Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP



 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Motor Behavioural Test: β-Caryophyllene Inhibits Chronic MPTP-Induced Defects in Motor Coordination via the CB2 Receptor
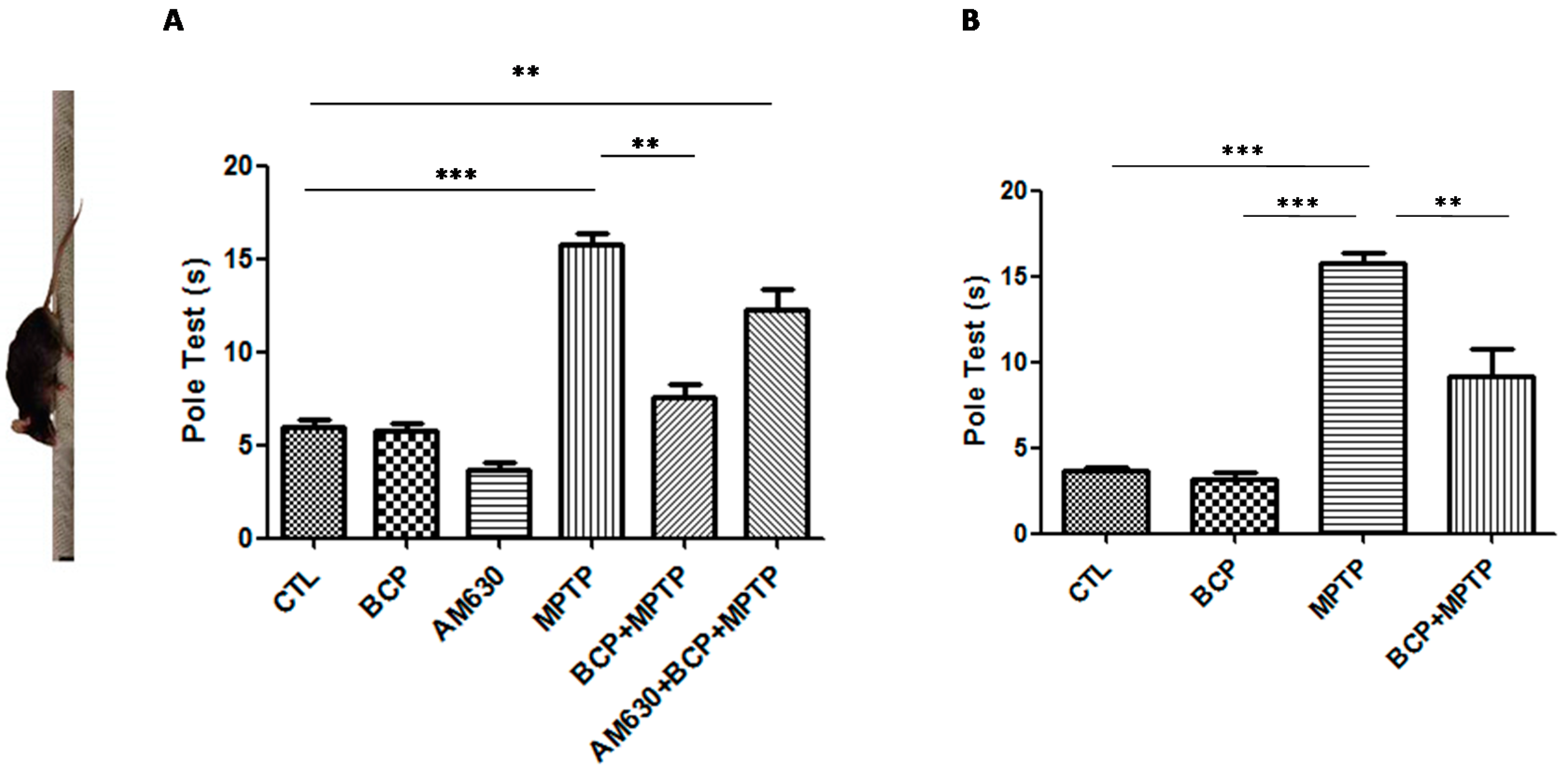
2.1.1. Pole Test Analysis
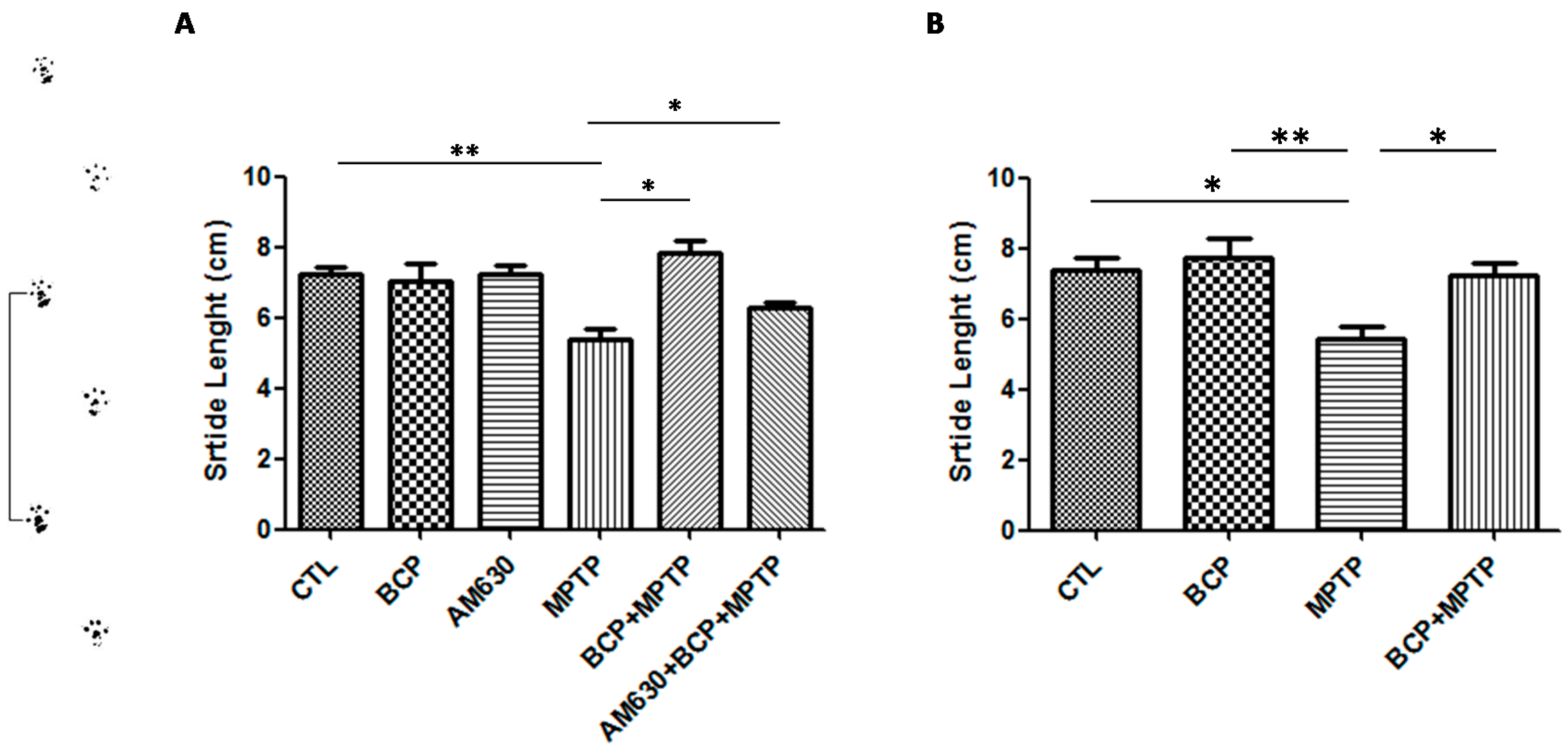
2.1.2. Gait Test Analysis
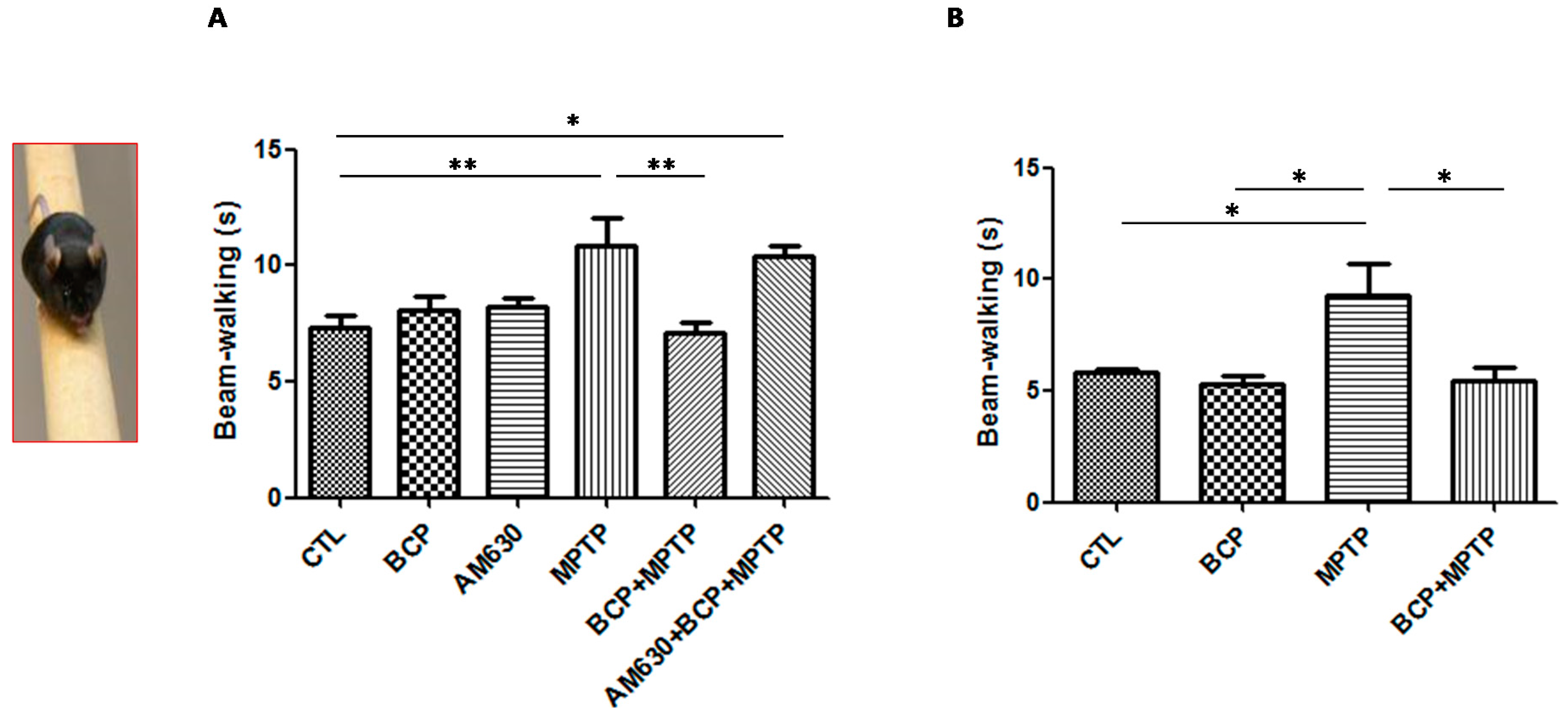
2.1.3. Beam Test
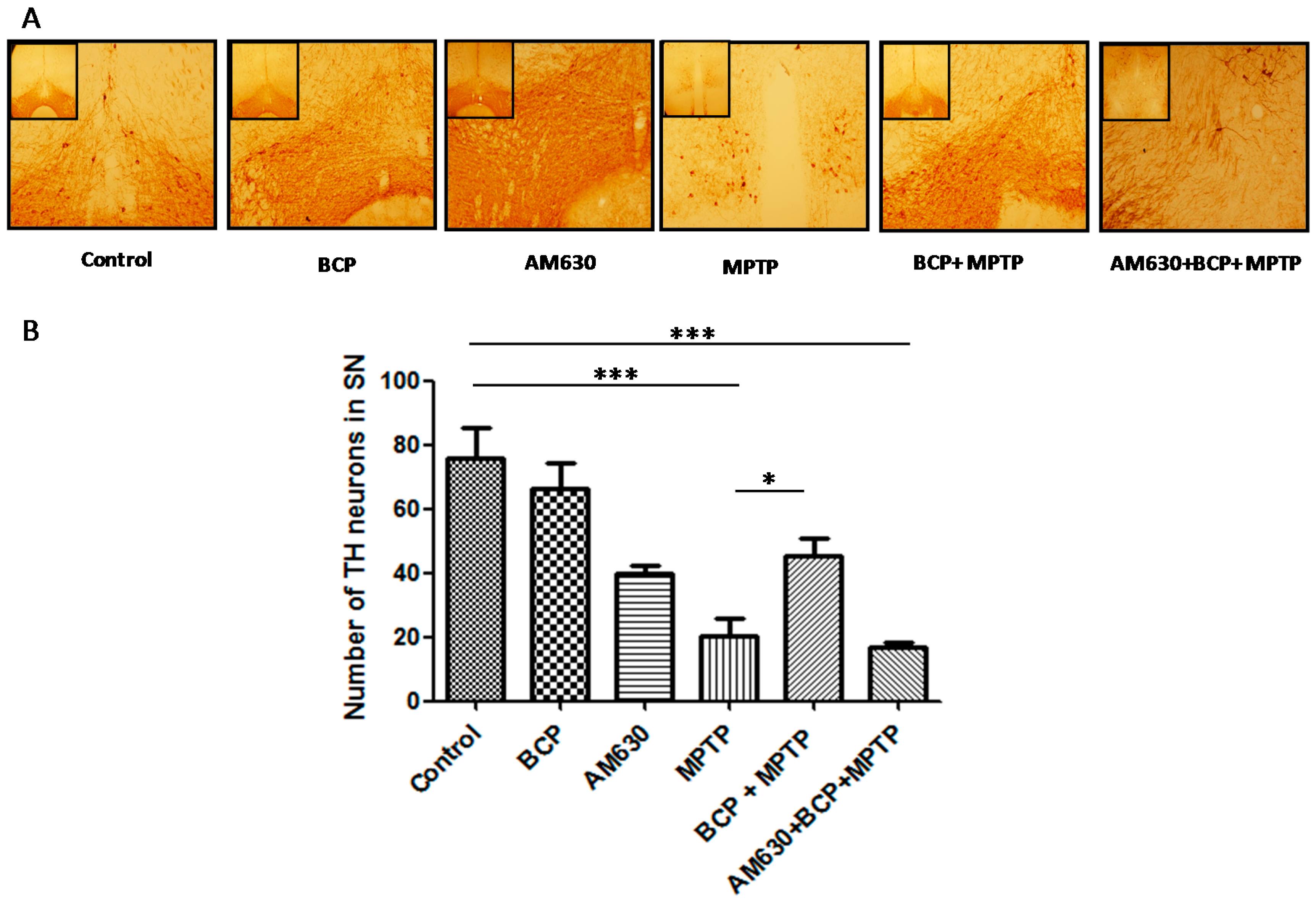
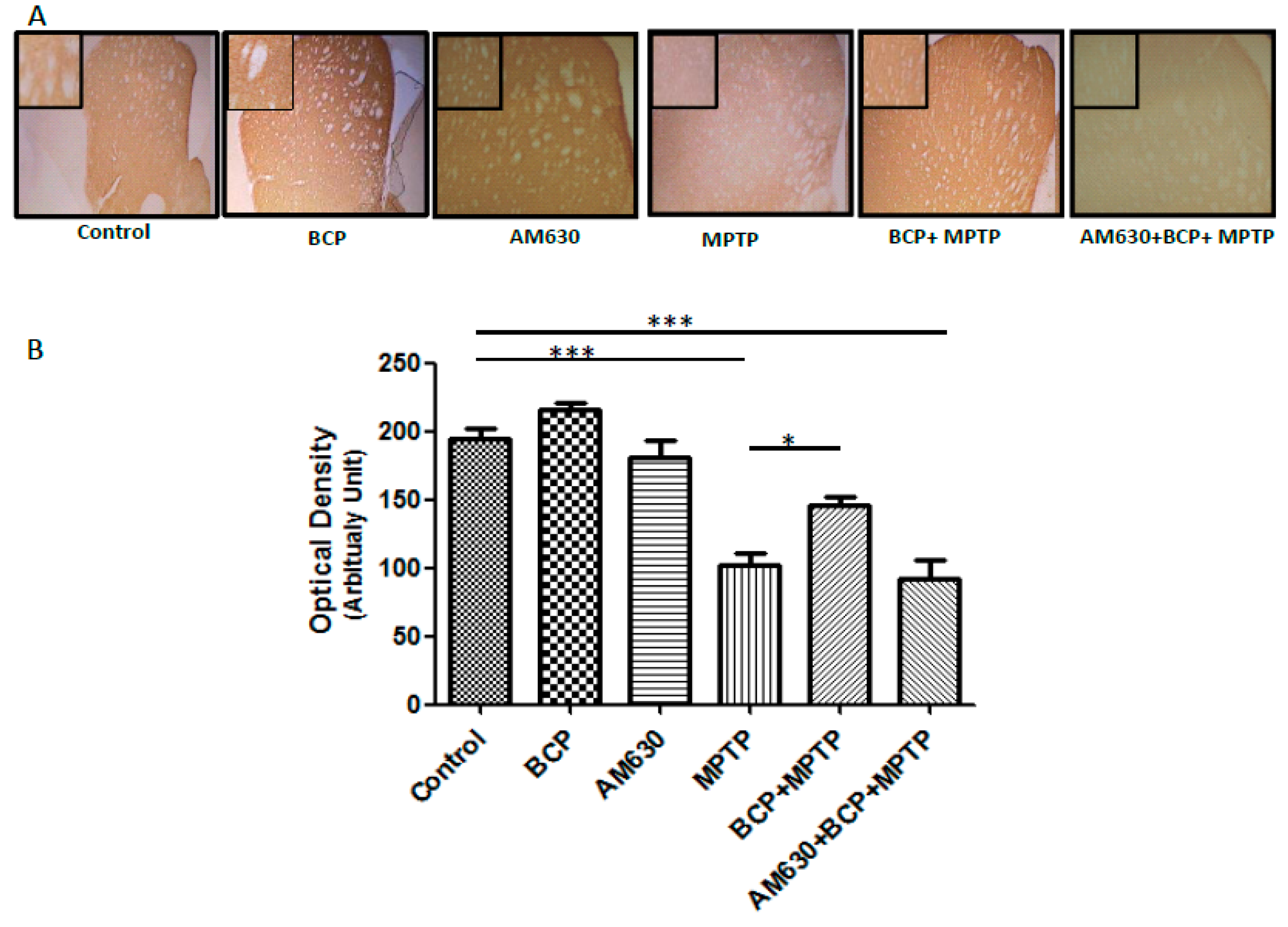
2.2. β-Caryophyllene Prevents Chronic MPTP-Induced Dopaminergic Neuron Loss in the SNpc and STR via the CB2 Receptor
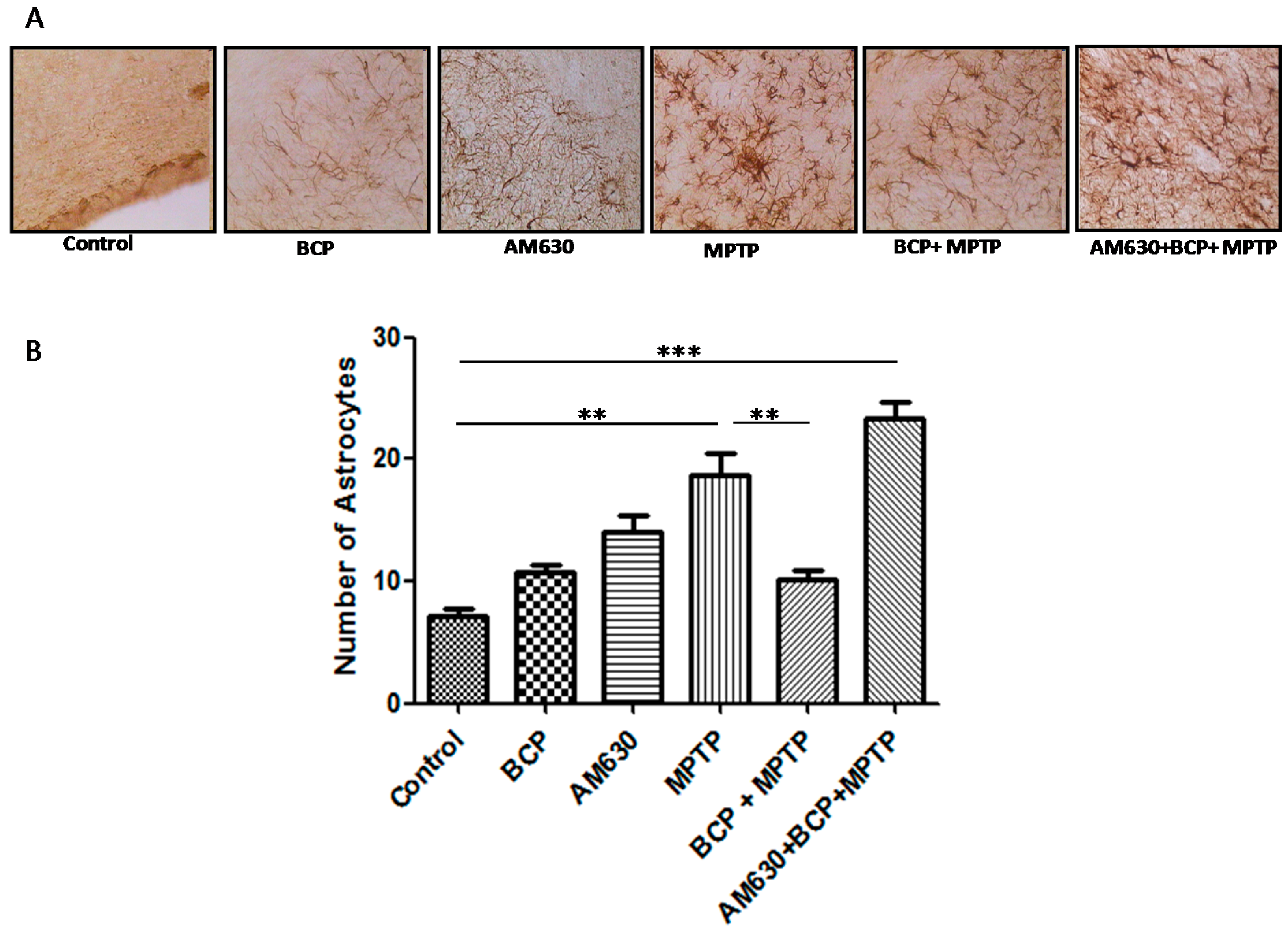
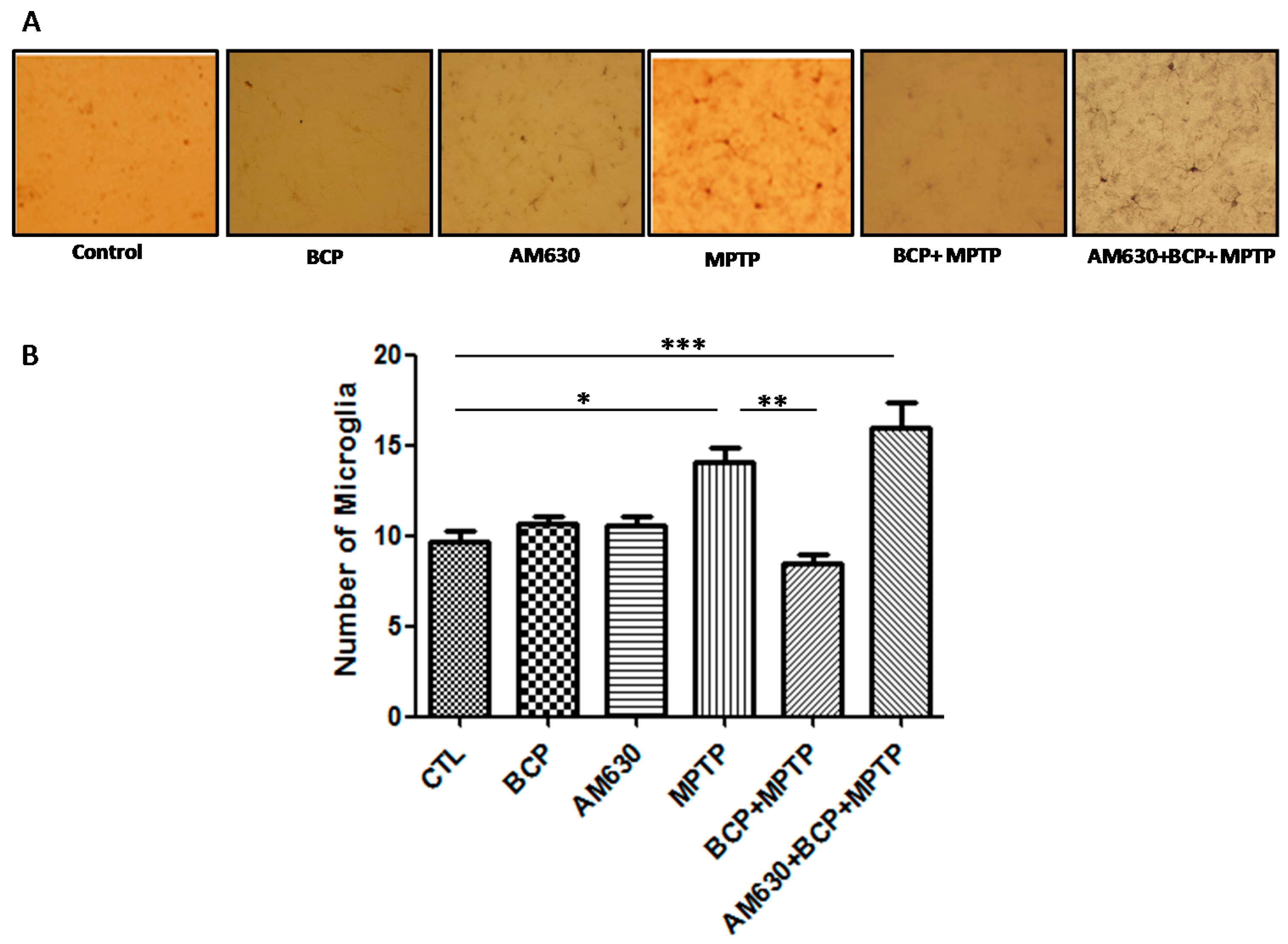
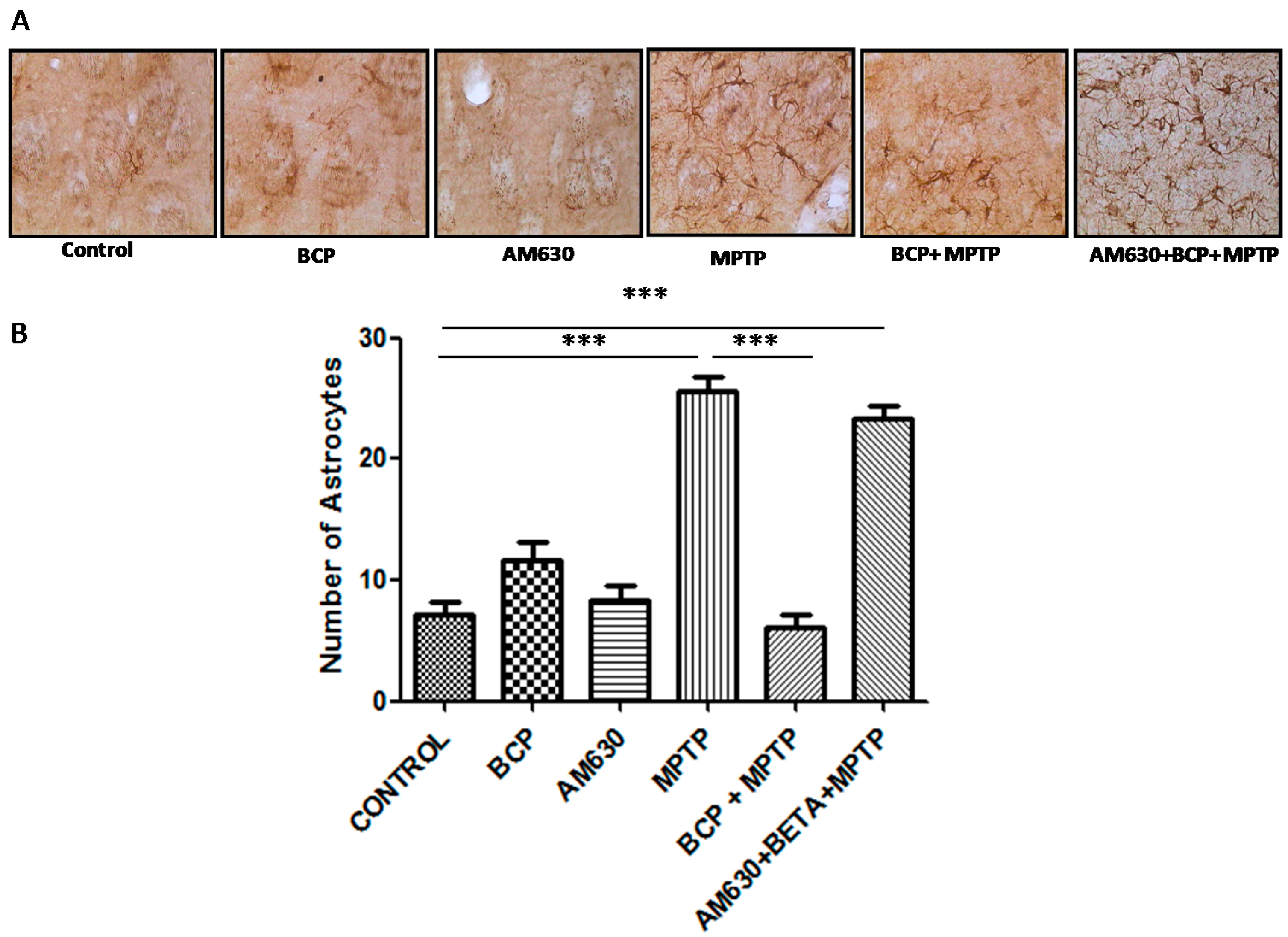
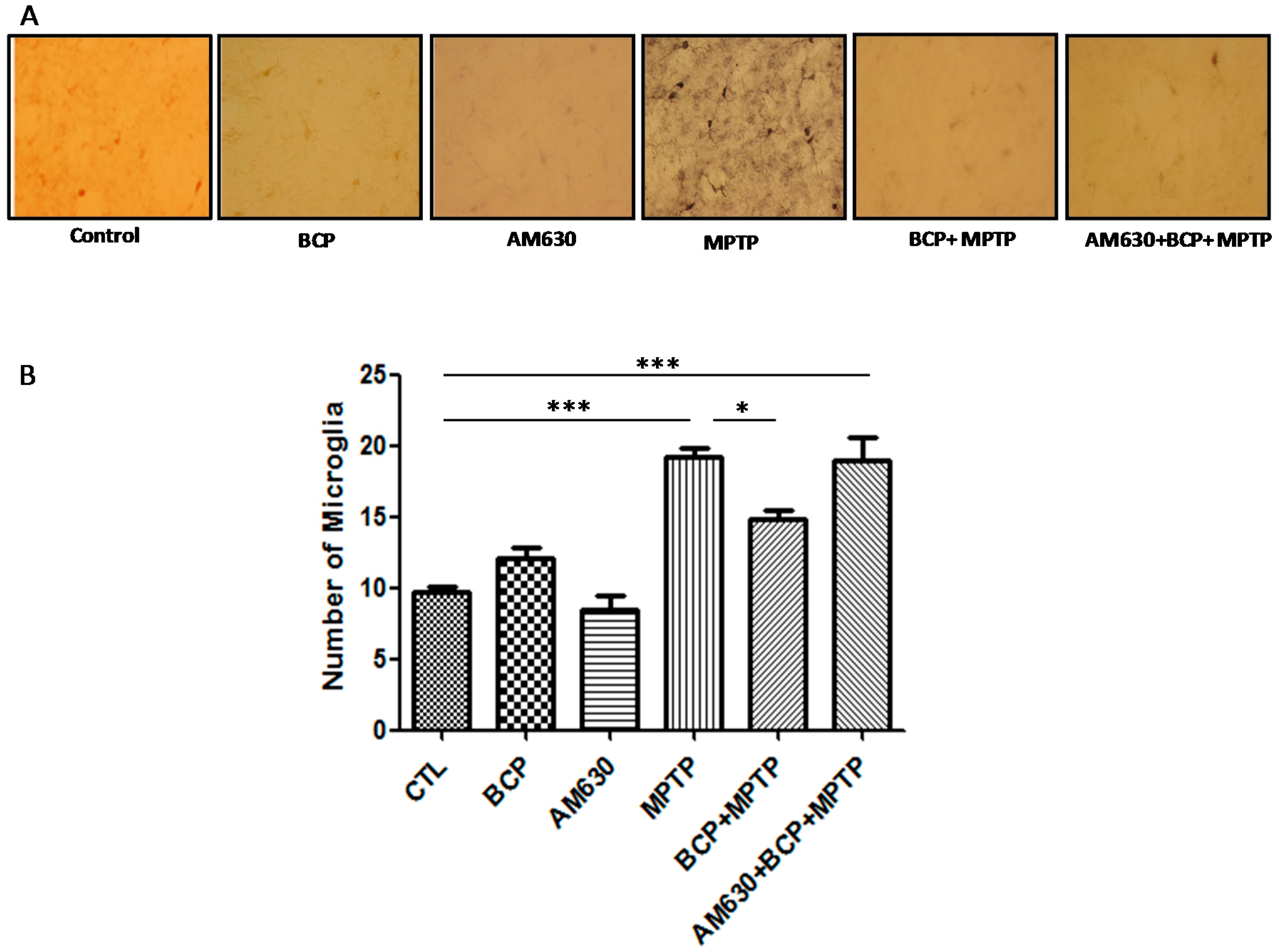
2.3. β-Caryophyllene Inhibits Chronic MPTP-Induced Astrocyte and Microglia Activation in the SNpc and STR via the CB2 Receptor
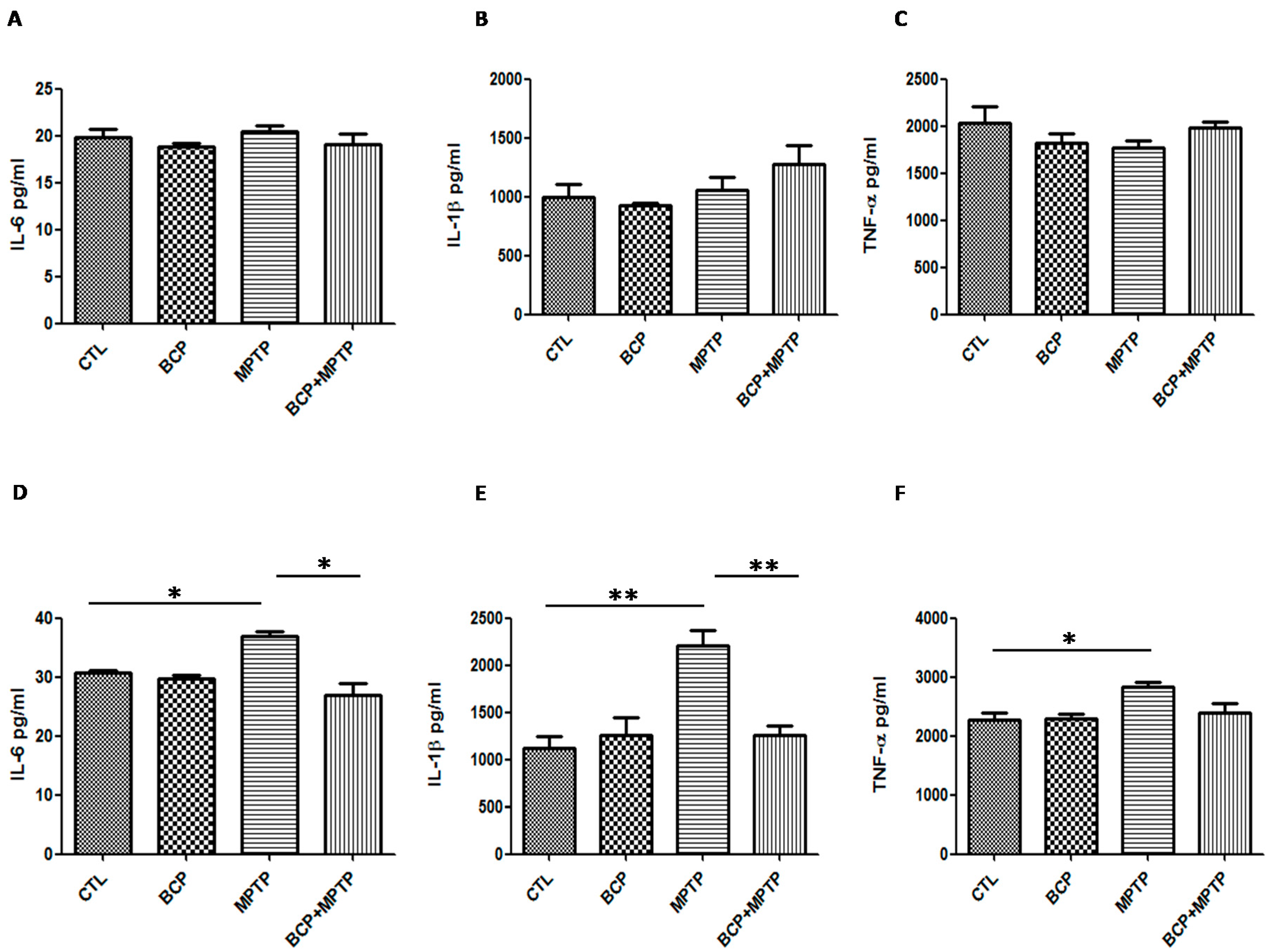
2.4. β-Caryophyllene Inhibits the Levels of Inflammatory Cytokines in the Nigrostriatal System
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drug Administration Schemes
4.3. Behavioural Studies
4.3.1. Pole Test
4.3.2. Gait Test
4.3.3. Beam Test
4.4. Measurement of the IL-1β, TNF-α and IL-6 Cytokines
4.5. Immunohistochemistry of Tyrosine Hydroxylase, GFAP and Iba
4.6. Tyrosine Hydroxylase Assessment
4.7. GFAP and Iba Assessment
5. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| BCP | β-caryophyllene |
| CB1R | type 1 cannabinoid receptor |
| CB2R | type 2 cannabinoid receptor |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| GFAP | glial fibrillary acidic protein |
| TH | tyrosine hydroxylase |
| CNS | central nervous system |
| NO | nitric oxide |
| ROS | oxygen species |
| GDNF | line-derived neurotrophic factor (GDNF) |
| CNTF | ciliary neurotrophic factor |
| SNpc | substantia nigra pars compacta |
| STR | striatum |
References
- Nutt, J.G.; Wooten, G.F. Diagnosis and initial management of Parkinson’s disease. N. Engl. J. Med. 2005, 353, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Jankovic, J. Genetic testing in Parkinson disease: Promises and pitfalls. Arch. Neurol. 2006, 63, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.; Noble, E.; Jones, D.; Burn, D. Hard to swallow: Dysphagia in Parkinson’s disease. Age Ageing 2006, 35, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Hong, J.S.; Zhang, W.; Liu, B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 2002, 22, 782–790. [Google Scholar] [PubMed]
- Chung, Y.C.; Kim, S.R.; Jin, B.K. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson’s disease. J. Immunol. 2010, 185, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–S120. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Pott-Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Bok, E.; Huh, S.H.; Park, J.Y.; Yoon, S.H.; Kim, S.R.; Kim, Y.S.; Maeng, S.; Park, S.H.; Jin, B.K. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 2011, 187, 6508–6517. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.A.; Griffin-Thomas, L. Emerging role of the cannabinoid receptor CB2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11:e3. [Google Scholar] [CrossRef] [PubMed]
- Zarruk, J.G.; Fernandez-Lopez, D.; Garcia-Yebenes, I.; Garcia-Gutierrez, M.S.; Vivancos, J.; Nombela, F.; Torres, M.; Burguete, M.C.; Manzanares, J.; Lizasoain, I.; et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 2012, 43, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Concannon, R.M.; Okine, B.N.; Finn, D.P.; Dowd, E. Differential upregulation of the cannabinoid CB2 receptor in neurotoxic and inflammation-driven rat models of Parkinson’s disease. Exp. Neurol. 2015, 269, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Martinez, A.A.; Seillier, A.; Koek, W.; Acosta, Y.; Fernández, E.; Strong, R.; Lutz, B.; Marsicano, G.; Roberts, J.L.; et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB2 receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Opdyke, D.L. Monographs on fragrance raw materials. Food Cosmet. Toxicol. 1973, 11, 1011–1081. [Google Scholar] [CrossRef]
- Lourens, A.C.; Reddy, D.; Baser, K.H.; Viljoen, A.M.; Van Vuuren, S.F. In vitro biological activity and essential oil composition of four indigenous South African Helichrysum species. J. Ethnopharmacol. 2004, 95, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Marimuthu, P.; de Heluani, C.S.; Catalan, C.A. Antioxidant and biocidal activities of Carumnigrum (seed) essential oil, oleoresin, and their selected components. J. Agric. Food Chem. 2006, 54, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kubo, I.; Chaudhuri, S.K.; Kubo, Y.; Sanchez, Y.; Ogura, T.; Saito, T.; Ishikawa, H.; Haraguchi, H. Cytotoxic and antioxidative sesquiterpenoids from Heterothecainuloides. Planta Med. 1996, 62, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, P.A.; Barry, B.W. Sesquiterpene components of volatile oils as skin penetration enhancers for the hydrophilic permeant 5-fluorouracil. J. Pharm. Pharmacol. 1994, 46, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.B.; Rangari, V.D. Phytochemical investigation and evaluation of anti-inflammatory and anti-arthritic activities of essential oil of Strobilanthus ixiocephala Benth. Indian J. Exp. Biol. 2003, 41, 890–894. [Google Scholar] [PubMed]
- Baricevic, D.; Sosa, S.; Della, L.R.; Tubaro, A.; Simonovska, B.; Krasna, A.; Zupancic, A. Topical anti-inflammatory activity of Salvia officinalis L. leaves: The relevance of ursolic acid. J. Ethnopharmacol. 2001, 75, 125–132. [Google Scholar] [CrossRef]
- Cho, J.Y.; Chang, H.J.; Lee, S.K.; Kim, H.J.; Hwang, J.K.; Chun, H.S. Amelioration of dextran sulfate sodium-induced colitis in mice by oral administration of β-caryophyllene, a sesquiterpene. Life Sci. 2007, 80, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Finazzi-Agrò, A.; Bernardi, G.; Maccarrone, M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol. Sci. 2007, 28, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisil, F.; Mein, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.Z.; Xie, X.Q.; Altmann, K.H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA. 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, H.; Suzuki, H.; Ueda, Y.; Kaya, T.; Inaba, T. In vitro andin vivopharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J. Pharmacol. Exp. Ther. 2001, 296, 420–425. [Google Scholar] [PubMed]
- Ibrahim, S.M.; Mix, E.; Böttcher, T.; Koczan, D.; Gold, R.; Rolfs, A.; Thiesen, H.J. Gene expression profiling of the nervous system in murine experimental autoimmune encephalomyelitis. Brain 2001, 124, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Batkai, S.; Osei-Hyiaman, D.; Pan, H.; El-Assal, O.; Rajesh, M.; Mukhopadhyay, P.; Hong, F.; Harvey-White, J.; Jafri, A.; Hasko, G.; et al. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J. 2007, 21, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, B.G.; Blázquez, C.; Gómez del Pulgar, T.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Wotherspoon, G.; Fox, A.; McIntyre, P.; Colley, S.; Bevan, S.; Winter, J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience 2005, 135, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Jean-Gilles, L.; Gran, B.; Constantinescu, C.S. Interaction between Cytokines, Cannabinoids and the Nervous System. Immunobiology 2010, 215, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Jackson-Lewis, V.; Guegan, C.; Wu, D.C.; Teismann, P.; Choi, D.K.; Tieu, K.; Przedborski, S. The role of glial cells in Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Cohen, O.; Choi, D.K.; Wu, D.C.; Marks, D.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Pathogenic role of glial cells in Parkinson’s disease. Mov. Disord. 2003, 18, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; O’Garra, A.; de Waal-Malefyt, R.; Vieira, P.; Mosmann, T.R. Interleukin-10. Annu. Rev. Immunol. 1993, 11, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Sieling, P.A.; Abrams, J.S.; Yamamura, M.; Salgame, P.; Bloom, B.R.; Rea, T.H.; Modlin, R.L. Immunosuppressive roles for IL-10 and IL-4 in human infection. In vitro modulation of T cell responses in leprosy. J. Immunol. 1993, 150, 5501–5510. [Google Scholar] [PubMed]
- Suzumura, A.; Takeuchi, H.; Zhang, G.; Kuno, R.; Mizuno, T. Roles of glia-derived cytokines on neuronal degeneration and regeneration. Ann. N. Y. Acad. Sci. 2006, 1088, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.J.; Castano, A.; Venero, J.L.; Cano, J.; Machado, A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol. Dis. 2000, 7, 429–447. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [PubMed]
- Mechoulam, R.; Spatz, M.; Shohami, E. Endocannabinoids and neuroprotection. Sci. STKE 2002, 129, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.; Breuer, A.; Mechoulam, R. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Aste-Amezaga, M.; Ma, X.; Sartori, A.; Trinchieri, G. Molecular mechanisms of the induction of IL-12 and its inhibition by IL-10. J. Immunol. 1998, 160, 5936–5944. [Google Scholar] [PubMed]
- Giralt, A.; Friedman, H.C.; Caneda-Ferron, B.; Urban, N.; Moreno, E.; Rubio, N.; Blanco, J.; Peterson, A.; Canals, J.M.; Alberch, J. BDNF regulation under GFAP promoter provides engineered astrocytes as a new approach for long-term protection in Huntington’s disease. Gene Ther. 2010, 17, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Houeland, G.; Romani, A.; Marchetti, C.; Amato, G.; Capsoni, S.; Cattaneo, A.; Marie, H. Transgenic mice with chronic NGF deprivation and Alzheimer’s disease-like pathology display hippocampal region-specific impairments in short- and long-term plasticities. J. Neurosci. 2010, 30, 13089–13094. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Yoo, K.Y.; Kim, D.W.; Lee, B.H.; Kang, T.C.; Choi, S.Y.; Han, B.H.; Kim, J.S.; Won, M.H. Ischemia-related changes of glial-derived neurotrophic factor and phosphatidylinositol 3-kinase in the hippocampus: Their possible correlation in astrocytes. Brain Res. 2006, 1072, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Gao, J.; Li, W.; Cai, D. Neurotrophic and neurorescue effects of echinacoside in the subacute MPTP mouse model of Parkinson’s disease. Brain Res. 2010, 1346, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.H.; Park, E.S.; Won, S.Y.; Lee, Y.A.; Kim, K.I.; Jeong, J.Y.; Baek, J.Y.; Cho, E.J.; Jin, M.; Chung, Y.C.; et al. TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson’s disease via CNTF. Brain 2015, 138, 3610–3622. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, R.E.; Sonsalla, P.K. The MPTP-treated mouse as a model of parkinsonism: How good is it? Neurochem. Int. 1992, 20, 299S–303S. [Google Scholar] [CrossRef]
- Fredriksson, A.; Eriksson, P.; Archer, T. MPTP-induced deficits in motor activity: Neuroprotective effects of the spintrapping agent, alpha-phenyl-tert-butyl-nitrone (PBN). J. Neural Transm. (Vienna) 1997, 104, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Rozas, G.; Lopez-Martin, E.; Guerra, M.J.; Labandeira-Garcia, J.L. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J. Neurosci. Methods 1998, 83, 165–175. [Google Scholar] [CrossRef]
- Spooren, W.P.; Vassout, A.; Waldmeier, P.; Gentsch, C. Differences in pre- and post-synaptic sensitivity to apomorphine between saline and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57BL/6 mice as reflected in climbing activity. Eur. J. Pharmacol. 1998, 353, 1–4. [Google Scholar] [CrossRef]
- Sedelis, M.; Schwarting, R.K.; Huston, J.P. Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef]
- Tillerson, J.L.; Caudle, W.M.; Reveron, M.E.; Miller, G.W. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp. Neurol. 2002, 178, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Cho, K.H.; Shin, M.S.; Lee, J.M.; Cho, H.S.; Kim, C.J.; Shin, D.H.; Yang, H.J. Berberine prevents nigrostriatal dopaminergic neuronal loss and suppresses hippocampal apoptosis in mice with Parkinson’s disease. Int. J. Mol. Med. 2014, 33, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Von-Bohlen Und Halbach, O. Modeling neurodegenerative diseases in vivo review. Neurodegener. Dis. 2005, 2, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Schintu, N.; Frau, L.; Ibba, M.; Garau, A.; Carboni, E.; Carta, A.R. Progressive dopaminergic degeneration in the chronic MPTPp mouse model of Parkinson’s disease. Neurotox. Res. 2009, 16, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Tillerson, J.L.; Miller, G.W. Grid performance test to measure behavioral impairment in the MPTP-treated-mouse model of parkinsonism. J. Neurosci. Methods 2003, 123, 189–200. [Google Scholar] [CrossRef]
- Kregiel, J.; Malek, N.; Popik, P.; Starowicz, K.; Rygula, R. Anandamide mediates cognitive judgement bias in rats. Neuropharmacology 2016, 101, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, H.; Yokoyama, H.; Kimoto, H.; Kato, H.; Araki, T. Biochemical alterations of the striatum in an MPTP-treated mouse model of Parkinson’s disease. Metab. Brain Dis. 2010, 25, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Antzoulatos, E.; Jakowec, M.W.; Petzinger, G.M.; Wood, R.I. Sex differences in motor behavior in the MPTP mouse model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2010, 95, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Fernagut, P.O.; Diguet, E.; Labattu, B.; Tison, F. A simple method to measure stride length as an index of nigrostriatal dysfunction in mice. J. Neurosci. Methods 2002, 113, 123–130. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: San Diego, CA, USA, 2013; pp. 31–231. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viveros-Paredes, J.M.; González-Castañeda, R.E.; Gertsch, J.; Chaparro-Huerta, V.; López-Roa, R.I.; Vázquez-Valls, E.; Beas-Zarate, C.; Camins-Espuny, A.; Flores-Soto, M.E. Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP. Pharmaceuticals 2017, 10, 60. https://doi.org/10.3390/ph10030060
Viveros-Paredes JM, González-Castañeda RE, Gertsch J, Chaparro-Huerta V, López-Roa RI, Vázquez-Valls E, Beas-Zarate C, Camins-Espuny A, Flores-Soto ME. Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP. Pharmaceuticals. 2017; 10(3):60. https://doi.org/10.3390/ph10030060
Chicago/Turabian StyleViveros-Paredes, Juan M., Rocio E. González-Castañeda, Juerg Gertsch, Veronica Chaparro-Huerta, Rocio I. López-Roa, Eduardo Vázquez-Valls, Carlos Beas-Zarate, Antoni Camins-Espuny, and Mario E. Flores-Soto. 2017. "Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP" Pharmaceuticals 10, no. 3: 60. https://doi.org/10.3390/ph10030060
APA StyleViveros-Paredes, J. M., González-Castañeda, R. E., Gertsch, J., Chaparro-Huerta, V., López-Roa, R. I., Vázquez-Valls, E., Beas-Zarate, C., Camins-Espuny, A., & Flores-Soto, M. E. (2017). Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP. Pharmaceuticals, 10(3), 60. https://doi.org/10.3390/ph10030060