
Modulation of TRP Channel Activity by Hydroxylation and Its Therapeutic Potential

{kind=link}
Abstract
:1. Introduction
2. Hydroxylation-Dependent Regulation of Hypoxic Gene Expression
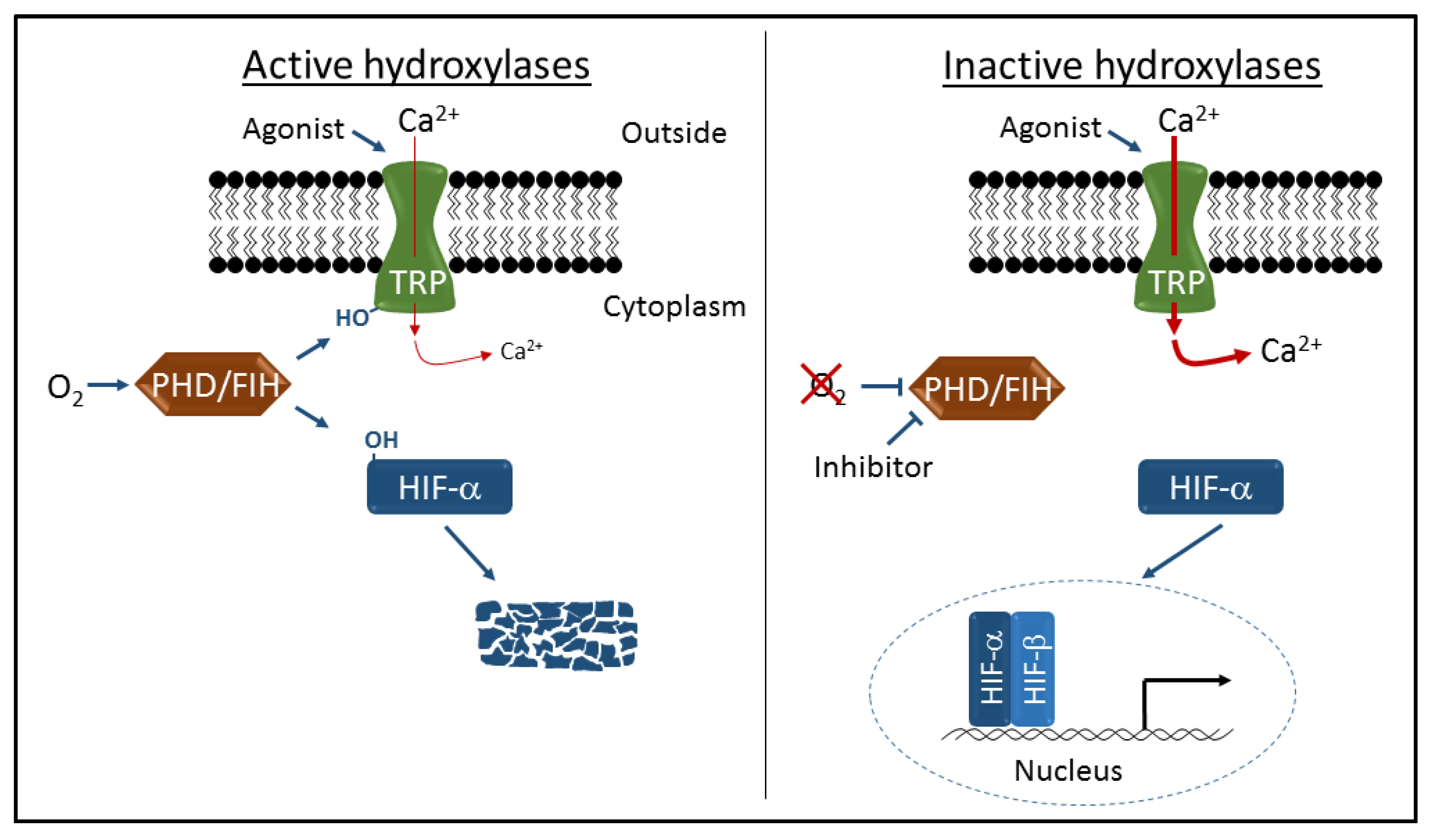
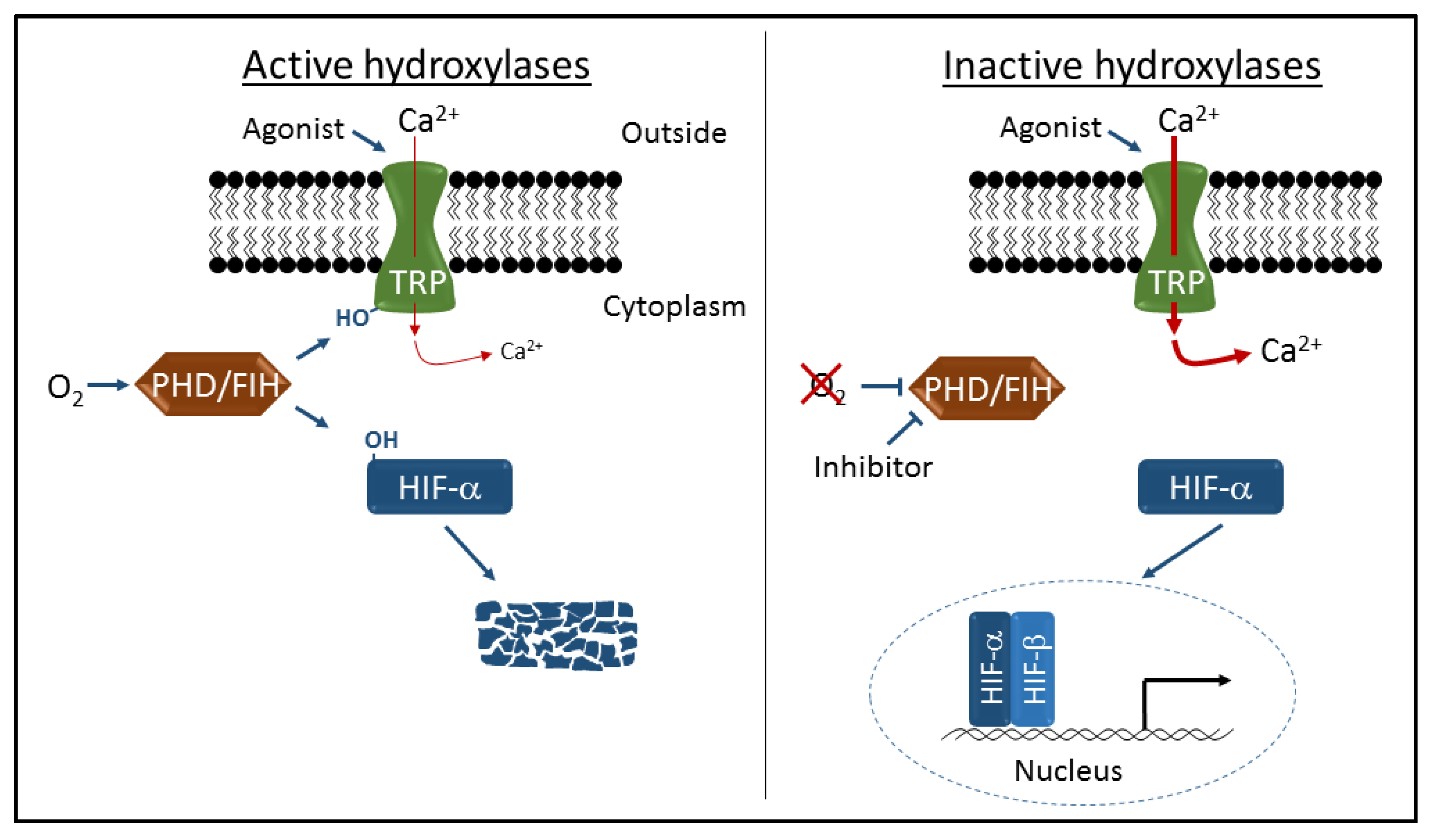
3. Hydroxylation-Dependent Regulation of TRP Channel Activity
4. Therapeutic Targeting of PHDs and FIH to Activate HIF
5. Therapeutic Targeting of PHDs and FIH to Modulate TRP Channel Activity
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brierley, S.M.; Hughes, P.A.; Page, A.J.; Kwan, K.Y.; Martin, C.M.; O’Donnell, T.A.; Cooper, N.J.; Harrington, A.M.; Adam, B.; Liebregts, T.; et al. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 2009, 137, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- Darby, W.G.; Grace, M.S.; Baratchi, S.; McIntyre, P. Modulation of TRPV4 by diverse mechanisms. Int. J. Biochem. Cell Biol. 2016, 78, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Kurokawa, T.; Mori, Y. Sensing of redox status by TRP channels. Cell Calcium 2016, 60, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Vrenken, K.S.; Jalink, K.; van Leeuwen, F.N.; Middelbeek, J. Beyond ion-conduction: Channel-dependent and -independent roles of TRP channels during development and tissue homeostasis. Biochim. Biophys. Acta 2016, 1863, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y. TRPs and pain. Semin. Immunopathol. 2016, 38, 277–291. [Google Scholar] [CrossRef] [PubMed]
- De Logu, F.; Patacchini, R.; Fontana, G.; Geppetti, P. TRP functions in the broncho-pulmonary system. Semin. Immunopathol. 2016, 38, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Derbenev, A.V.; Zsombok, A. Potential therapeutic value of TRPV1 and TRPA1 in diabetes mellitus and obesity. Semin. Immunopathol. 2016, 38, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wu, B.M.; Yao, H.W.; Meng, X.M.; Huang, C.; Ni, M.M.; Li, J. Novel insights into TRPM7 function in fibrotic diseases: A potential therapeutic target. J. Cell. Physiol. 2015, 230, 1163–1169. [Google Scholar] [PubMed]
- Voolstra, O.; Huber, A. Post-translational modifications of TRP channels. Cells 2014, 3, 258–287. [Google Scholar] [CrossRef] [PubMed]
- Karttunen, S.; Duffield, M.; Scrimgeour, N.R.; Squires, L.; Lim, W.L.; Dallas, M.L.; Scragg, J.L.; Chicher, J.; Dave, K.A.; Whitelaw, M.L.; et al. Oxygen-dependent hydroxylation by FIH regulates the TRPV3 ion channel. J. Cell Sci. 2015, 128, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kuwaki, T.; Kiyonaka, S.; Numata, T.; Kozai, D.; Mizuno, Y.; Yamamoto, S.; Naito, S.; Knevels, E.; Carmeliet, P.; et al. TRPA1 underlies a sensing mechanism for O2. Nat. Chem. Biol. 2011, 7, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Loenarz, C.; Schofield, C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 2011, 36, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Peet, D.; Linke, S. Regulation of HIF: Asparaginyl hydroxylation. Novartis Found. Symp. 2006, 272, 37–49. [Google Scholar] [PubMed]
- Nguyen, T.L.; Duran, R.V. Prolyl hydroxylase domain enzymes and their role in cell signaling and cancer metabolism. Int. J. Biochem. Cell Biol. 2016, 80, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Webb, J.D.; Ratcliffe, P.J. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann. N. Y. Acad. Sci. 2009, 1177, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Janke, K.; Brockmeier, U.; Kuhlmann, K.; Eisenacher, M.; Nolde, J.; Meyer, H.E.; Mairbaurl, H.; Metzen, E. Factor inhibiting HIF-1 (FIH-1) modulates protein interactions of apoptosis-stimulating p53 binding protein 2 (ASPP2). J. Cell Sci. 2013, 126, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.; Crifo, B.; Halligan, D.N.; et al. FIH regulates cellular metabolism through hydroxylation of the deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed]
- Laursen, W.J.; Bagriantsev, S.N.; Gracheva, E.O. TRPA1 channels: Chemical and temperature sensitivity. Curr. Top. Membr. 2014, 74, 89–112. [Google Scholar] [PubMed]
- Moilanen, L.J.; Hamalainen, M.; Nummenmaa, E.; Ilmarinen, P.; Vuolteenaho, K.; Nieminen, R.M.; Lehtimaki, L.; Moilanen, E. Monosodium iodoacetate-induced inflammation and joint pain are reduced in trpa1 deficient mice-potential role of TRPA1 in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- DeBerry, J.J.; Schwartz, E.S.; Davis, B.M. TRPA1 mediates bladder hyperalgesia in a mouse model of cystitis. Pain 2014, 155, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Kremeyer, B.; Lopera, F.; Cox, J.J.; Momin, A.; Rugiero, F.; Marsh, S.; Woods, C.G.; Jones, N.G.; Paterson, K.J.; Fricker, F.R.; et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 2010, 66, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Nassini, R.; Materazzi, S.; Benemei, S.; Geppetti, P. The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev. Physiol. Biochem. Pharmacol. 2014, 167, 1–43. [Google Scholar] [PubMed]
- Pokorski, M.; Takeda, K.; Sato, Y.; Okada, Y. The hypoxic ventilatory response and TRPA1 antagonism in conscious mice. Acta Physiol. 2014, 210, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Nakamura, S.; Zhao, M.; So, K.; Inoue, K.; Numata, T.; Takahashi, N.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; et al. Cold sensitivity of TRPA1 is unveiled by the prolyl hydroxylation blockade-induced sensitization to ROS. Nat. Commun. 2016, 7, 12840. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Kolodziejczyk, K.; Kougioumtzidou, E.; Attwell, D. Proton-gated Ca(2+)-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 2016, 529, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Peier, A.M.; Reeve, A.J.; Andersson, D.A.; Moqrich, A.; Earley, T.J.; Hergarden, A.C.; Story, G.M.; Colley, S.; Hogenesch, J.B.; McIntyre, P.; et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 2002, 296, 2046–2049. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Gunthorpe, M.J.; Kelsell, R.E.; Hayes, P.D.; Reilly, P.; Facer, P.; Wright, J.E.; Jerman, J.C.; Walhin, J.P.; Ooi, L.; et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 2002, 418, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ramsey, I.S.; Kotecha, S.A.; Moran, M.M.; Chong, J.A.; Lawson, D.; Ge, P.; Lilly, J.; Silos-Santiago, I.; Xie, Y.; et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 2002, 418, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Broad, L.M.; Mogg, A.J.; Eberle, E.; Tolley, M.; Li, D.L.; Knopp, K.L. TRPV3 in drug development. Pharmaceuticals 2016, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Hu, H. Thermally activated TRPV3 channels. Curr. Top. Membr. 2014, 74, 325–364. [Google Scholar] [PubMed]
- Yang, M.; Chowdhury, R.; Ge, W.; Hamed, R.B.; McDonough, M.A.; Claridge, T.D.; Kessler, B.M.; Cockman, M.E.; Ratcliffe, P.J.; Schofield, C.J. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 2011, 278, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Fedele, A.O.; Linke, S.; Balrak, W.; Lisy, K.; Whitelaw, M.L.; Peet, D.J. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 2006, 281, 22575–22585. [Google Scholar] [CrossRef] [PubMed]
- Bleymehl, K.; Perez-Gomez, A.; Omura, M.; Moreno-Perez, A.; Macias, D.; Bai, Z.; Johnson, R.S.; Leinders-Zufall, T.; Zufall, F.; Mombaerts, P. A sensor for low environmental oxygen in the mouse main olfactory epithelium. Neuron 2016, 92, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yang, F.; Liu, S.; Colton, C.K.; Wang, C.; Cui, Y.; Cao, X.; Zhu, M.X.; Sun, C.; Wang, K.; et al. Heteromeric heat-sensitive transient receptor potential channels exhibit distinct temperature and chemical response. J. Biol. Chem. 2012, 287, 7279–7288. [Google Scholar] [CrossRef] [PubMed]
- Dann, C.E., 3rd; Bruick, R.K.; Deisenhofer, J. Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 15351–15356. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.M.; Hewitson, K.S.; McNeill, L.A.; Seibel, J.F.; Schlemminger, I.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J. Biol. Chem. 2003, 278, 1802–1806. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, S.J.; Jeong, D.G.; Lee, S.M.; Ryu, S.E. Structure of human FIH-1 reveals a unique active site pocket and interaction sites for HIF-1 and von hippel-lindau. J. Biol. Chem. 2003, 278, 7558–7563. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Li, V.; Flashman, E.; Chowdhury, R.; Mohr, C.; Lienard, B.M.; Zondlo, J.; Oldham, N.J.; Clifton, I.J.; Lewis, J.; et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. USA 2006, 103, 9814–9819. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Schofield, C.J.; Ratcliffe, P.J. Hypoxia-inducible factor prolyl-hydroxylase: Purification and assays of PHD2. Methods Enzymol. 2007, 435, 25–42. [Google Scholar] [PubMed]
- Linke, S.; Hampton-Smith, R.J.; Peet, D.J. Characterization of ankyrin repeat-containing proteins as substrates of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible transcription factor. Methods Enzymol. 2007, 435, 61–85. [Google Scholar] [PubMed]
- Chan, M.C.; Holt-Martyn, J.P.; Schofield, C.J.; Ratcliffe, P.J. Pharmacological targeting of the HIF hydroxylases—A new field in medicine development. Mol. Asp. Med. 2016, 47–48, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Flagg, S.C.; Martin, C.B.; Taabazuing, C.Y.; Holmes, B.E.; Knapp, M.J. Screening chelating inhibitors of HIF-prolyl hydroxylase domain 2 (PHD2) and factor inhibiting hif (FIH). J. Inorg. Biochem. 2012, 113, 25–30. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; McNeill, L.A.; Tilliet, M.; Papamicael, C.A.; Chen, Q.Y.; Banerji, B.; Hewitson, K.S.; Schofield, C.J. Selective inhibition of factor inhibiting hypoxia-inducible factor. J. Am. Chem. Soc. 2005, 127, 7680–7681. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.; Roux, D.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006, 66, 3688–3698. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Leffler, A.; Niedermirtl, F.; Babes, A.; Zimmermann, K.; Filipovic, M.R.; Izydorczyk, I.; Eberhardt, M.; Kichko, T.I.; Mueller-Tribbensee, S.M.; et al. TRPA1 and substance p mediate colitis in mice. Gastroenterology 2011, 141, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Szallasi, A. Transient receptor potential (TRP) channels: A clinical perspective. Br. J. Pharmacol. 2014, 171, 2474–2507. [Google Scholar] [PubMed]
- Zhang, N.; Fu, Z.; Linke, S.; Chicher, J.; Gorman, J.J.; Visk, D.; Haddad, G.G.; Poellinger, L.; Peet, D.J.; Powell, F.; et al. The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab. 2010, 11, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Aijima, R.; Wang, B.; Takao, T.; Mihara, H.; Kashio, M.; Ohsaki, Y.; Zhang, J.Q.; Mizuno, A.; Suzuki, M.; Yamashita, Y.; et al. The thermosensitive TRPV3 channel contributes to rapid wound healing in oral epithelia. FASEB J. 2015, 29, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.X.; Hu, M.S.; Esquivel, M.; Liang, G.Y.; Rennert, R.C.; McArdle, A.; Paik, K.J.; Duscher, D.; Gurtner, G.C.; Lorenz, H.P.; et al. The role of hypoxia-inducible factor in wound healing. Adv. Wound Care 2014, 3, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.Y.; Huang, Y.; Kwan, H.Y.; Chung, H.Y.; Yao, X. Activation of transient receptor potential vanilloid 3 channel suppresses adipogenesis. Endocrinology 2015, 156, 2074–2086. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagarajan, Y.; Rychkov, G.Y.; Peet, D.J. Modulation of TRP Channel Activity by Hydroxylation and Its Therapeutic Potential. Pharmaceuticals 2017, 10, 35. https://doi.org/10.3390/ph10020035
Nagarajan Y, Rychkov GY, Peet DJ. Modulation of TRP Channel Activity by Hydroxylation and Its Therapeutic Potential. Pharmaceuticals. 2017; 10(2):35. https://doi.org/10.3390/ph10020035
Chicago/Turabian StyleNagarajan, Yagnesh, Grigori Y. Rychkov, and Daniel J. Peet. 2017. "Modulation of TRP Channel Activity by Hydroxylation and Its Therapeutic Potential" Pharmaceuticals 10, no. 2: 35. https://doi.org/10.3390/ph10020035
APA StyleNagarajan, Y., Rychkov, G. Y., & Peet, D. J. (2017). Modulation of TRP Channel Activity by Hydroxylation and Its Therapeutic Potential. Pharmaceuticals, 10(2), 35. https://doi.org/10.3390/ph10020035