Redesigning an Electrochemical MIP Sensor for PFOS: Practicalities and Pitfalls

 ,
,  ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials, Electrochemical Apparatus and Characterization Techniques
2.2. Preparation for Molecularly Imprinted Polymer (MIP) and Non-Imprinted Polymer (NIP) Sensors: From Synthesis to Template Removal
2.3. Electrochemical Characterization and Performance of MIP and NIP Sensors
3. Results and Discussion
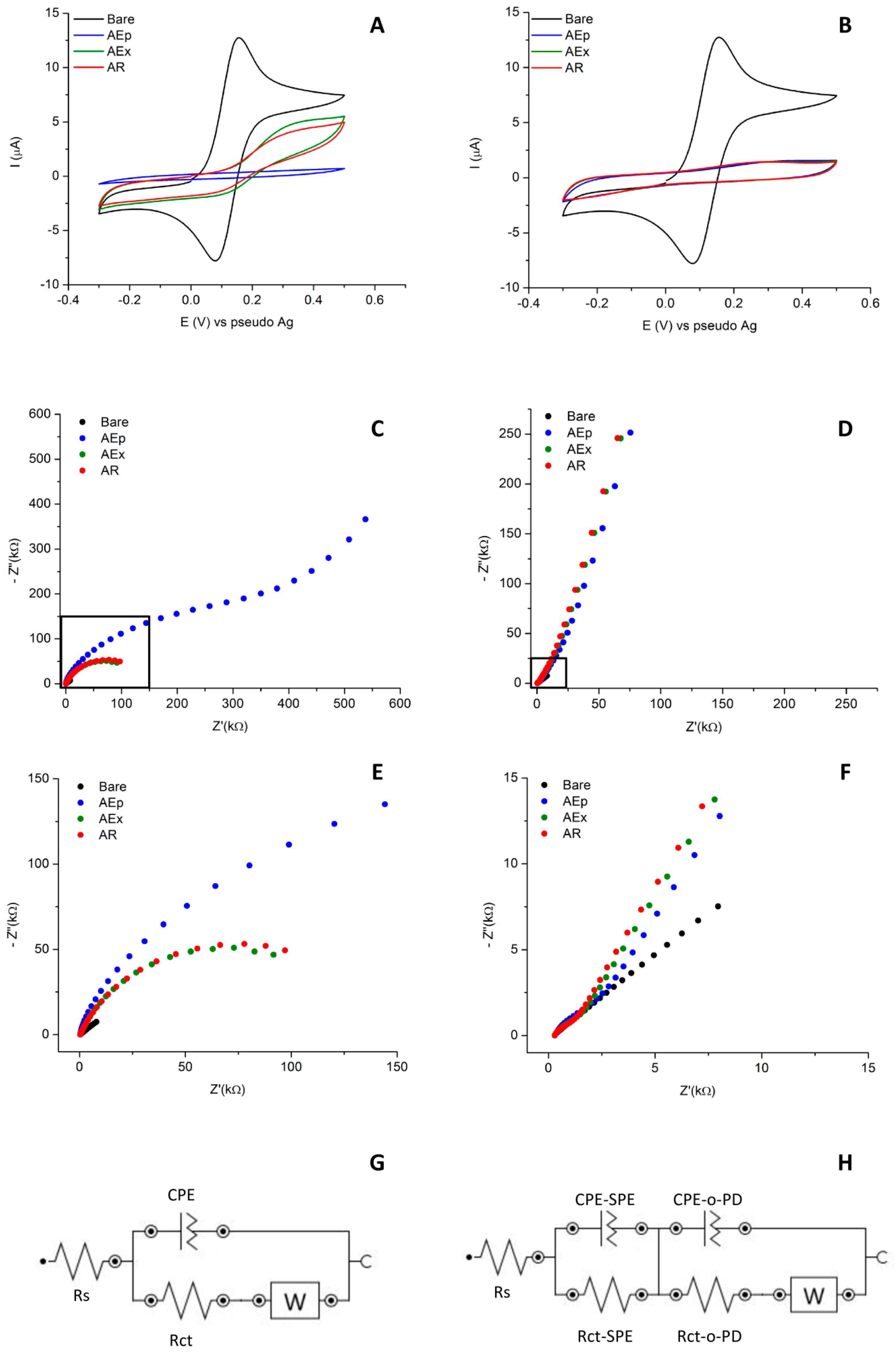
3.1. Electrochemical Characterization of MIP-Gold Screen-Printed Electrodes (Au-SPE)
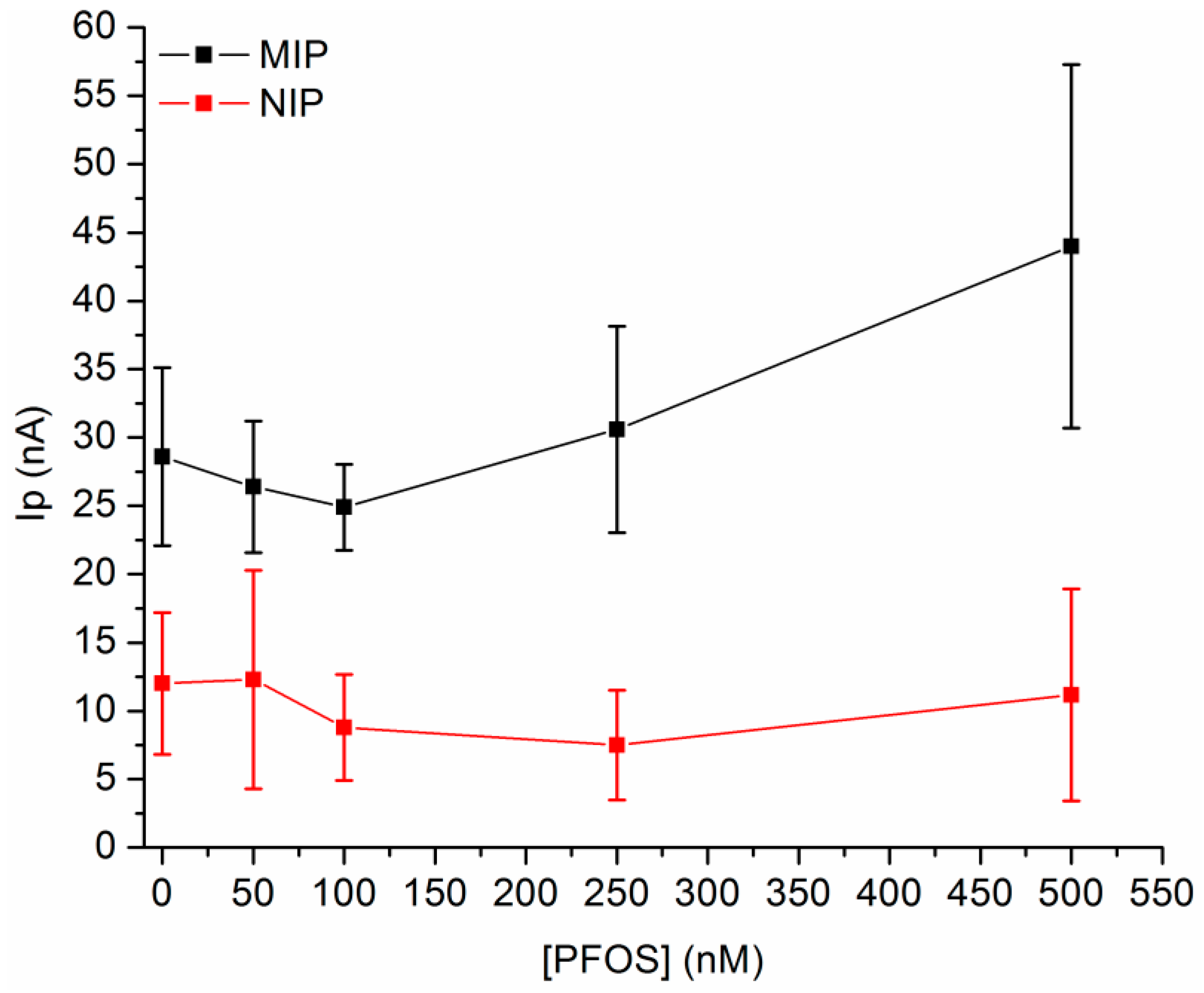
3.2. Performance of MIP-Au-SPE
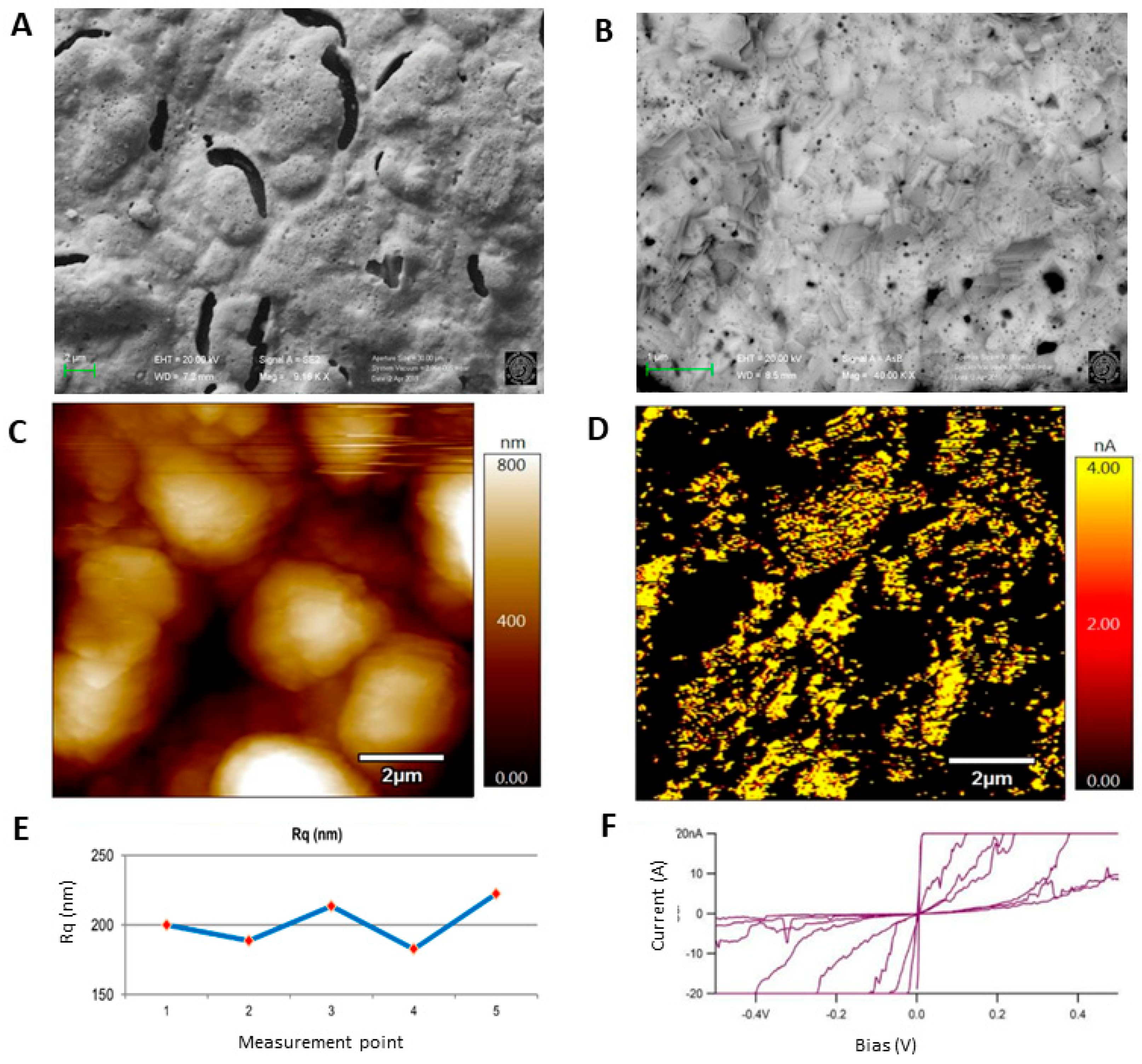
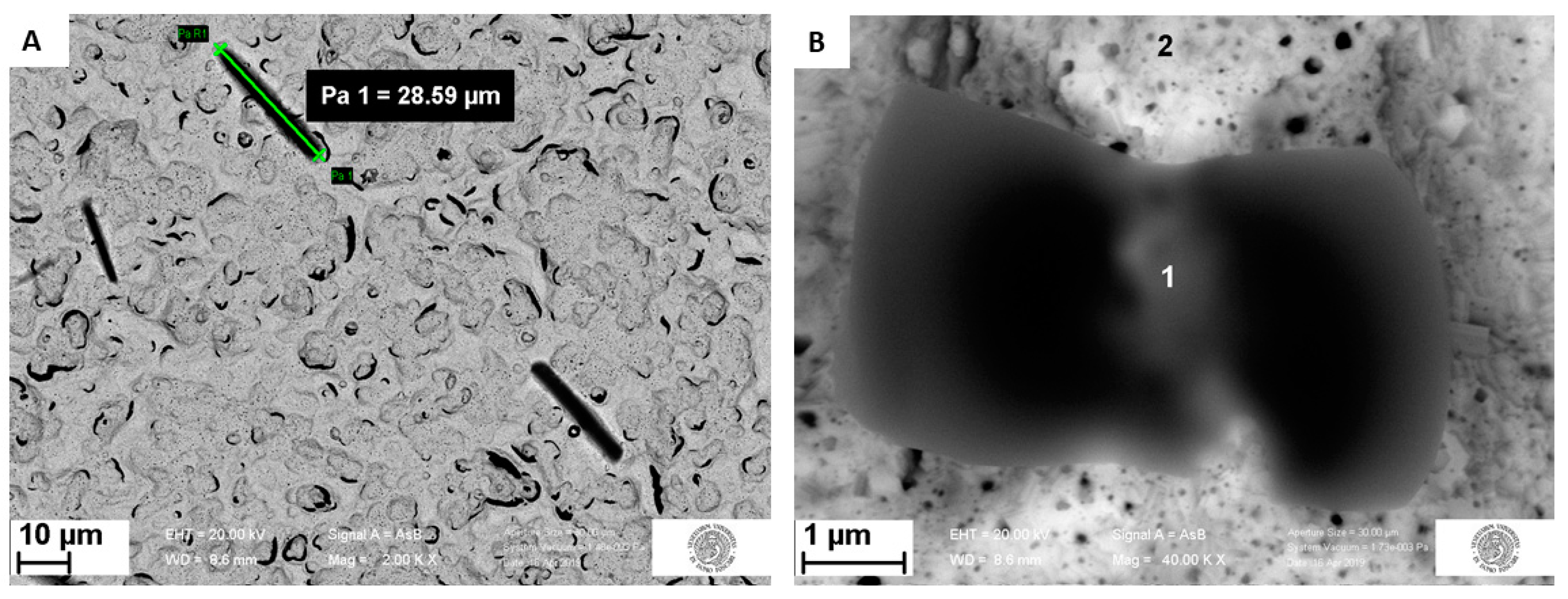
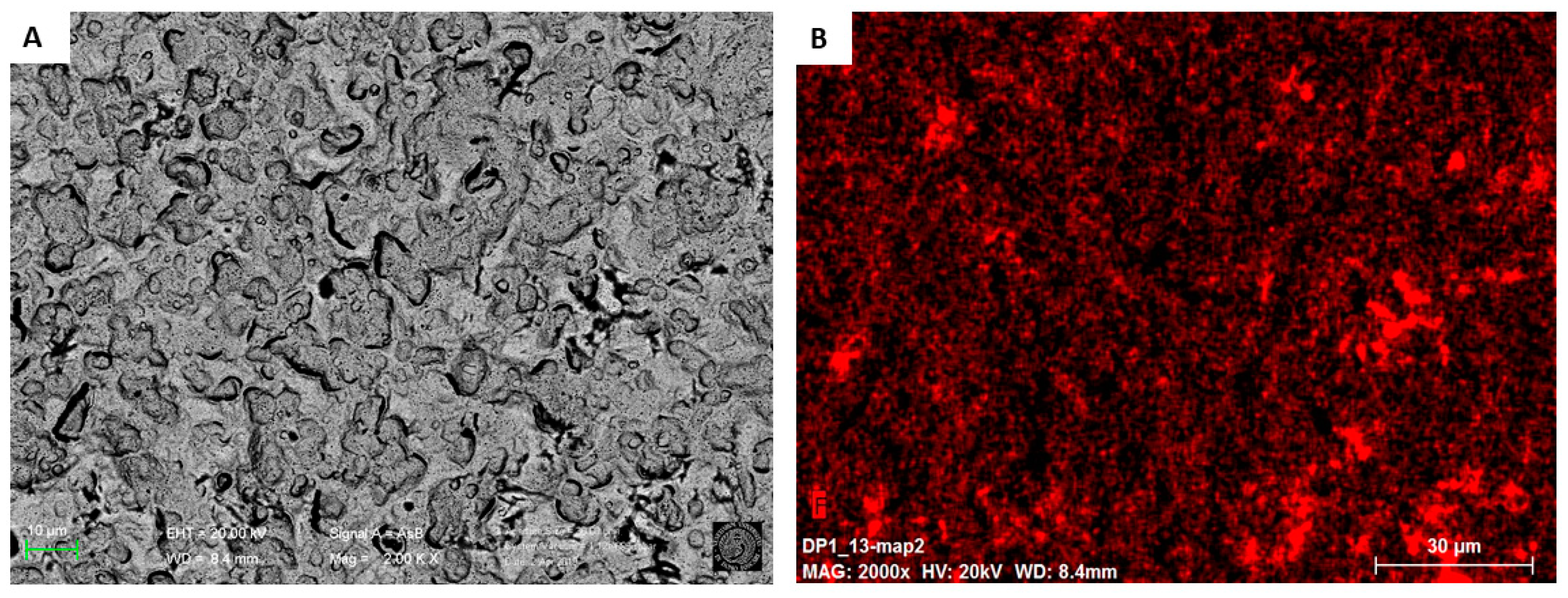
3.3. Morphological Characterization
3.3.1. Surface Analysis of Unmodified Au-SPE
3.3.2. Surface Analysis of MIP and NIP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moro, G.; De Wael, K.; Moretto, L.M. Challenges in the electrochemical (bio)sensing of nonelectroactive food and environmental contaminants. Curr. Opin. Electrochem. 2019, 16, 57–65. [Google Scholar] [CrossRef]
- Suryanarayanan, V.; Wu, C.-T.; Ho, K.-C. Molecularly imprinted electrochemical sensors. Electroanalysis 2010, 22, 1795–1811. [Google Scholar] [CrossRef]
- Lahcen, A.A.; Amine, A. Recent advances in electrochemical sensors based on molecularly imprinted polymers and nanomaterials. Electroanalysis 2019, 31, 188–201. [Google Scholar] [CrossRef]
- Crapnell, R.D.; Hudson, A.; Foster, C.W.; Eersels, K.; van Grinsven, B.; Cleij, T.J.; Banks, C.E.; Peeters, M. Recent advances in electrosynthesized molecularly imprinted polymer sensing platforms for bioanalyte detection. Sensors 2019, 19, 1204. [Google Scholar] [CrossRef] [PubMed]
- Moro, G.; Bottari, F.; Sleegers, N.; Florea, A.; Cowen, T.; Moretto, L.M.; Piletsky, S.; De Wael, K. Conductive imprinted polymers for the direct electrochemical detection of β-lactam antibiotics: The case of cefquinome. Sens. Actuators B Chem. 2019, 297, 126786. [Google Scholar] [CrossRef]
- Sharma, P.S.; Pietrzyk-Le, A.; D’Souza, F.; Kutner, W. Electrochemically synthesized polymers in molecular imprinting for chemical sensing. Anal. Bioanal. Chem. 2012, 402, 3177–3204. [Google Scholar] [CrossRef]
- Luo, J.; Huang, J.; Wu, Y.; Sun, J.; Wei, W.; Liu, X. Synthesis of hydrophilic and conductive molecularly imprinted polyaniline particles for the sensitive and selective protein detection. Biosens. Bioelectron. 2017, 94, 39–46. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, T.; Xu, J.; Xue, C. Recent advances of molecularly imprinted polymer-based sensors in the detection of food safety hazard factors. Biosens. Bioelectron. 2019, 141, 111447. [Google Scholar] [CrossRef]
- Ahmad, O.S.; Bedwell, T.S.; Esen, C.; Garcia-Cruz, A.; Piletsky, S.A. Molecularly imprinted polymers in electrochemical and optical sensors. Trends Biotechnol. 2018, 37, 294–309. [Google Scholar] [CrossRef]
- Liu, S.; Yang, R.; Yin, N.; Wang, Y.L.; Faiola, F. Environmental and human relevant PFOS and PFOA doses alter human mesenchymal stem cell self-renewal, adipogenesis and osteogenesis. Ecotoxicol. Environ. Saf. 2019, 169, 564–572. [Google Scholar] [CrossRef]
- Zeng, Z.; Song, B.; Xiao, R.; Zeng, G.; Gong, J.; Chen, M.; Xu, P.; Zhang, P.; Shen, M.; Yi, H. Assessing the human health risks of perfluorooctane sulfonate by in vivo and in vitro studies. Environ. Int. 2019, 126, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Karimian, N.; Stortini, A.M.; Moretto, L.M.; Costantino, C.; Bogialli, S.; Ugo, P. Electrochemosensor for Trace Analysis of Perfluorooctanesulfonate in Water Based on a Molecularly Imprinted Poly(o-phenylenediamine) Polymer. ACS Sens. 2018, 3, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- US EPA. PFOA &PFOS Drinking Water Health Advisories; EPA: Washington, DC, USA, 2016; pp. 1–4.
- Lattach, Y.; Fourati, N.; Zerrouki, C.; Fougnion, J.M.; Garnier, F.; Pernelle, C.; Remita, S. Molecularly imprinted surface acoustic wave sensors: The synergy of electrochemical and gravimetric transductions in chemical recognition processes. Electrochim. Acta 2012, 73, 36–44. [Google Scholar] [CrossRef]
- Turco, A.; Corvaglia, S.; Mazzotta, E. Electrochemical sensor for sulfadimethoxine based on molecularly imprinted polypyrrole: Study of imprinting parameters. Biosens. Bioelectron. 2015, 63, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Jin, J.; Chen, G.Z. A comparative study on electrochemical co-deposition and capacitance of composite films of conducting polymers and carbon nanotubes. Electrochim. Acta 2007, 53, 525–537. [Google Scholar] [CrossRef]
- Fomo, G.; Waryo, T.; Feleni, U.; Baker, P.; Iwuoha, E. Electrochemical Polymerization-Functional Polymers; Jafar Mazumder, M.A., Sheardown, H., Al-Ahmed, A., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Wang, H.; Yuan, L.; Zhu, H.; Jin, R.; Xing, J. Comparative study of capsaicin molecularly imprinted polymers prepared by different polymerization methods. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 157–164. [Google Scholar] [CrossRef]
- Boukamp, B.A. Practical application of the Kramers-Kronig transformation on impedance measurements in solid state electrochemistry. Solid State Ion. 1993, 62, 131–141. [Google Scholar] [CrossRef]
- Agarwal, P. Application of Measurement Models to Impedance Spectroscopy. J. Electrochem. Soc. 1995, 142, 4159. [Google Scholar] [CrossRef]
- Losito, I.; Palmisano, F.; Zambonin, P.G. O-Phenylenediamine Electropolymerization By Cyclic Voltammetry Combined with Electrospray Ionization-Ion Trap Mass Spectrometry. Anal. Chem. 2003, 75, 4988–4995. [Google Scholar] [CrossRef]
- Musiani, M.M. Characterization of electroactive polymer layers by electrochemical impedance spectroscopy (EIS). Electrochim. Acta 1990, 35, 1665–1670. [Google Scholar] [CrossRef]
- Deslouis, C.; Musiani, M.M.; Tribollet, B. Free-Standing Membranes for the Study of Electrochemical Reactions Occurring at Conducting Polymer/Electrolyte Interfaces. J. Phys. Chem. 1996, 100, 8994–8999. [Google Scholar] [CrossRef]
- Gadelmawla, E.S.; Koura, M.M.; Maksoud, T.M.A.; Elewa, I.M.; Soliman, H.H. Roughness parameters. J. Mater. Process. Technol. 2002, 123, 133–145. [Google Scholar] [CrossRef]
- Milano, F.; Giotta, L.; Chirizzi, D.; Papazoglou, S.; Kryou, C.; De Bartolomeo, A.; De Leo, V.; Guascito, M.R.; Zergioti, I. Phosphate modified screen printed electrodes by lift treatment for glucose detection. Biosensors 2018, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.; Newbury, D.E.; Echlin, P.; Joy, D.C.; Fiori, C.; Lifshin, E. Scanning Electron Microscopy and X-Ray Microanalysis, 4th ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Reimer, L. Scanning Electron Microscopy. Physics of Image Formation and Microanalysis, 2nd ed.; Springer: New York, NY, USA, 1998. [Google Scholar]
- Czapik, A.; Gdaniec, M. A new polymorph of benzene-1,2-diamine: Isomorphism with 2-amino-phenol and two-dimensional iso-structurality of polymorphs. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2010, 66, 198–201. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moro, G.; Cristofori, D.; Bottari, F.; Cattaruzza, E.; De Wael, K.; Moretto, L.M. Redesigning an Electrochemical MIP Sensor for PFOS: Practicalities and Pitfalls. Sensors 2019, 19, 4433. https://doi.org/10.3390/s19204433
Moro G, Cristofori D, Bottari F, Cattaruzza E, De Wael K, Moretto LM. Redesigning an Electrochemical MIP Sensor for PFOS: Practicalities and Pitfalls. Sensors. 2019; 19(20):4433. https://doi.org/10.3390/s19204433
Chicago/Turabian StyleMoro, Giulia, Davide Cristofori, Fabio Bottari, Elti Cattaruzza, Karolien De Wael, and Ligia Maria Moretto. 2019. "Redesigning an Electrochemical MIP Sensor for PFOS: Practicalities and Pitfalls" Sensors 19, no. 20: 4433. https://doi.org/10.3390/s19204433
APA StyleMoro, G., Cristofori, D., Bottari, F., Cattaruzza, E., De Wael, K., & Moretto, L. M. (2019). Redesigning an Electrochemical MIP Sensor for PFOS: Practicalities and Pitfalls. Sensors, 19(20), 4433. https://doi.org/10.3390/s19204433