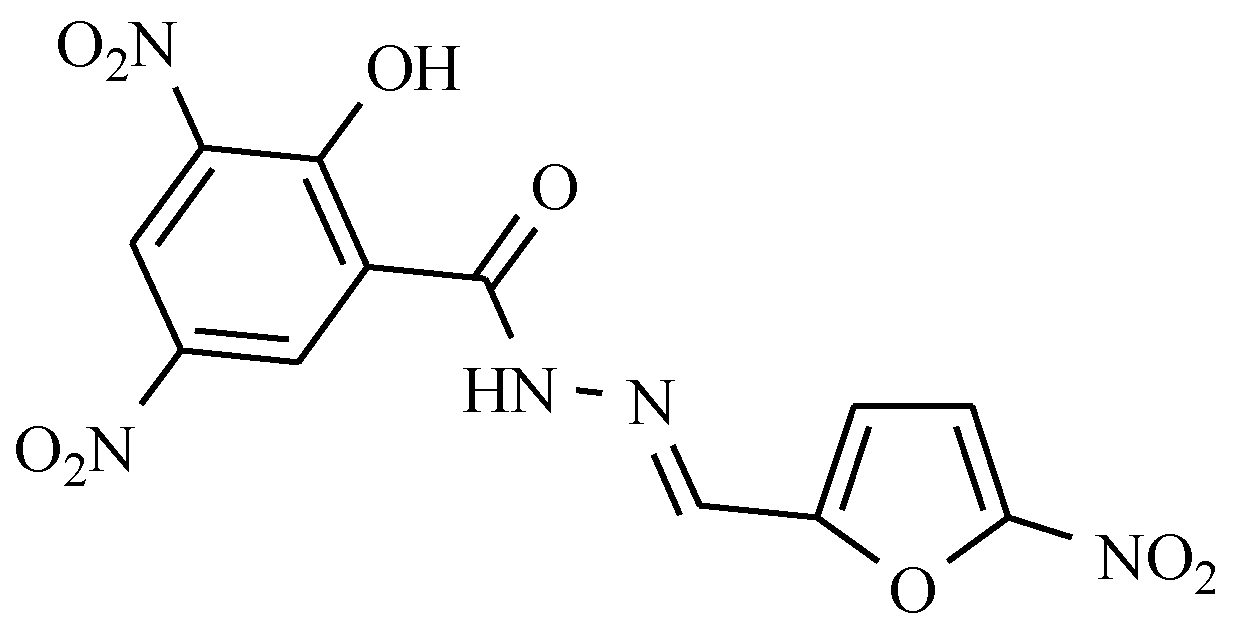
7.6. Others Antibacterial Nitro Drugs
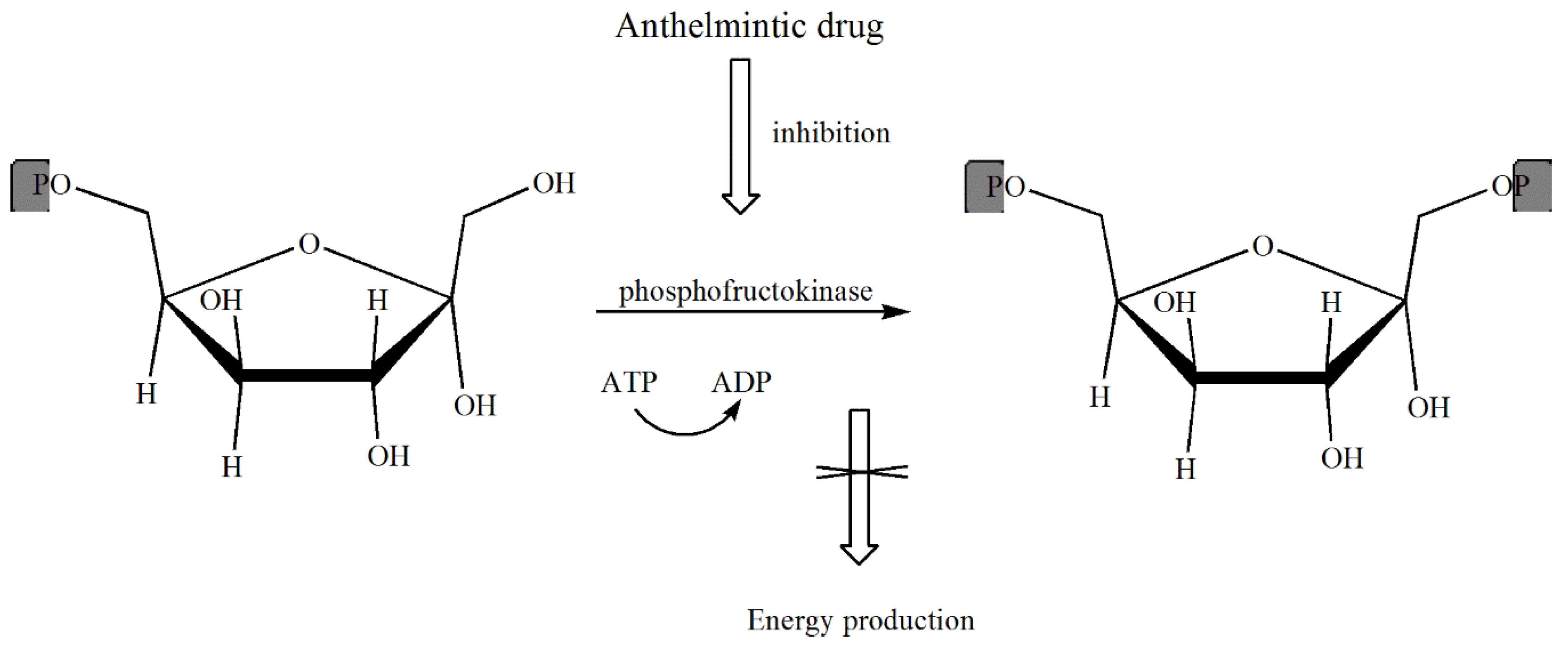
8. Anthelmintics
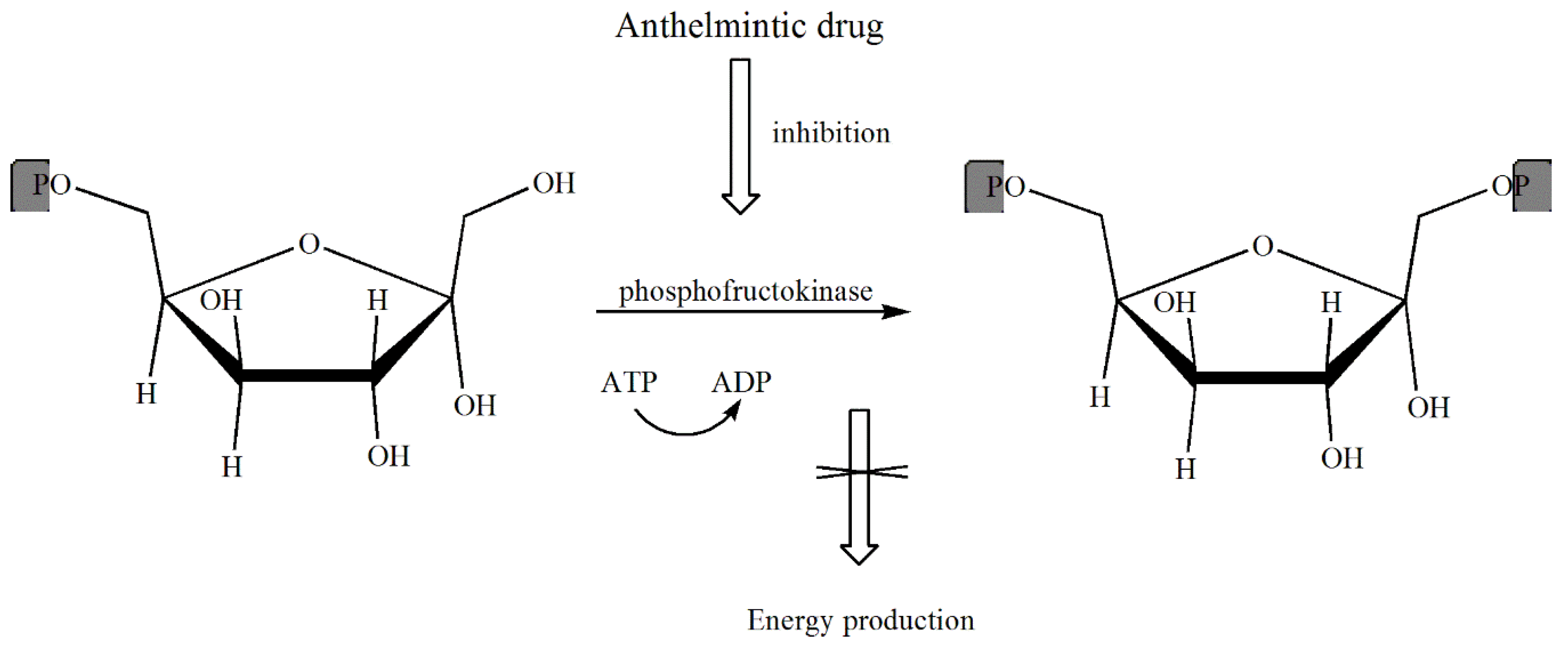
The Mechanisms of Action of Anthelmintic Drugs
9. Antiprotozoal Drugs
9.1. Nitroimidazole Derivatives
9.2. 5-Nitrofuran Derivatives
9.3. 2-Nitroimidazole Derivatives
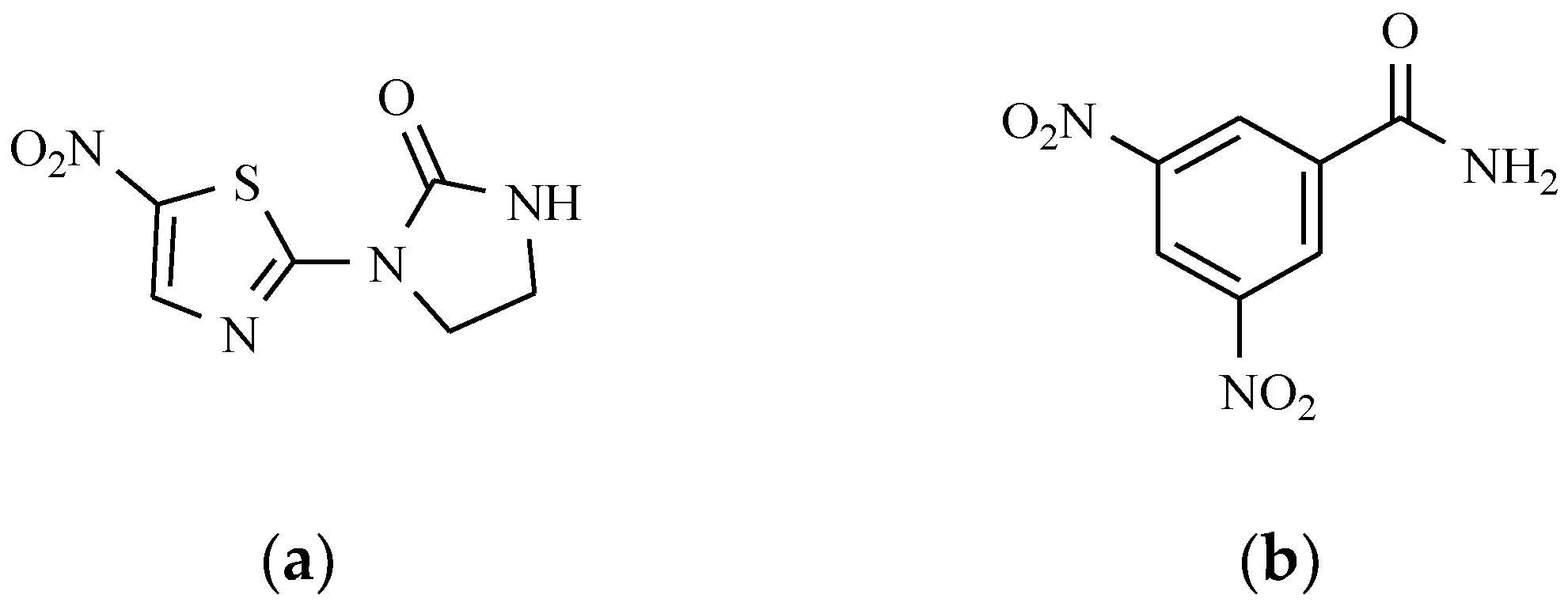
9.4. 5-Nitrothiazole Derivatives
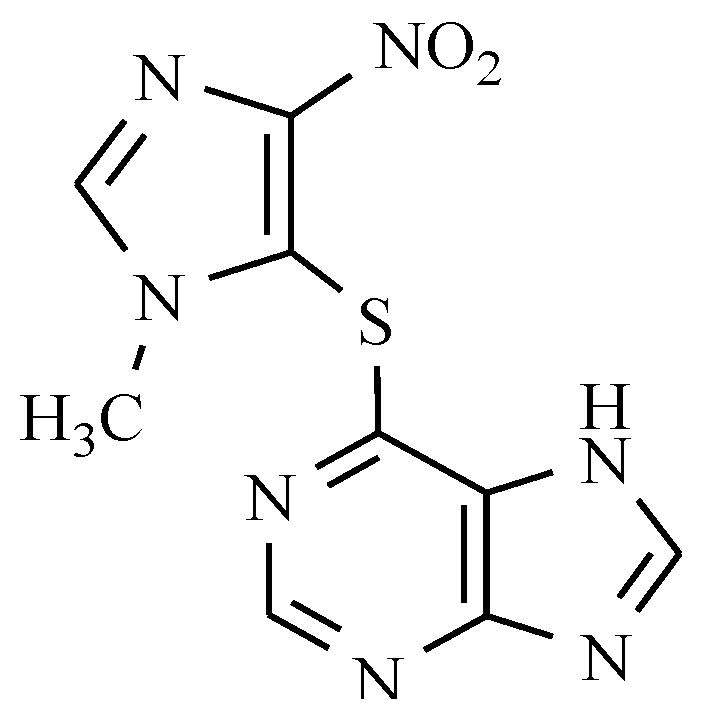
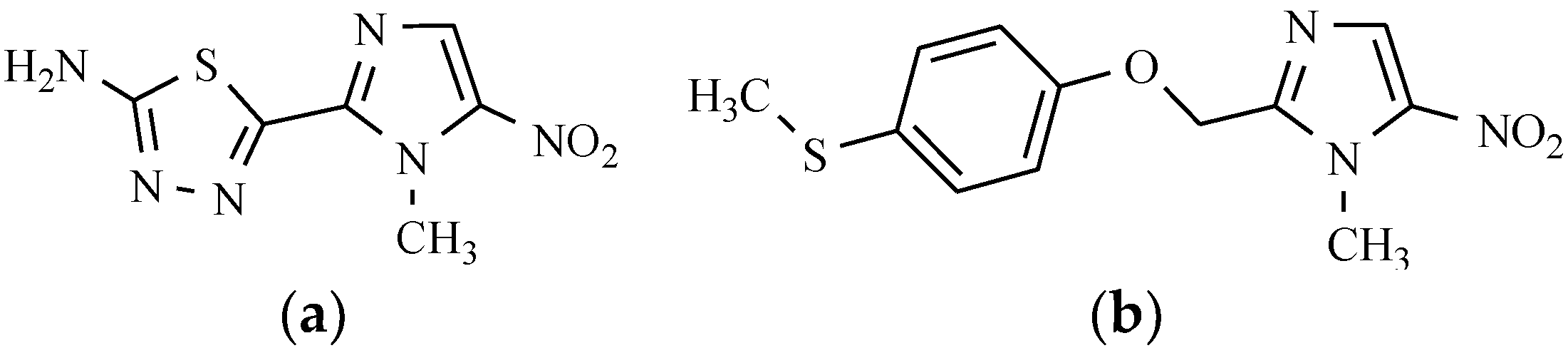
9.5. Others Antiprotozoal Drugs
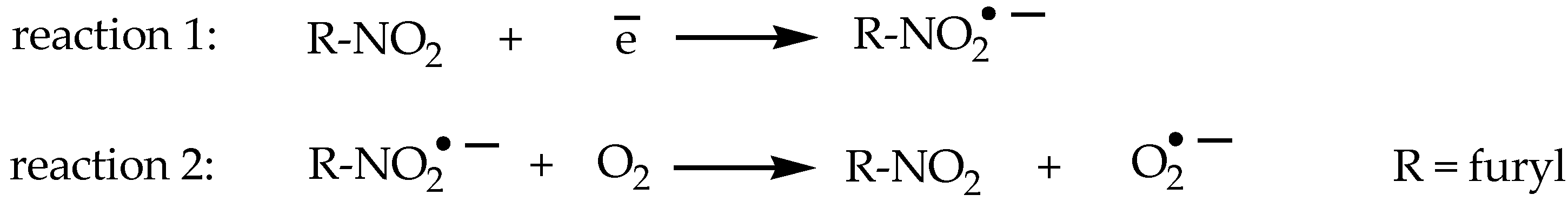
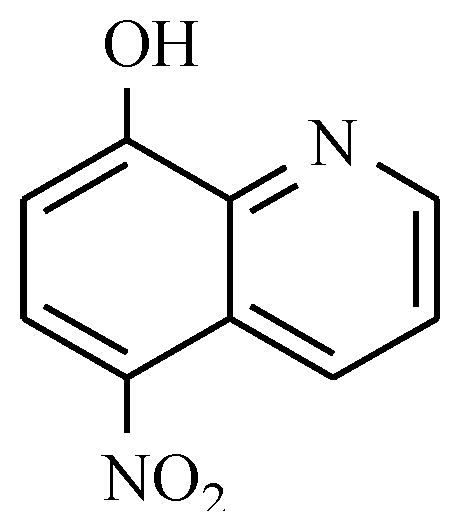
10. Radiosensitizers
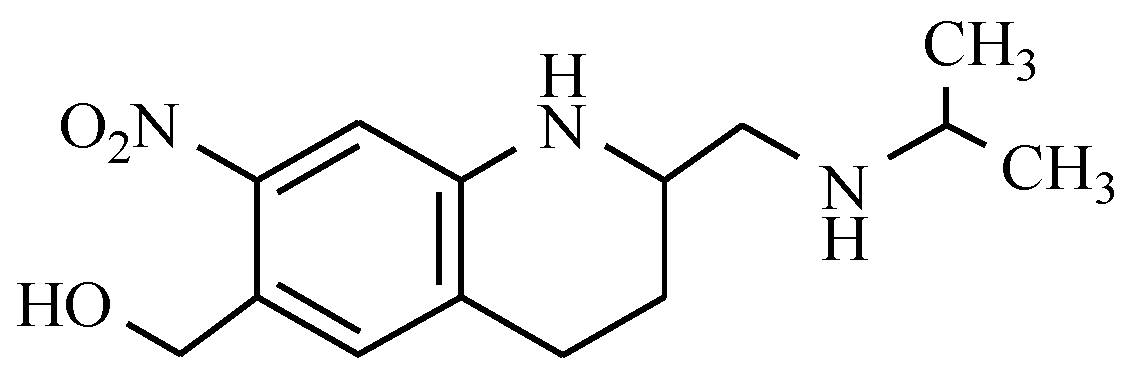
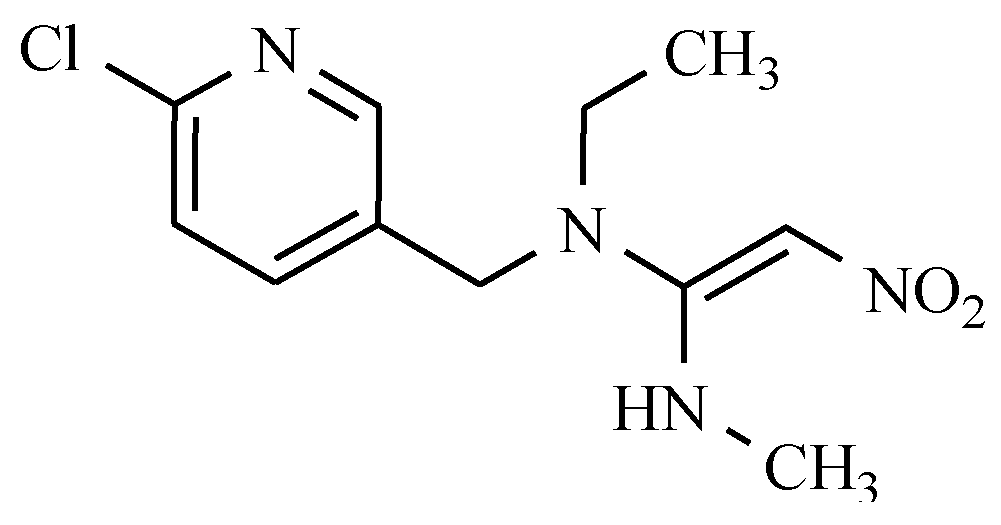
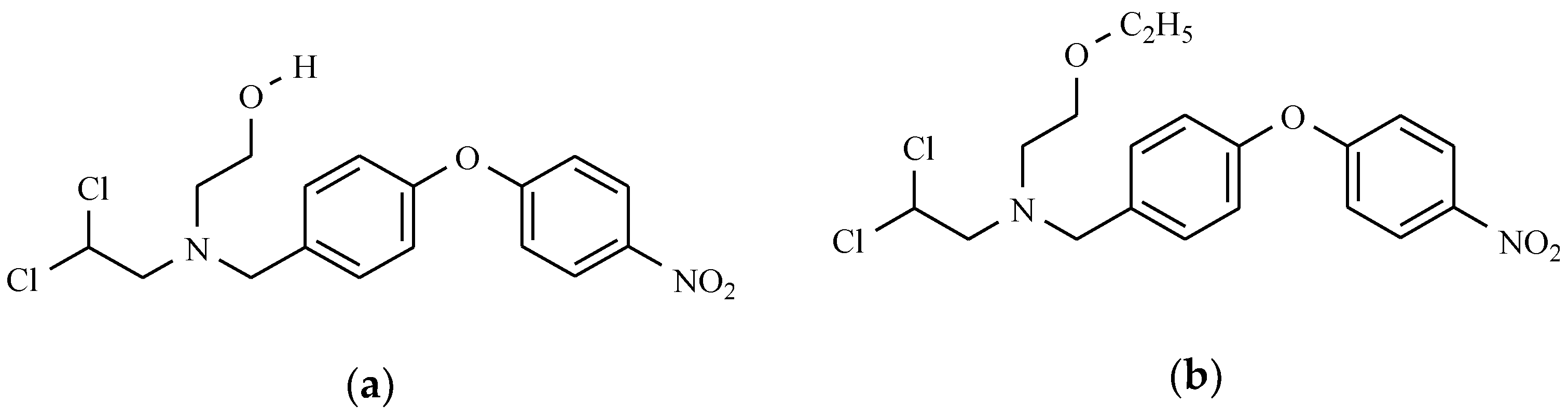
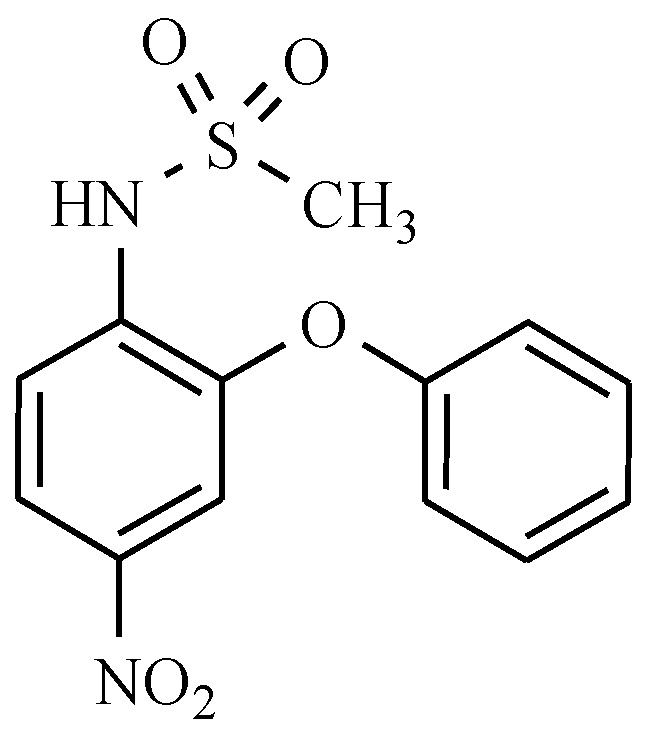
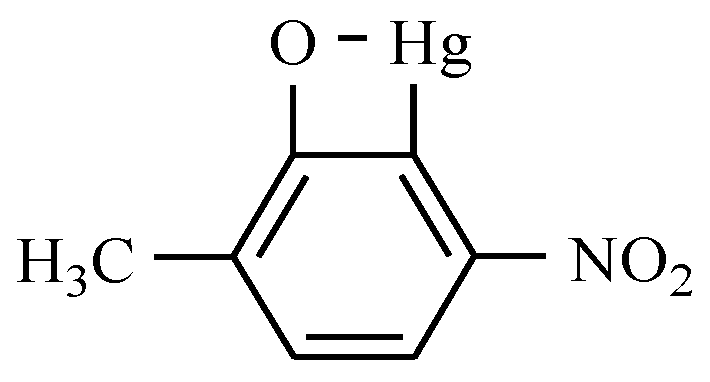
11. Drugs with Others Effects
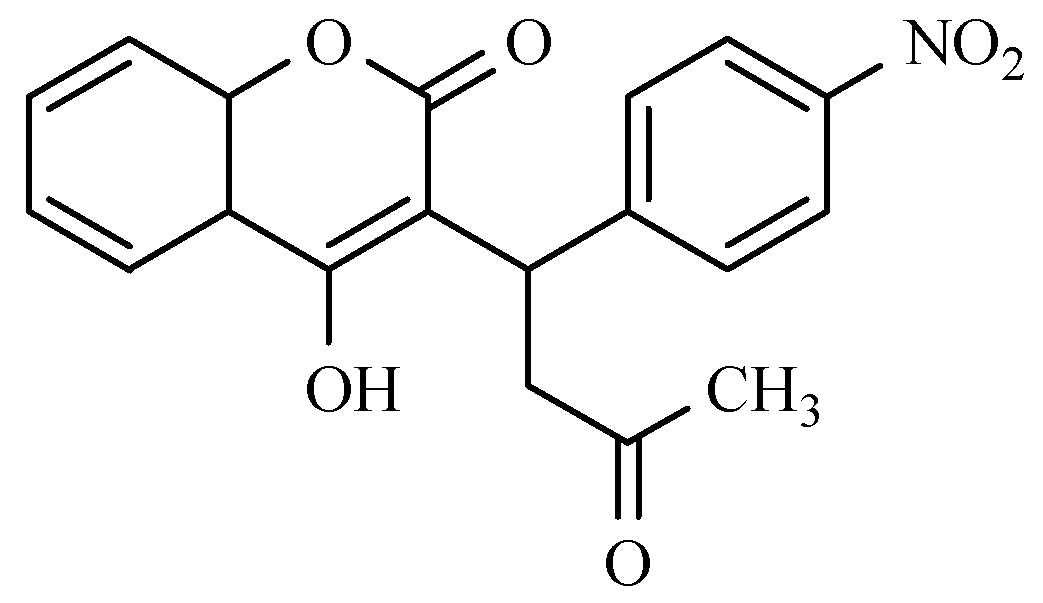
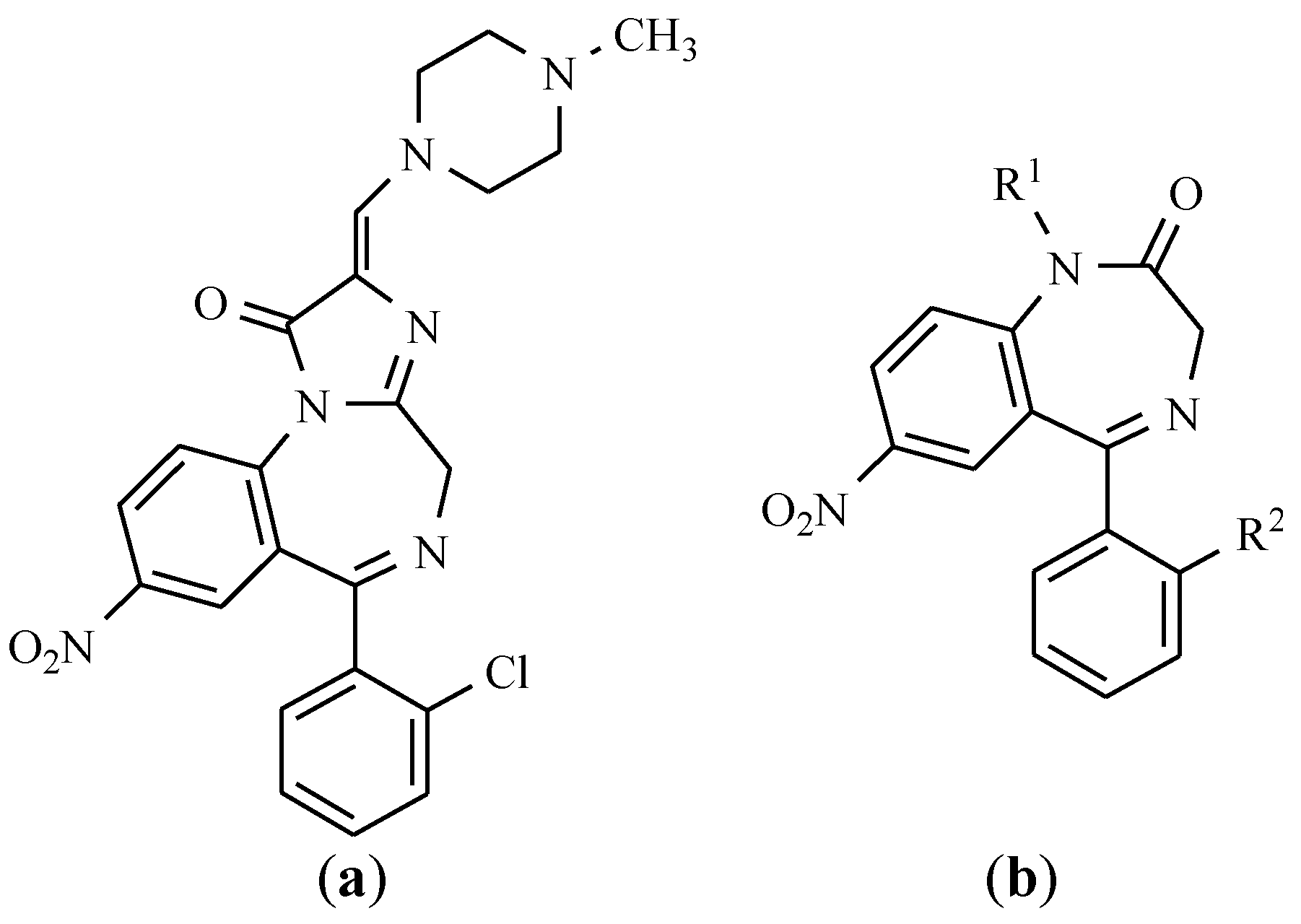
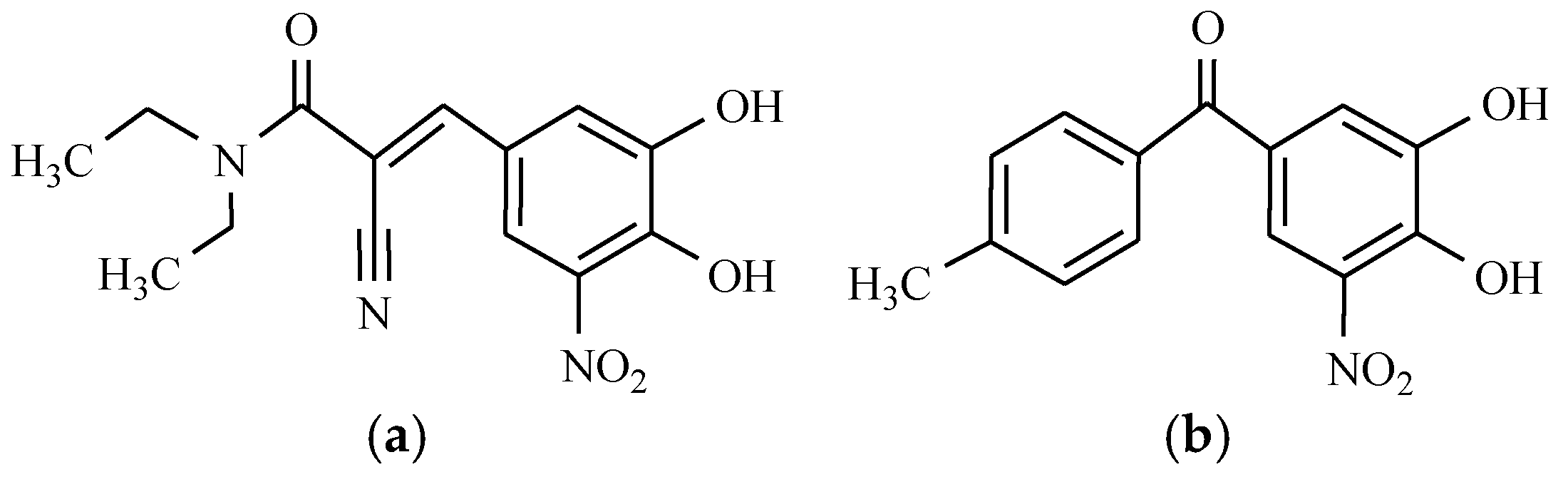
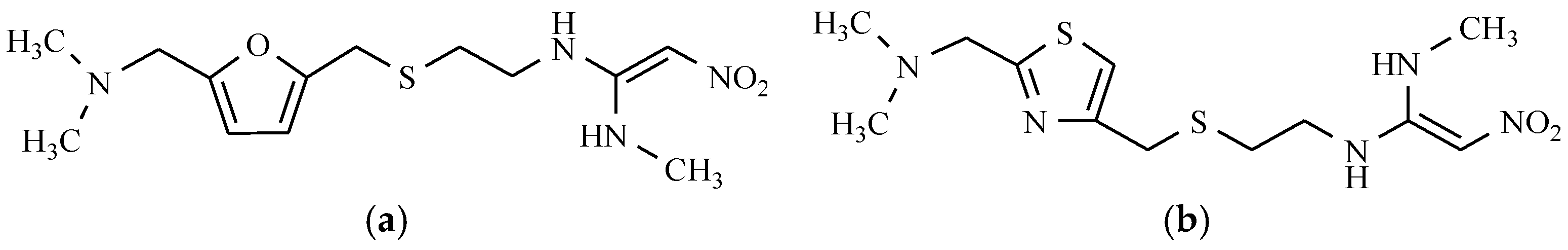
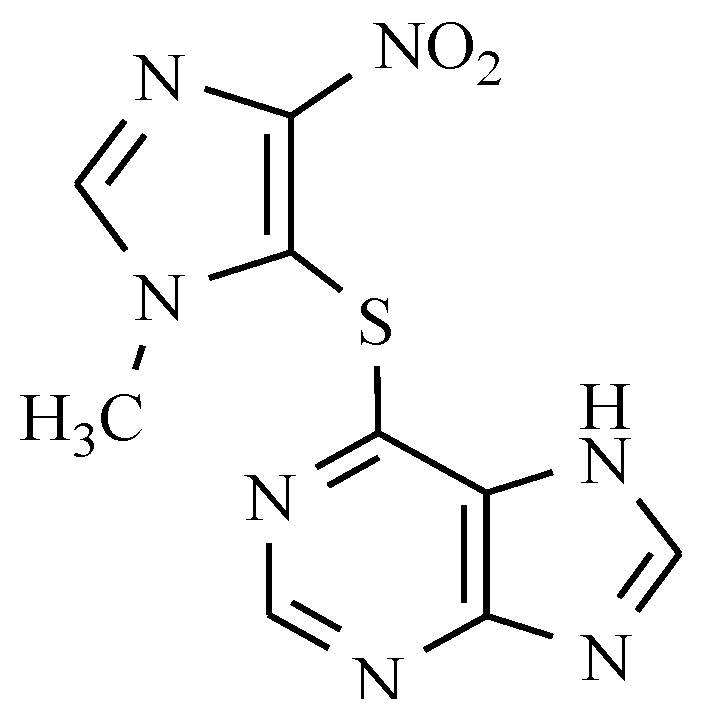
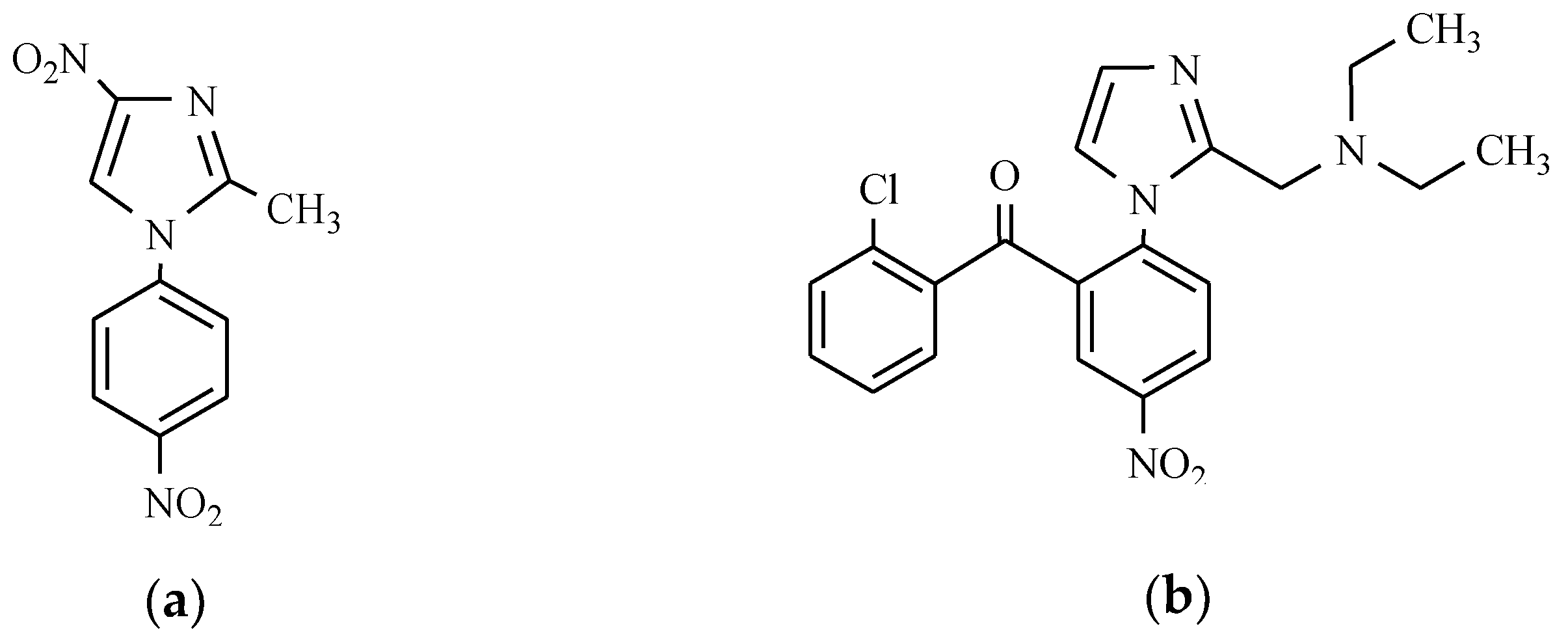
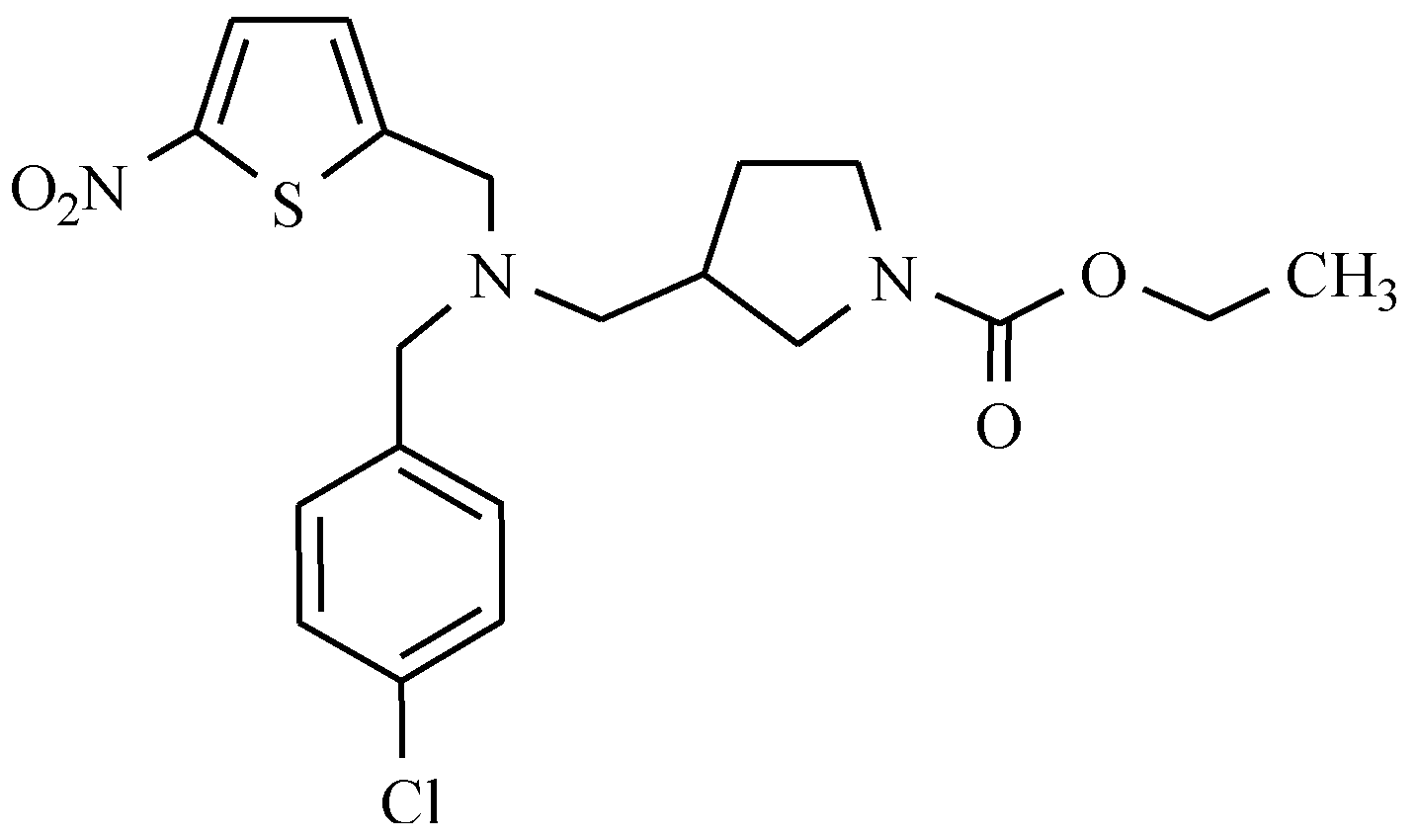
12. Drugs of the Future
13. Conclusions
Author Contributions
Conflicts of Interest
References
- Williams, M. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 15th ed.; Royal Society of Chemisry: Cambridge, UK, 2013. [Google Scholar]
- Brayfield, A. Martindale: The Complete Drug Reference, 39th ed.; Pharmaceutical Press: London, UK, 2017. [Google Scholar]
- PDR Staff. 2017 Physicians’ Desk Reference, 71st ed.; PDR Network: Montvale, NJ, USA, 2016. [Google Scholar]
- Klabunde, R.E. Cardiovascular Physiology Concepts. Available online: www.cvpharmacology.com (accessed on 23 June 2017).
- Isaeva, E.V. Effect of nifedipine in high concentrations on inhibitory synaptic transmission. Neurophys 1999, 31, 63–65. [Google Scholar] [CrossRef]
- Metra, M.; Nodari, S.; Nordio, G.; Bonandi, L.; Raddino, R.; Feroldi, P.; Dei Cas, L.; Visioli, O. A randomized double-blind crossover study of nicardipine and nifedipine in patients with angina pectoris and concomitant essential hypertension. Cardiovasc. Drug Ther. 1988, 1, 513–521. [Google Scholar] [CrossRef]
- Van Geijn, H.P.; Lenglet, J.E.; Bolte, A.C. Nifedipine trials: effectiveness and safety aspects. BJOG Int. J. Obstet. Gynaecol. 2005, 112, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Van Schie, R.M.F.; Wessels, J.A.M.; le Cessie, S.; de Boer, A.; Schalekamp, T.; van der Meer, F.J.M.; Verhoef, T.I. Loading and maintenance dose algorithms for phenprocoumon and acenocoumarol using patient characteristics and pharmacogenetic data. Eur. Heart J. 2011, 32, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Trailokya, A.; Hiremath, J.S.; Sawhney, J.P.S.; Mishra, Y.K.; Kanhere, V.; Srinivasa, R.; Tiwaskar, M. Acenocoumarol: A review of anticoagulant efficacy and safety. JAPI 2016, 64, 88–93. [Google Scholar] [PubMed]
- Mets, M.A.J.; Volkerts, E.R.; Olivier, B.; Verster, J.C. Effect of hypnotic drugs on body balance and standing steadiness. Sleep Med. Rev. 2010, 14, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Maltby, J.R.; Hamilton, R.C.; Phillips, R. Comparison of flunitrazepam and thiopentone for induction of general anaesthesia. Can. Anesth. J. Soc. 1980, 27, 331–337. [Google Scholar] [CrossRef]
- Jenner, P.; Pratt, J.A.; Marsden, C.D. Mechanism of action of clonazepam in myoclonus in relation to effects on GABA and 5-HT. Adv. Neurol. 1986, 43, 629–643. [Google Scholar] [PubMed]
- Buscemi, N.; Vandermeer, B.; Friesen, C.; Bialy, L.; Tubman, M.; Ospina, M.; Klassen, T.P.; Witmans, M. The efficacy and safety of drug treatments for chronic insomnia in adults: A meta-analysis of RCTs. J. Gen. Int. Med. 2007, 22, 1335–1350. [Google Scholar] [CrossRef] [PubMed]
- Gordin, A.; Kaakkola, S.; Teräväinen, H. Clinical advantages of COMT inhibition with entacapone—A review. J. Neural Trans. 2004, 111, 1343–1363. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.D. Tolcapone: Review of its pharmacology and use as adjunctive therapy in patients with Parkinson’s disease. Clin. Interv. Aging 2009, 4, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Leegwater-Kim, J.; Waters, C. Role of tolcapone in the treatment of Parkinson’s disease. Expert Rev. Neurother. 2007, 7, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Futagami, S.; Shimpuku, M.; Kawagoe, T.; Kusunoki, M.; Ueki, N.; Miyake, K.; Iwakiri, K.; Sakamoto, C. Nizatidine administration improves clinical symptoms and gastric emptying of the patients with functional dyspepsia accompanying with impaired gastric emptying. Gastroenterology 2011, 140, S230. [Google Scholar] [CrossRef]
- Law, R.; Maltepe, C.; Bozzo, P.; Einarson, A. Treatment of the heartbum and acid reflux associated with nausea and vomiting during pregnancy. Can. Fam. Phys. 2010, 56, 143–144. [Google Scholar]
- Koskenpato, J.; Punkkinen, J.N.; Kairemo, K.; Färkkilä, M. Nizatidine and gastric emptying in functional dyspepsia. Dig. Dis. Sci. 2008, 53, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Uemura, H.; Harada, M.; Miura, T.; Moriyama, M.; Fukuoka, H.; Kitami, K.; Hosaka, M. Inhibition of PSA flare in prostate cancer patients by administration of flutamide for 2 weeks before initiation of treatment of slow-releasing LH-RH agonist. Int. J. Clin. Oncol. 2001, 6, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Sufrin, G.; Coffey, D.S. Flutamide. Mechanism of action of a new nonsteroidal antiandrogen. Investig. Urol. 1976, 13, 429–434. [Google Scholar]
- Ask, K.; Décologne, N.; Ginies, C.; La, M.; Boucher, J.L.; Holmec, J.A.; Pelczar, H.; Camus, P. Metabolism of nilutamide in rat lung. Biochem. Pharmacol. 2006, 71, 377–385. [Google Scholar] [CrossRef] [PubMed]
- La Mantia, L.; Mascoli, N.; Milanese, C. Azathioprine. Safety profile in multiple sclerosis patients. Neurol. Sci. 2007, 28, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Swerlick, R.A.; McCall, C.O. Azathioprine in dermatology: the past, the present, and the future. J. Am. Acad. Dermatol. 2006, 55, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.; Bombardelli, E.; Manzotti, C.; Battaglia, A.; Samori, C. Camptothecin derivatives with antitumor activity. EP Patent 2010/38 EP 2044078 B1, 22 September 2010. [Google Scholar]
- Clark, J.W. Rubitecan. Exp. Opin. Investig. Drugs 2006, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Bains, A.; Buna, D.; Hoag, N.A. A retrospective review assessing the efficacy and safety of nitrofurantoin in renal impairment. Can. Pharm. J. 2009, 142, 248–252. [Google Scholar] [CrossRef]
- Cunha, B.A.; Schoch, P.E.; Hage, J.R. Nitrofurantoin: preferred empiric therapy for community-acquired lower urinary tract infections. Mayo Clin. Proc. 2011, 86, 1243–1244. [Google Scholar] [CrossRef] [PubMed]
- El-Zaher, A.A.; Mahrouse, M.A. A validated spectrofluoremetric method for the determination of nifuroxazide through coumarin formation using experimental design. Chem. Cent. J. 2013, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, M.J.; Smith, A.; Heckelman, P.E. Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 13th ed.; Merck: Whitehouse Station, NJ, USA, 2001. [Google Scholar]
- Gagliardi, S.; Consonni, S.; Ronzoni, A.; Bulgheroni, A.; Ceriani, D. Nifuratel Sulfoxide for Use in the Treatment of Bacterial Infections. EP Patent EP 2797914 B1, 16 September 2015. [Google Scholar]
- Yan, X.D.; Zhang, L.J.; Wang, J.P. Residue depletion of nitrovin in chicken after oral administration. J. Agric. Food Chem. 2011, 59, 3414–3419. [Google Scholar] [CrossRef] [PubMed]
- Bot, C.; Hall, B.S.; Alvarez, G.; Di Maio, R.; González, M.; Cerecetto, H.; Wilkinson, S.R. Evaluating nitrofurans as trypanocidal agents. Antimicrob. Agents Chemother. 2013, 57, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Boshoff, H. Nitroimidazoles for the treatment of TB: Past, present, and future. Future Med. Chem. 2011, 3, 1427–1454. [Google Scholar] [CrossRef] [PubMed]
- Mital, A. Synthetic nitroimidazoles: Biological activities and mutagenicity relationships. Sci. Pharm. 2009, 77, 497–520. [Google Scholar] [CrossRef]
- Schwebke, J.R.; Desmond, R.A. Tinidazole vs metronidazole for the treatment of bacterial vaginosis. Am. J. Obstet. Gynecol. 2011, 204, 211.e1–211.e6. [Google Scholar] [CrossRef] [PubMed]
- Ratnaparkhi, M.P.; Dhiwar, S.B.; Gurav, R.R.; Bhore, S.S. Formulation and In-Vitro Characterization of Nimorazole Mouth Dissolving Tablets. Res. J. Pharm. Biol. Chem. Sci. 2012, 3, 303–308. [Google Scholar]
- Patel, P.; Patel, K.; Bhatt, K.; Patel, S. New improved RP-HPLC method for determination of norfloxacin and ornidazole in their combined dosage form. Int. J. Res. Pharm. Biomed. Sci. 2011, 2, 710–713. [Google Scholar]
- Ashtekar, D.R.; Costa-Pereira, R.; Nagarajan, K.; Vishvanathan, N.; Bhatt, A.D.; Rittel, W. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1993, 37, 183–186. [Google Scholar] [CrossRef] [PubMed]
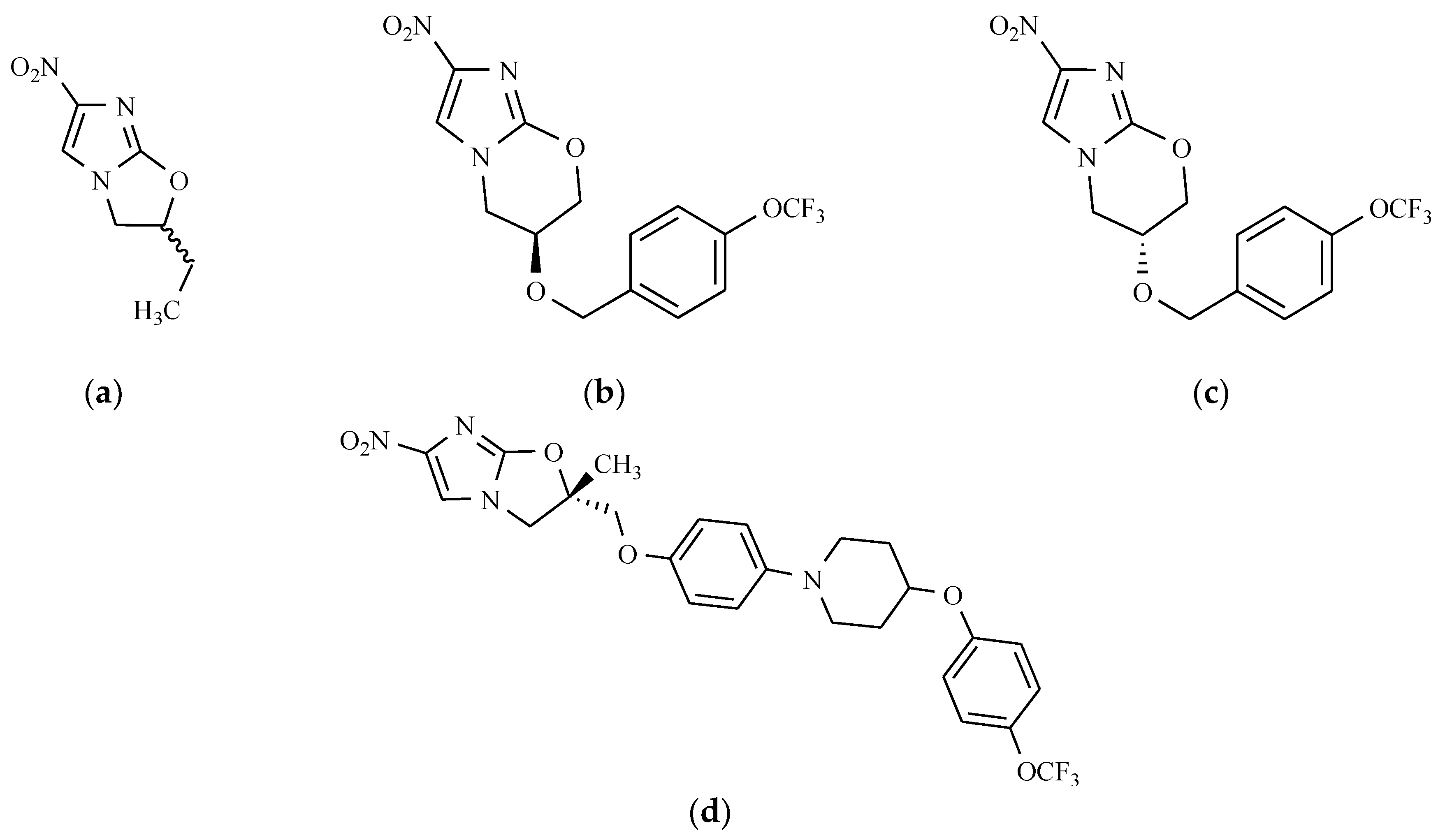
- Barry, C.E., 3rd; Boshoff, H.I.M.; Dowd, C.S. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr. Pharm. Des. 2004, 10, 3239–3269. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC 67683. A nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Pontali, E.; Centis, R.; D’Ambrosio, L.; Migliori, G.B. Delamanid (OPC 67683) for treatment of multi-drug resistant tuberculosis. Expert Rev. Anti-Infect. Ther. 2015, 13, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Roberts, A.J.; Norval, S.; Patterson, S.; Foth, B.J.; Berriman, M.; Read, K.D.; Fairlamb, A.H. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in leishmania. PLOS Pathog. 2016, 12, e1005971. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Dawson, R.; Hanckom, M.; Narunsky, K.; Venter, A.; Hittel, N.; Geiter, L.J.; Wells, C.D.; Paccaly, A.J.; Donald, P.R. Early bactericidal activity of delamanid (OPC-67683) in smear-positive pulmonary tuberculosis patients. Int. J. Tuberc. Lung Dis. 2011, 15, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Warrener, P.; Van Devanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Manjunatha, U.; Boshoff, H.I.M.; Ha, Y.H.; Niyomrattanakit, P.; Ledwidge, R.; Dowd, C.S.; Lee, I.Y.; Kim, P.; Zhang, L.; et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 2008, 322, 1392–1395. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Zhang, L.; Manjunatha, U.H.; Singh, R.; Patel, S.; Jiricek, J.; Keller, T.H.; Boshoff, H.I.M.; Barry, C.E., 3rd; Dowd, C.S. Structure-activity relationships of antitubercular nitroimidazoles. I. Structural features associated with aerobic and anaerobic activities of 4- and 5-nitroimidazoles. J. Med. Chem. 2009, 52, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Kang, S.; Boshoff, H.I.M.; Jiricek, J.; Collins, M.; Singh, R.; Manjunatha, U.H.; Niyomrattanakit, P.; Zhang, L.; Goodwin, M.; et al. Structure-activity relationships of antitubercular nitroimidazoles. 2. Determinants of aerobic activity and quantitative structure-activity relationships. J. Med. Chem. 2009, 52, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Naber, K.G.; Niggemann, H.; Stein, G.; Stein, G. Review of the literature and individual patients’ data meta-analysis on efficacy and tolerance of nitroxoline in the treatment of uncomplicated urinary tract infections. BMC Infect. Dis. 2014, 14, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, S.; Yang, D.; Pan, K.; Li, L.; Yuan, S. Preclinical pharmacodynamic evaluation of antibiotic nitroxoline for anticancer drug repurposing. Oncol. Lett. 2016, 11, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Mitrović, A.; Sosič, I.; Kos, Š.; Tratar, U.L.; Breznik, B.; Kranjc, S.; Mirković, B.; Gobec, S.; Lah, T.; Serša, G.; et al. Addition of 2-(ethylamino)acetonitrile group to nitroxoline results in significantly improved anti-tumor activity in vitro and in vivo. Oncotarget 2017, 8, 59136–59147. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, N.; Kourelis, T.G.; Mamos, P.; Magoulas, G.E.; Kalpaxis, D.L. Insights into the chloramphenicol inhibition effect on peptidyl transferase activity, using two new analogs of the drug. Open Enzyme Inhib. J. 2011, 4, 1–10. [Google Scholar] [CrossRef]
- Liaqat, J.; Sumbal, F.; Sabri, A.N. Tetracycline and chloramphenicol efficiency against selected biofilm forming bacteria. Curr. Microbiol. 2009, 59, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Bustos, J.A.; Rodriguez, S.; Jimenez, J.A.; Moyano, L.M.; Castillo, Y.; Ayvar, V.; Allan, J.C.; Craig, P.S.; Gonzales, A.E.; Gilman, R.H.; et al. Detection of Taenia solium taeniasis coproantigen is an early indicator of treatment failure for taeniasis. Clin. Vaccine Immunol. 2012, 19, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Salvana, E.M.T.; King, C.H. Schistosomiasis in travelers and immigrants. Curr. Inf. Dis. Rep. 2008, 10, 42–49. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Foth, B.J.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Mitrowska, K. Przyczyny i skutki zakazu stosowania 5-nitroimidazoli u zwierząt, których tkanki lub produkty przeznaczone są do spożycia przez ludzi. Med. Weter. 2015, 71, 736–742. [Google Scholar]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Ruiz-Postigo, J.A.; Samo, M.; Jannin, J.G. Monitoring the use of nifurtimox-eflornithine combination therapy (NECT) in the treatment of second stage gambiense human African trypanosomiasis. Res. Rep. Trop. Med. 2012, 3, 93–101. [Google Scholar] [CrossRef]
- Mullokandov, E.; Ahn, J.; Szalkiewicz, A.; Babayeva, M. Protein binding drug-drug interaction between warfarin and tizoxanide in human plasma. Austin J. Pharmacol. Ther. 2014, 2, 1038. [Google Scholar]
- Raviraj, J.; Bokkasam, V.K.; Kumar, V.S.; Reddy, U.S.; Suman, V. Radiosensitizers, radioprotectors, and radiation mitigators. Indian J. Dent. Res. 2014, 25, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Asquith, J.C.; Foster, J.L.; Willson, R.L.; Ings, R.; McFadzean, J.A. Metronidazole (“Flagyl”), a radiosensitizer of hypoxic cells. Br. J. Radiol. 1974, 47, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.D.; Greenstock, C.L.; Reuvers, A.P.; McDonald, E.; Dunlop, I. Radiation chemical studies with nitrojurazone as related to its mechanism of radiosensitization. Radiat. Res. 1973, 53, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Guichard, M.; Malaise, E.P. Radiosensitizing effects of misonidazole and SR 2508 on a human melanoma transplanted in nude mice: influence on repair of potentially lethal damage. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 465–468. [Google Scholar] [CrossRef]
- Dische, S.; Saunders, M.I.; Lee, M.E.; Adams, G.E.; Flockhart, I.R. Clinical testing of the radiosensitizer Ro 07–0582: experience with multiple doses. Br. J. Cancer 1977, 35, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Cosmatos, D.; Marcial, V.A.; Fu, K.K.; Rotman, M.; Cooper, J.S.; Ortiz, H.G.; Beitler, J.J.; Abrams, R.A.; Curran, W.J. Results of an RTOG phase III trial (RTOG 85–27) comparing radiotherapy plus etanidazole with radiotherapy alone for locally advanced head and neck carcinomas. Int. J. Radiat. Oncol. Biol. Phys. 1995, 32, 567–576. [Google Scholar] [CrossRef]
- Overgaard, J.; Hansen, H.S.; Overgaard, M.; Bastholt, L.; Berthelsen, A. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck cancer Study (DAHANCA) Protocol 5–85. Radiother. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Kendall, G.C.; Mokhonova, E.I.; Moran, M.; Sejbuk, N.E.; Wang, D.W.; Silva, O.; Wang, R.T.; Martinez, L.; Lu, Q.L.; Damoiseaux, R.; et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci. Transl. Med. 2012, 4, 164ra160. [Google Scholar] [CrossRef] [PubMed]
- Muehlschlegel, S.; Rordorf, G.; Bodock, M.; Sims, J.R. Dantrolene mediates vasorelaxation in cerebrial vasoconstriction: a case series. Neurocrit. Care 2009, 10, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Oo, Y.W.; Gomez-Hurtado, N.; Walweel, K.; van Helden, D.F.; Imtiaz, M.S.; Knollmann, B.C.; Laver, D.R. Essential role of calmodulin in RyR inhibition by dantrolene. Mol. Pharmacol. 2015, 88, 57–63. [Google Scholar] [CrossRef] [PubMed]
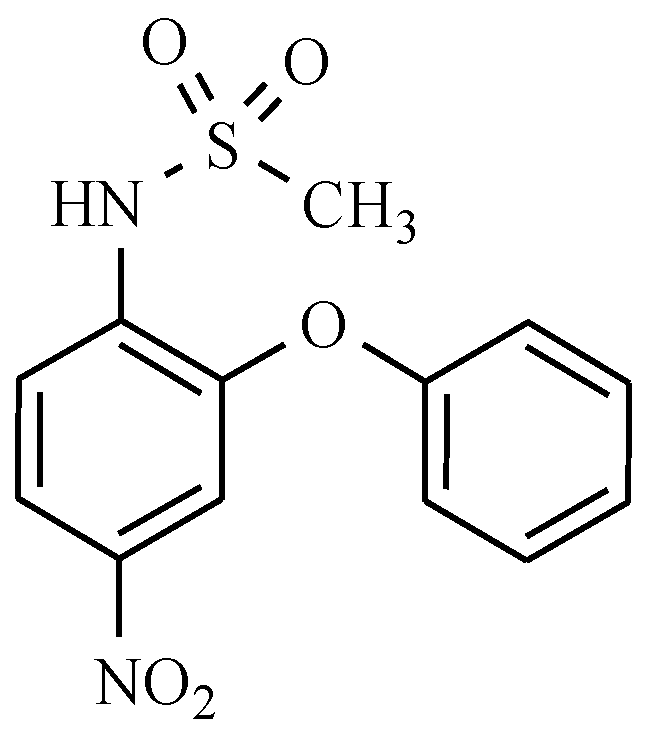
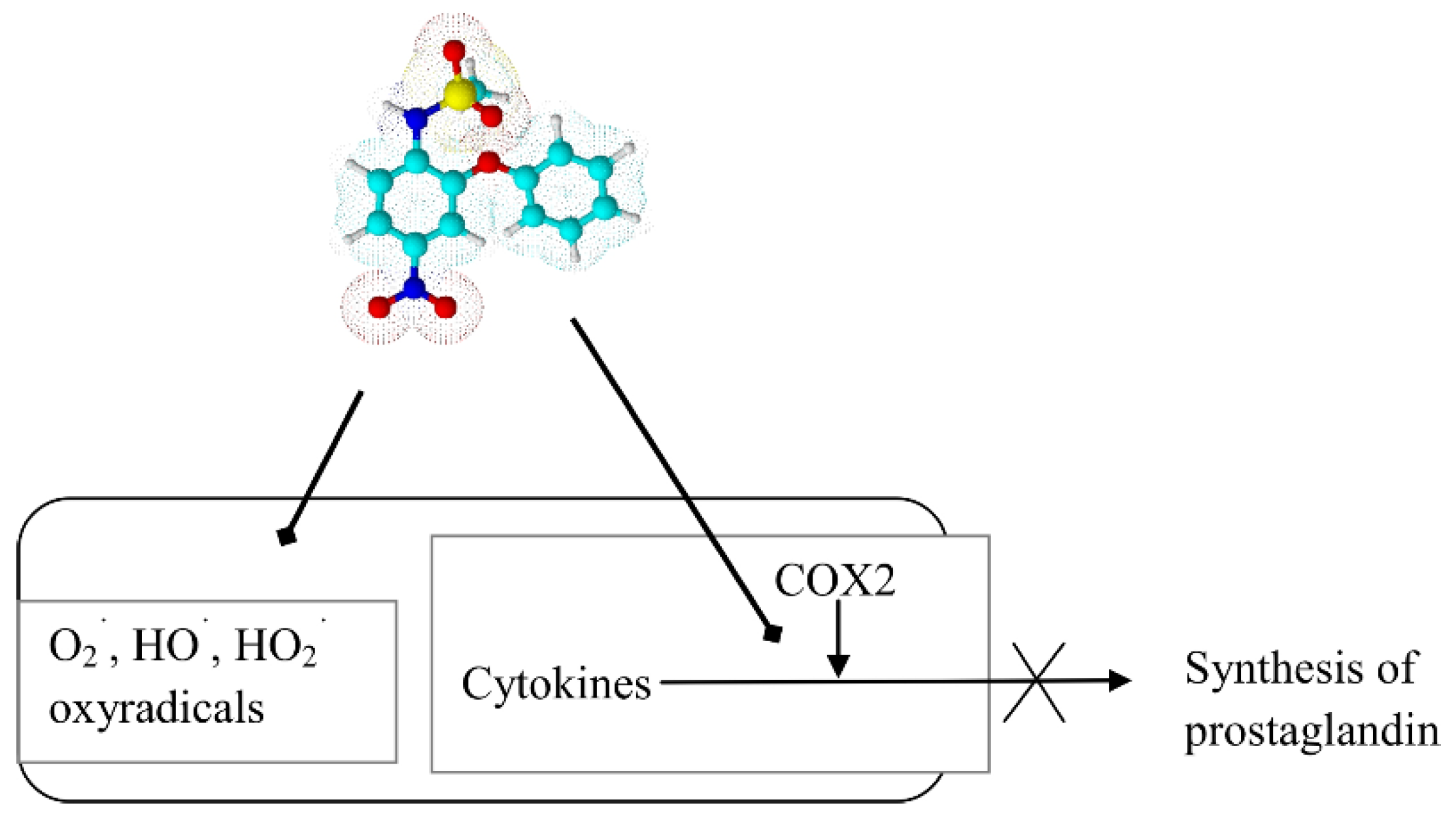
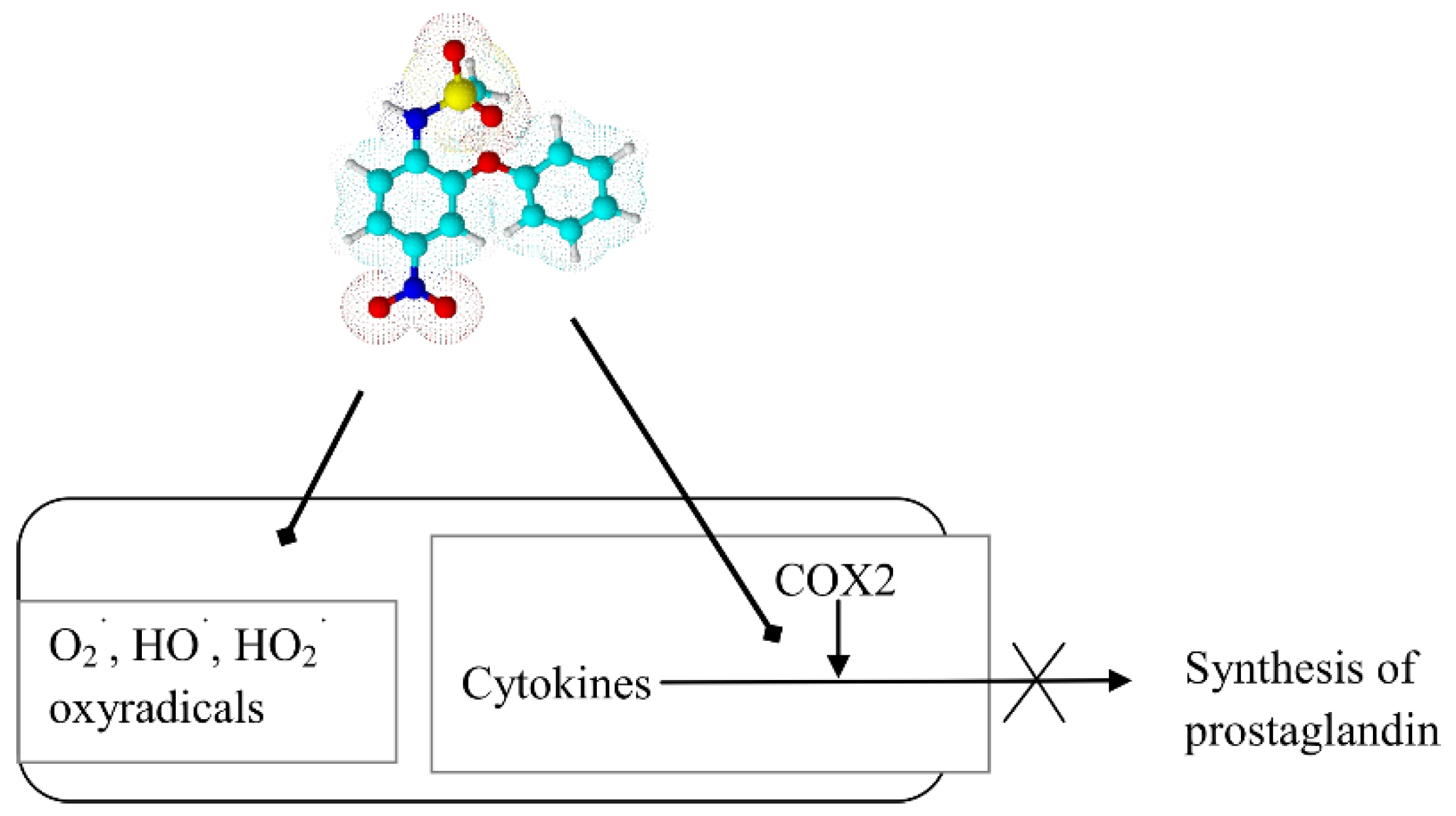
- Rainsford, K.D. Nimesulide—A multifactorial approach to inflammation and pain. Curr. Med. Res. Opin. 2006, 22, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Al-Abd, A.M.; Al-Abbasi, F.A.; Nofal, S.M.; Khalifa, A.E.; Williams, R.O.; El-Eraky, W.I.; Nagy, A.A.; Abdel-Nain, A.B. Nimesulide improves the symptomatic and disease modifying effects of leflunomide in collagen induced arthritis. PLoS ONE 2014, 9, e111843. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.R.; Rangwala, S.M.; Shapiro, J.S.; Rich, A.S.; Rhoades, B.; Qi, Y.; Wang, J.; Rajala, M.W.; Pocai, A.; Scherer, P.E.; et al. Regulation of fasted blood glucose by resistin. Science 2004, 303, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Scherer, P.E. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 2002, 13, 84–89. [Google Scholar] [CrossRef]
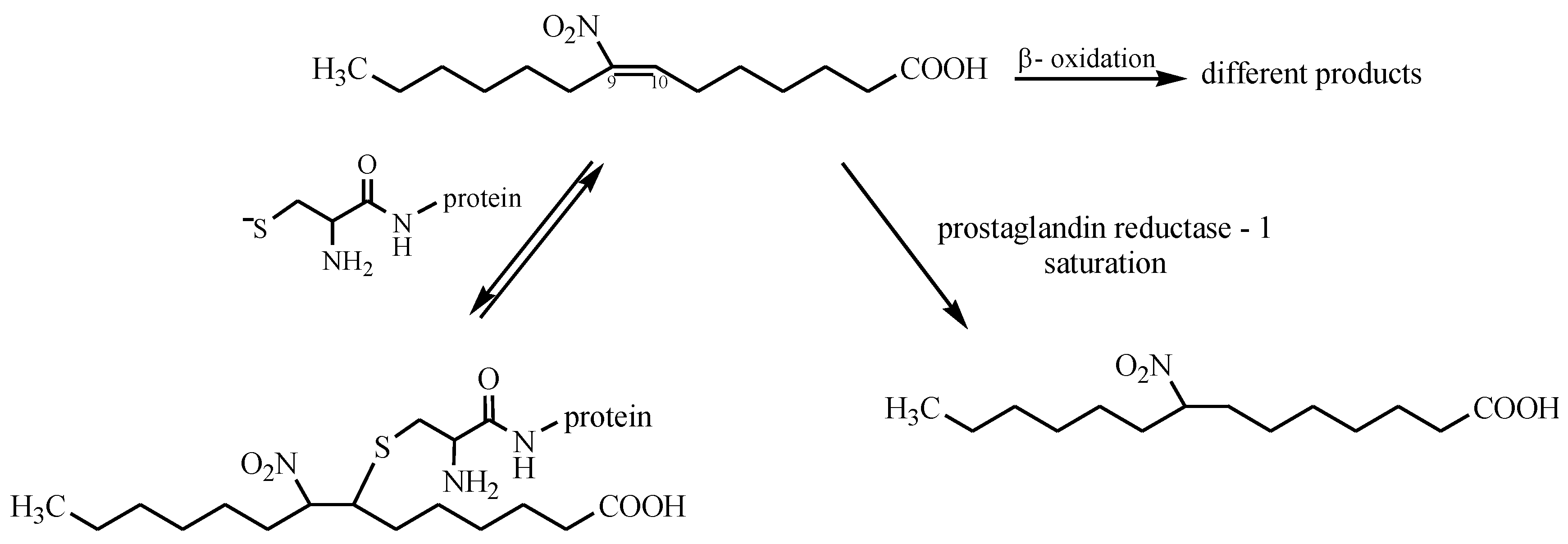
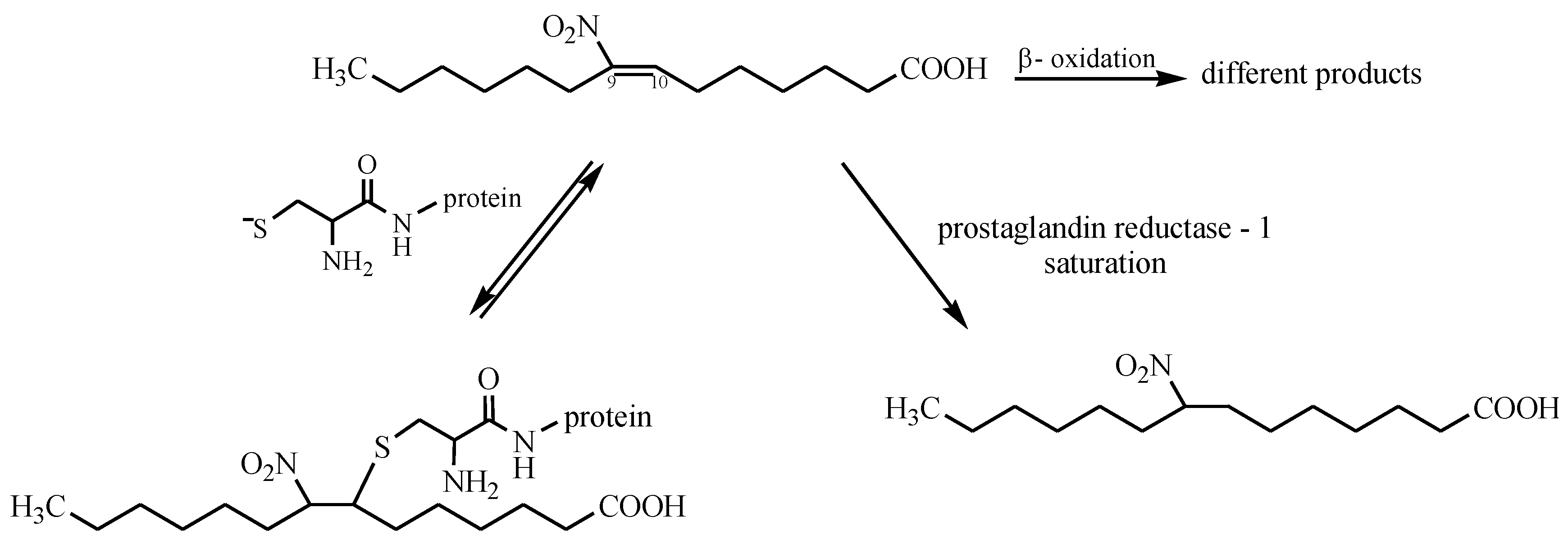
- Villacorta, L.; Gao, Z.; Schopfer, F.J.; Freeman, B.A.; Chen, Y.E. Nitro-fatty acids in cardiovascular regulation and diseases: Characteristics and molecular mechanisms. Front. Biosci. 2016, 21, 873–889. [Google Scholar] [CrossRef]
- Pereira, C.; Ferreira, N.R.; Rocha, B.S.; Barbosa, R.M.; Laranjinha, J. The redox interplay between nitrite and nitric oxide: From the gut to the brain. Redox Biol. 2013, 1, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, H. Nitro-fatty acids: novel anti-inflammatory lipid mediators. Braz. J. Med. Biol. Res. 2013, 46, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.; Turell, L.; Vitturi, D.A.; Coitino, E.L.; Lebrato, L.; Moller, M.N.; Sagasti, C.; Salvatore, S.R.; Woodcock, S.R.; Schopfer, F.J. Thiol addition to conjugated nitrolinoleic acid. FASEB J. 2017, 31 (Supl. 1), 605.1. [Google Scholar]
- Freeman, B.A.; Baker, P.R.S.; Schopfer, F.J.; Woodcock, S.R.; Napolitano, A.; d’Ischia, M. Nitro-fatty acid formation and signaling. J. Biol. Chem. 2008, 283, 15515–15519. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, V.; Schopfer, F.J.; Khoo, N.K.H.; Rudolph, T.K.; Cole, M.P.; Woodcock, S.R.; Bonacci, G.; Groeger, A.L.; Golin-Bisello, F.; Chen, C.S.; et al. Nitro-fatty acid metabolome: saturation, desaturation, β-oxidation, and protein adduction. J. Biol. Chem. 2009, 284, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Mata-Perez, C.; Sanchez-Calvo, B.; Begara-Moralez, J.C.; Padilla, M.N.; Valderrama, R.; Corpas, F.J.; Barroso, J.B. Nitric oxide release from nitro-fatty acids in Arabidopsis roots. Plant Signal Behav. 2016, 11, e1154255. [Google Scholar] [CrossRef] [PubMed]
- Villacorta, L.; Chang, L.; Salvatore, S.R.; Ichikawa, T.; Zhang, J.; Petrovic-Djergovic, D.; Jia, L.; Carlsen, H.; Schopfer, F.J.; Freeman, B.A.; et al. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signaling in lipid rafts. Cardiovasc. Res. 2013, 98, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Wynne, B.M.; Chiao, C.W.; Webb, R.C. Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J. Am. Soc. Hypertens. 2009, 3, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Yang, T. Protection of nitro-fatty acids against kidney diseases. Am. J. Physiol. Renal. Physiol. 2016, 310, F697–F704. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.C.; Bosquesi, P.L.; dos Santos, J.L. A prodrug approach to improve the physico-chemical properties and decrease the genotoxicity of nitro compounds. Curr. Pharm. Des. 2011, 17, 3515–3526. [Google Scholar] [CrossRef] [PubMed]
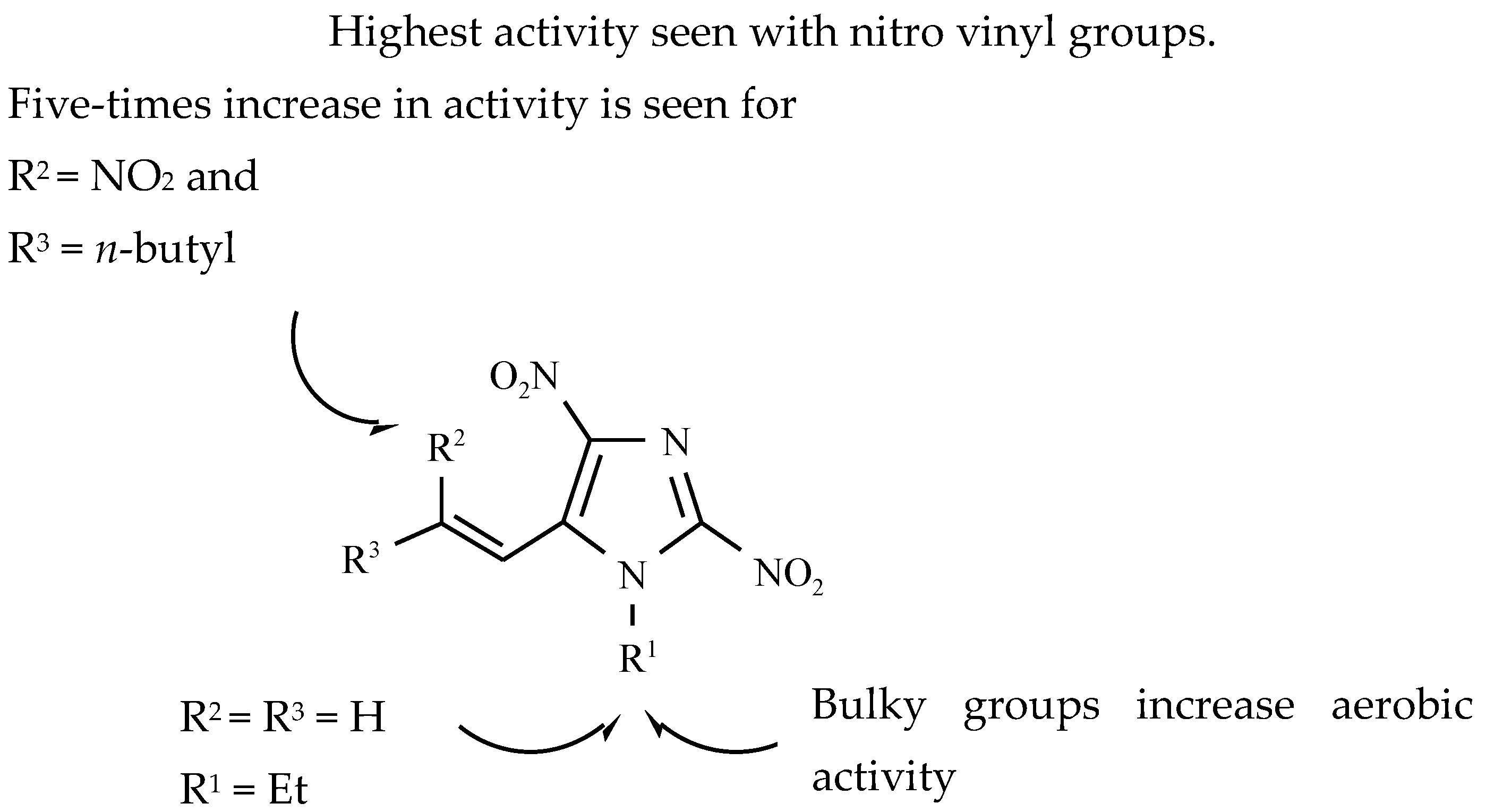

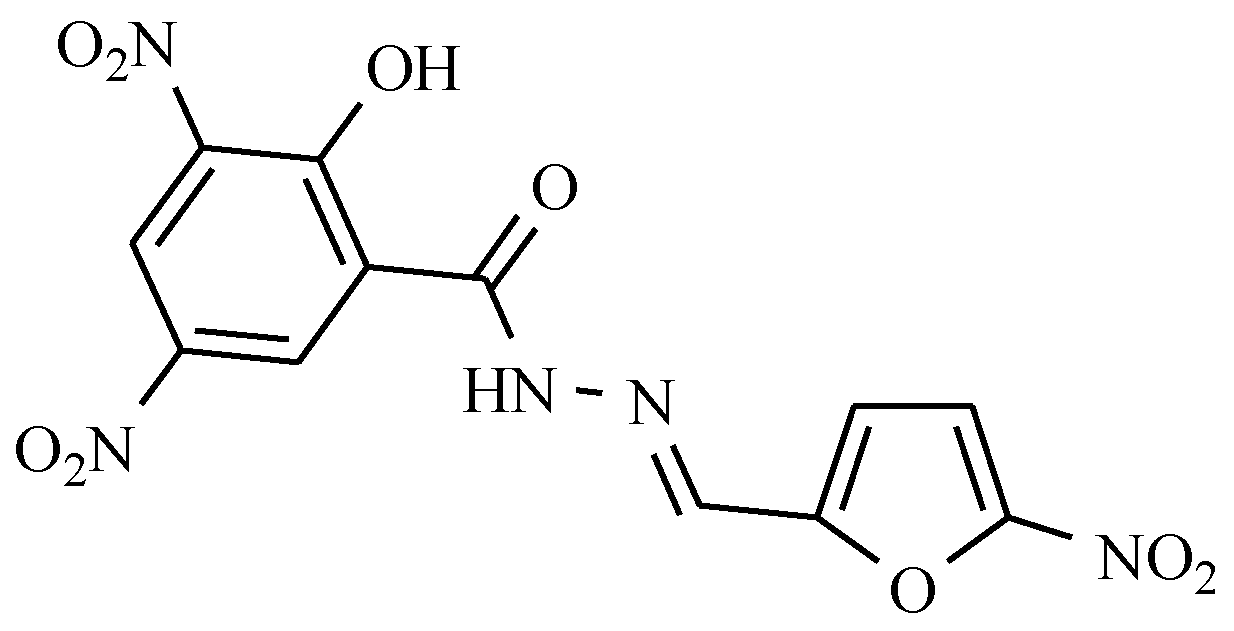
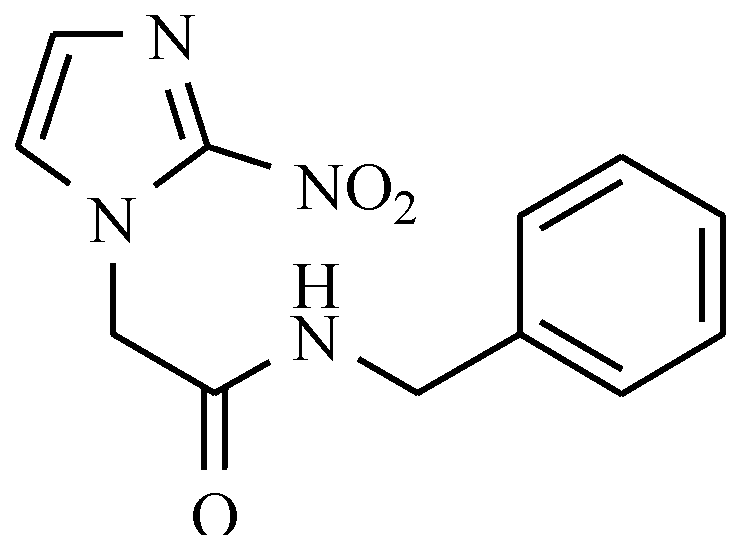
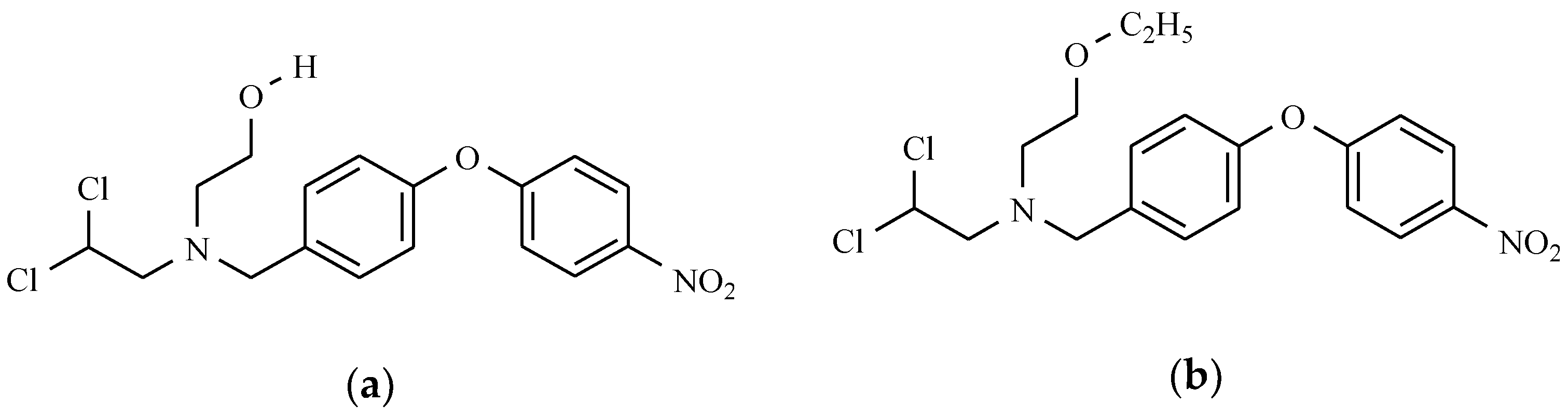
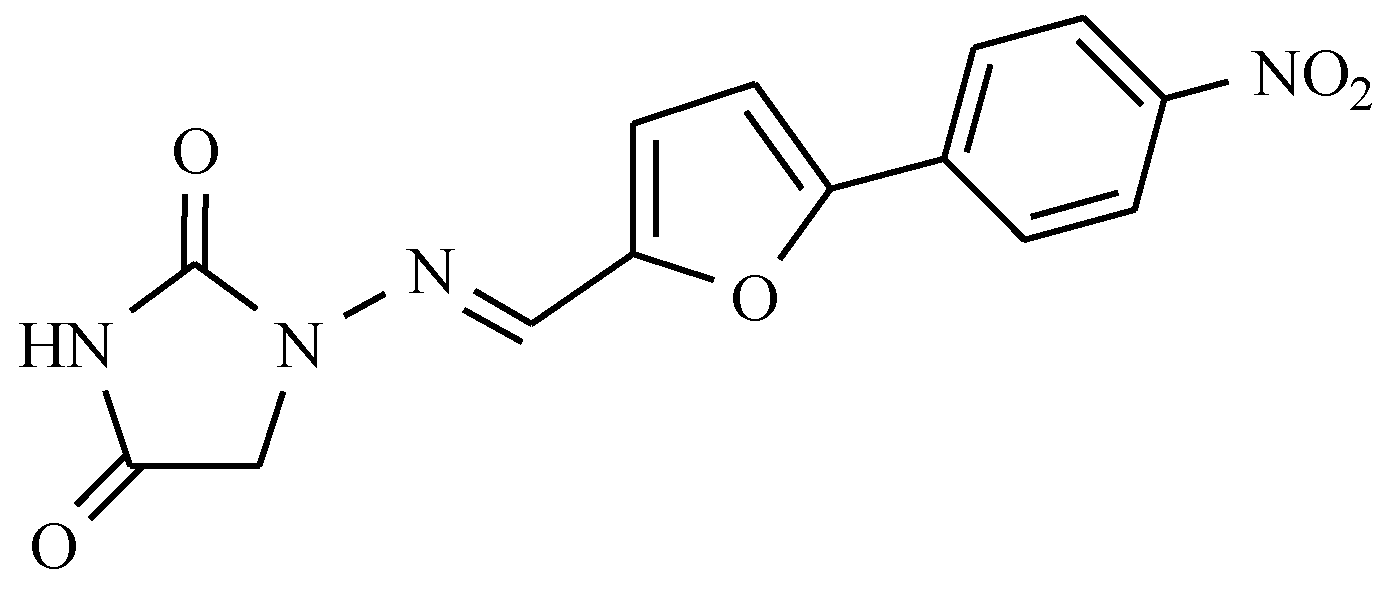

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
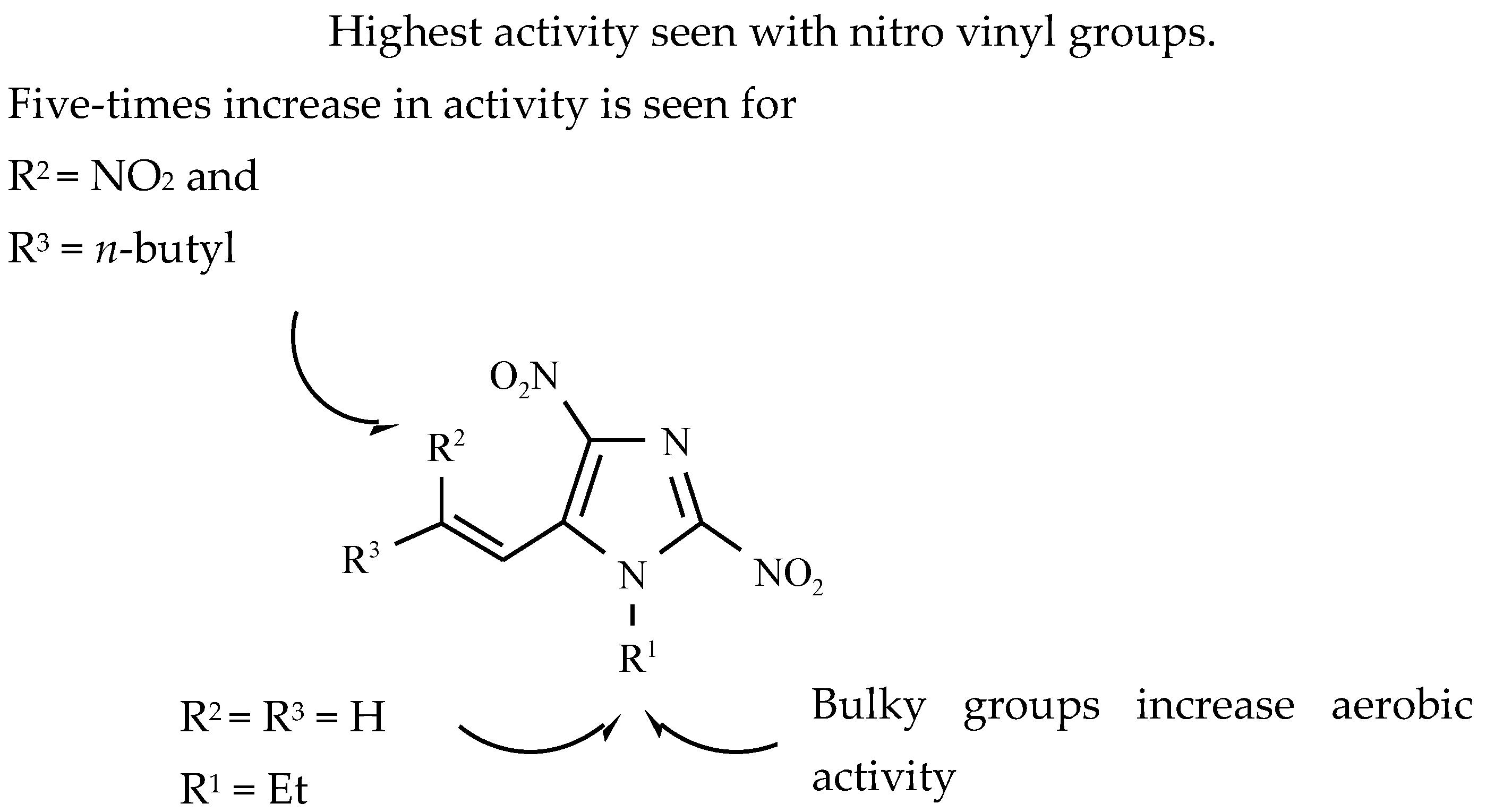{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Structure | Derivative | Nitro Group Position | R1 | R2 | R3 |
|---|---|---|---|---|---|
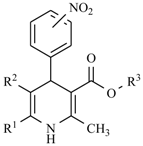 | Aranidipine | ortho | -CH3 | 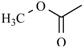 | 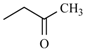 |
| Barnidipine | meta | -CH3 | |  | |
| Benidipine | meta | -CH3 | | 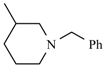 | |
| Cylnidipine | meta | -CH3 | 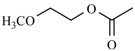 | 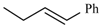 | |
| Efonidipine | meta | -CH3 | 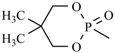 |  | |
| Nifedipine | ortho | -CH3 | | -CH3 | |
| Nicardipine | meta | -CH3 | | | |
| Nilvadipine | meta | -CN | |  | |
| Nisoldipine | ortho | -CH3 | | 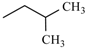 | |
| Nitrendipine | meta | -CH3 | | -CH2CH3 | |
| Pranidipine | meta | -CH3 | | | |
| Nimodipine | meta | -CH3 | 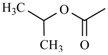 | 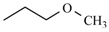 | |
| Manidipine | ortho | -CH3 | | 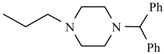 | |
| Falnidipine | ortho | -CH3 | | 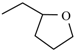 |
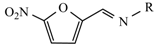 | 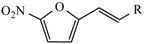 | ||
|---|---|---|---|
| Compound | R | Compound | R |
| Nitrofurantoin | 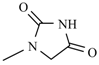 | Furazidine | 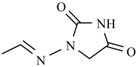 |
| Furazolidone | 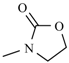 | Nifurzide | 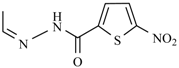 |
| Nifuroxime | -OH | Nitrovin | 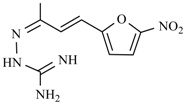 |
| Nifurtoinol | 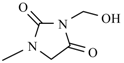 | Nifurmazole | 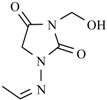 |
| Nifuratel | 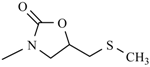 | Nifurizone | 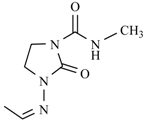 |
| Nifuradene | 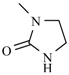 | Nifurvidine | 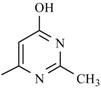 |
| Nifurimide | 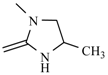 | Nifuralide | 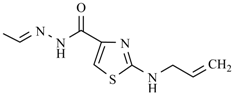 |
| Nifurtimox | 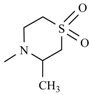 | Nifurpirinol | 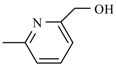 |
| Furaltadone | 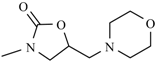 | Nifurprazine |  |
| Nifurfoline |  | ||
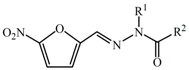 | 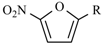 | |||
|---|---|---|---|---|
| Compound | R1 | R2 | Compound | R |
| Nifuroxazide | -H | 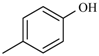 | Nifurthiazole | 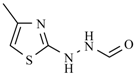 |
| Nifurpipone | -H | 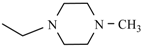 | Nifuratrone | 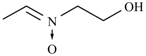 |
| Nifuraldezone | -H | 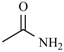 | Furazolium | 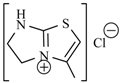 |
| Nifurethazone | -(CH2)2N(CH3)2 | -NH2 | Nifuroquine | 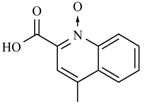 |
| Nitrofural | -H | -NH2 | ||
| Nidroxyzone | -(CH2)2OH | -NH2 | ||
| Nihydrazone | -H | -CH3 | ||
| General Structure | Derivative | R1 | R2 |
|---|---|---|---|
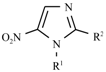 | Metronidazole |  | -CH3 |
| Nimorazole | 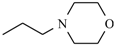 | -H | |
| Ornidazole |  | -CH3 | |
| Secnidazole | 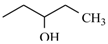 | -CH3 | |
| Tinidazole | 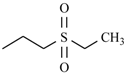 | -CH3 | |
| Ipronidazole | -CH3 | 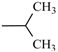 | |
| Azanidazole | -CH3 | 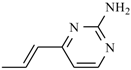 | |
| Megazole | -CH3 | 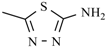 | |
| Propenidazole | -CH3 |  |
| Main Structure Caption | Derivative | R |
|---|---|---|
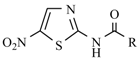 | Aminitrazole | -CH3 |
| Nithiazide | -NH-CH2-CH3 | |
| Tenonitrozole |  | |
| Tizoxanide | 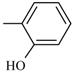 | |
| Nitazoxanide | 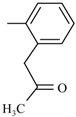 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olender, D.; Żwawiak, J.; Zaprutko, L. Multidirectional Efficacy of Biologically Active Nitro Compounds Included in Medicines. Pharmaceuticals 2018, 11, 54. https://doi.org/10.3390/ph11020054
Olender D, Żwawiak J, Zaprutko L. Multidirectional Efficacy of Biologically Active Nitro Compounds Included in Medicines. Pharmaceuticals. 2018; 11(2):54. https://doi.org/10.3390/ph11020054
Chicago/Turabian StyleOlender, Dorota, Justyna Żwawiak, and Lucjusz Zaprutko. 2018. "Multidirectional Efficacy of Biologically Active Nitro Compounds Included in Medicines" Pharmaceuticals 11, no. 2: 54. https://doi.org/10.3390/ph11020054
APA StyleOlender, D., Żwawiak, J., & Zaprutko, L. (2018). Multidirectional Efficacy of Biologically Active Nitro Compounds Included in Medicines. Pharmaceuticals, 11(2), 54. https://doi.org/10.3390/ph11020054