Evidence of Reactivity in the Membrane for the Unstable Monochloramine during MIMS Analysis

 ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Ammonia Permeation
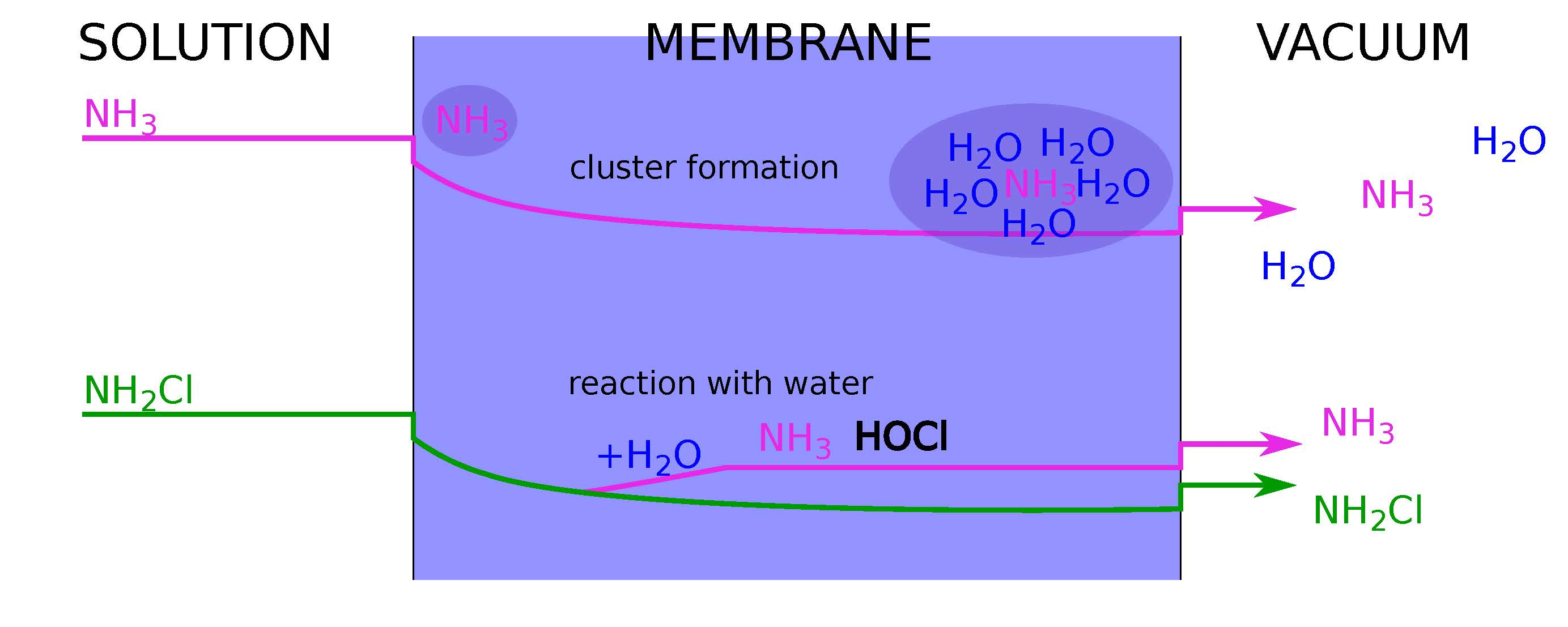
2.1.1. Diffusion and Enrichment
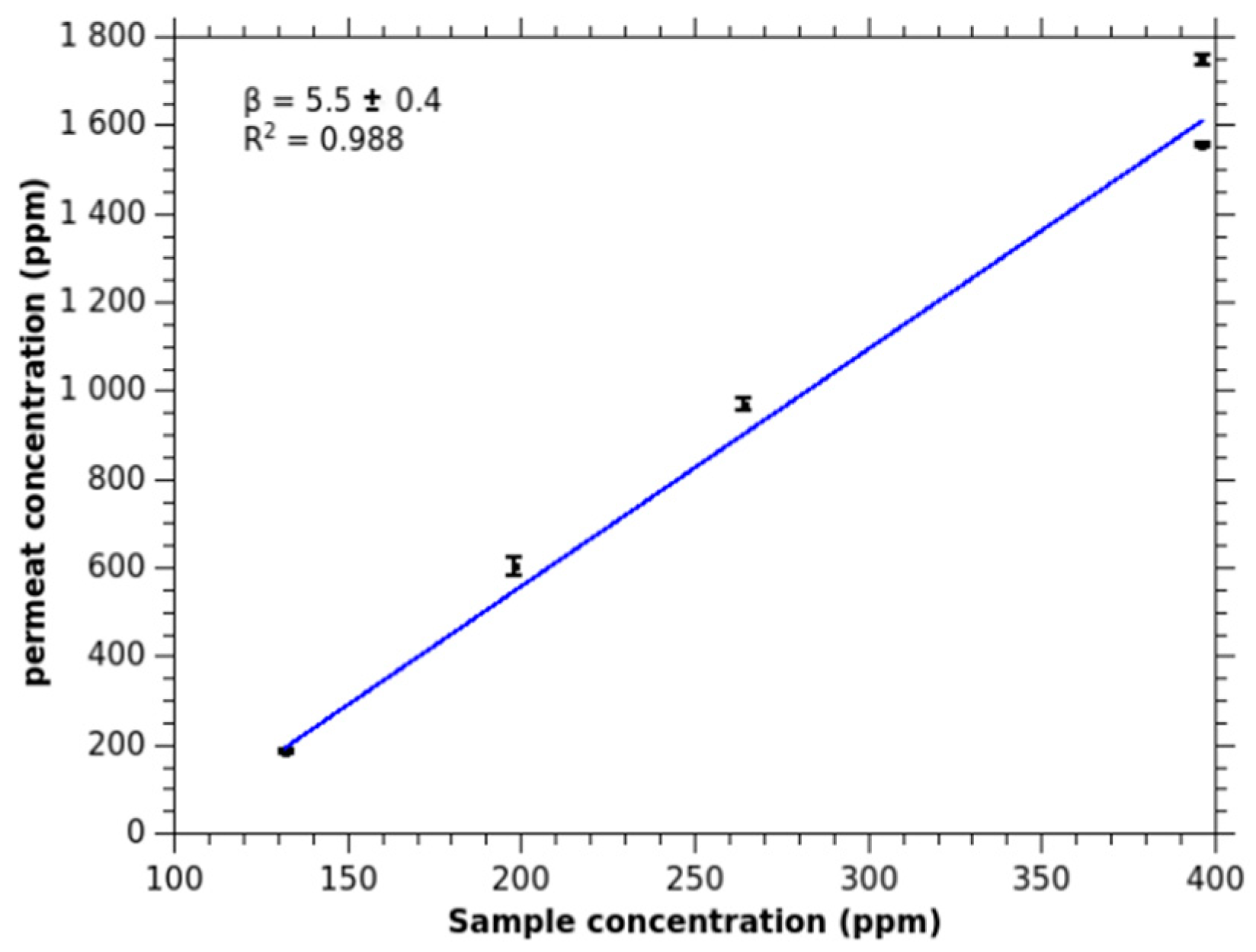
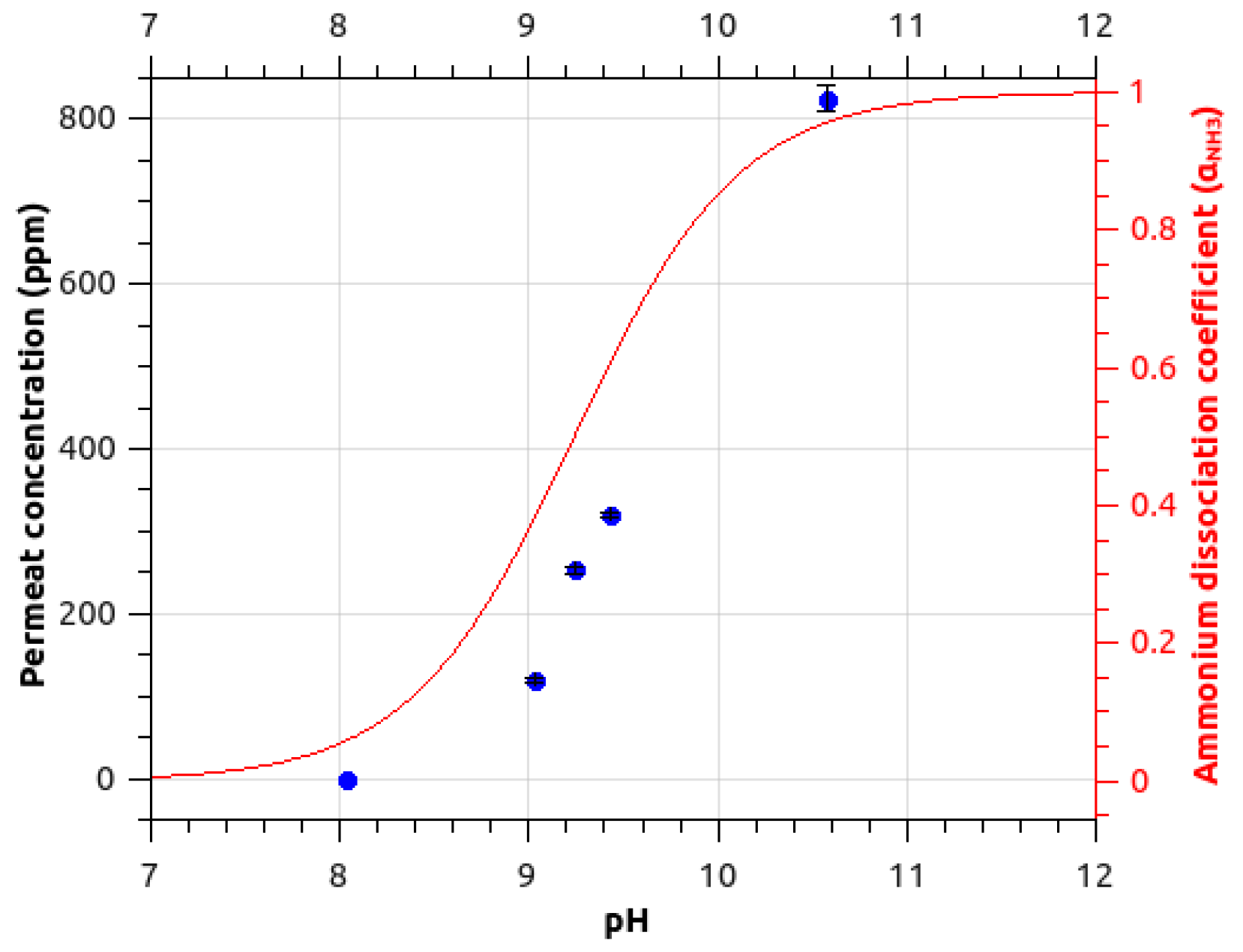
2.1.2. pH Influence on Ammonia Signal
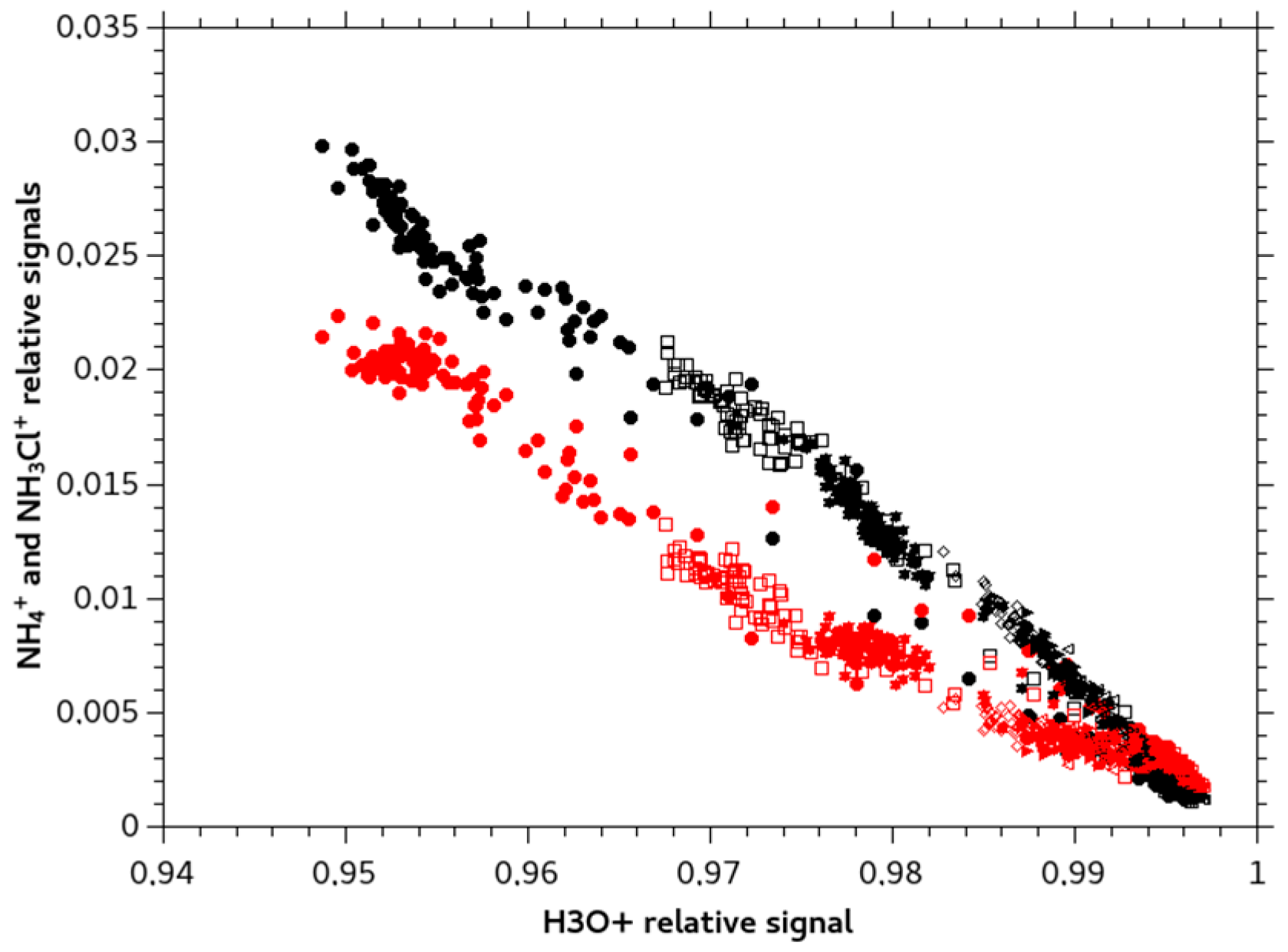
2.2. Monochloramine Permeation
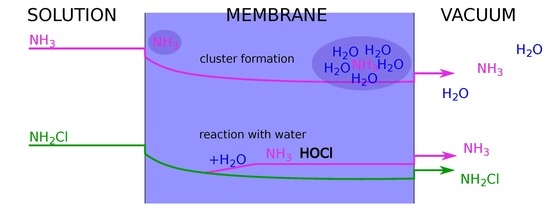
3. Origin of the Observed NH4+ ion Signal for NH2Cl Solutions
- at the working pH, dissociation is partial and presence of m/z 18.034 is due to the transfer of ammonia in the PDMS membrane;
- reactivity of the protonated monochloramine occurs in the ICR cell, either by dissociation or secondary reaction;
- dissociation of the neutral monochloramine happens somehow in-between the solubilization step in the PDMS membrane and ionization in the ICR cell.
3.1. Is It Due to Permeation of NH3 from the Feed Solution?
3.2. Is It Due to Ion-Molecule Reactivity?
3.3. Is Neutral Monochloramine Reacting in the Vacuum Chamber?
4. Materials and Methods
4.1. Chloramine and Ammonia Solutions
4.2. Membrane Inlet
4.3. Compact Low-Field FT-ICR Mass Spectrometer and Chemical Ionization
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MIMS | Membrane Inlet Mass Spectrometry |
| PDMS | Polydimethylsiloxane |
| VOC | Volatile Organic Compound |
| EI | Electronic Ionization |
| CI | Chemical Ionization |
| FT-ICR | Fourier Transform Ion Cyclotron Resonance |
| SIFT | Selected Ion Flow Tube |
| PTFE | PolyTetraFluoroEthylene |
| PTR | Proton Transfer Reaction |
References
- Bier, M.E.; Cooks, R.G. Membrane interface for selective introduction of volatile compounds directly into the ionization chamber of a mass spectrometer. Anal. Chem. 1987, 59, 597–601. [Google Scholar] [CrossRef]
- Krogh, E.; Gill, C.G. Membrane introduction mass spectrometry (MIMS): A versatile tool for direct, real-time chemical measurements. J. Mass Spectrom. 2014, 49, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Eschenbach, W.; Lewicka-Szczebak, D.; Stange, C.F.; Dyckmans, J.; Well, R. Measuring 15N Abundance and Concentration of Aqueous Nitrate, Nitrite, and Ammonium by Membrane Inlet Quadrupole Mass Spectrometry. Anal. Chem. 2017, 89, 6076–6081. [Google Scholar] [CrossRef] [PubMed]
- Ketola, R.A.; Virkki, V.T.; Ojala, M.; Komppa, V.; Kotiaho, T. Comparison of different methods for the determination of volatile organic compounds in water samples. Talanta 1997, 44, 373–382. [Google Scholar] [CrossRef]
- Short, R.T.; Fries, D.P.; Kerr, M.L.; Lembke, C.E.; Toler, S.K.; Wenner, P.G.; Byrne, R.H. Underwater mass spectrometers for in situ chemical analysis of the hydrosphere. J. Am. Soc. Mass Spectrom. 2001, 12, 676–682. [Google Scholar] [CrossRef]
- Hemond, H.; Camilli, R. NEREUS: Engineering concept for an underwater mass spectrometer. TrAC Trends Anal. Chem. 2002, 21, 526–533. [Google Scholar] [CrossRef]
- Louarn, E.; Hamrouni, A.; Colbeau-Justin, C.; Bruschi, L.; Lemaire, J.; Heninger, M.; Mestdagh, H. Characterization of a membrane inlet interfaced with a compact chemical ionization FT-ICR for real-time and quantitative VOC analysis in water. Int. J. Mass Spectrom. 2013, 353, 26–35. [Google Scholar] [CrossRef]
- Davey, N.G.; Bell, R.J.; Krogh, E.T.; Gill, C.G. A membrane introduction mass spectrometer utilizing ion-molecule reactions for the on-line speciation and quantitation of volatile organic molecules. Rapid Commun. Mass Spectrom. 2015, 29, 2187–2194. [Google Scholar] [CrossRef]
- Blake, R.S.; Monks, P.S.; Ellis, A.M. Proton-Transfer Reaction Mass Spectrometry. Chem. Rev. 2009, 109, 861–896. [Google Scholar] [CrossRef]
- Dehon, C.; Gaüzère, E.; Vaussier, J.; Heninger, M.; Tchapla, A.; Bleton, J.; Mestdagh, H. Quantitative analysis of a complex mixture using proton transfer reaction in an FTICR mass spectrometer. Int. J. Mass Spectrom. 2008, 272, 29–37. [Google Scholar] [CrossRef]
- Soulard, M.; Bloc, F.; Hatterer, A. Diagrams of existence of chloramines and bromamines in aqueous solution. J. Chem. Soc. Dalton Trans. 1981, 2300–2310. [Google Scholar] [CrossRef]
- Kinani, S.; Richard, B.; Souissi, Y.; Bouchonnet, S. Analysis of inorganic chloramines in water. TrAC Trends Anal. Chem. 2012, 33, 55–67. [Google Scholar] [CrossRef]
- Kotiaho, T.; Lister, A.K.; Hayward, M.J.; Cooks, R.G. On-line monitoring of chloramine reactions by membrane introduction mass spectrometry. Talanta 1991, 38, 195–200. [Google Scholar] [CrossRef]
- Shang, C.; Blatchley, E.R. Differentiation and Quantification of Free Chlorine and Inorganic Chloramines in Aqueous Solution by MIMS. Environ. Sci. Technol. 1999, 33, 2218–2223. [Google Scholar] [CrossRef]
- Weaver, W.A.; Li, J.; Wen, Y.; Johnston, J.; Blatchley, M.R.; Blatchley, E.R., III. Volatile disinfection by-product analysis from chlorinated indoor swimming pools. Water Res. 2009, 43, 3308–3318. [Google Scholar] [CrossRef] [PubMed]
- Riter, L.S.; Takáts, Z.; Charles, L.; Cooks, R.G. High surface area membrane introduction mass spectrometry for analysis of volatile and semi-volatile organic compounds in air. Rapid Commun. Mass Spectrom. 2001, 15, 1520–1524. [Google Scholar] [CrossRef]
- Senthilmohan, S.T.; Kettle, A.J.; McEwan, M.J.; Dummer, J.; Edwards, S.J.; Wilson, P.F.; Epton, M.J. Detection of monobromamine, monochloramine and dichloramine using selected ion flow tube mass spectrometry and their relevance as breath markers. Rapid Commun. Mass Spectrom. 2008, 22, 677–681. [Google Scholar] [CrossRef]
- Hu, W.-P.; Langford, V.S.; McEwan, M.J.; Milligan, D.B.; Storer, M.K.; Dummer, J.; Epton, M.J. Monitoring chloramines and bromamines in a humid environment using selected ion flow tube mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 1744–1748. [Google Scholar] [CrossRef]
- Blyler, L.L.; Lieberman, R.A.; Cohen, L.G.; Ferrara, J.A.; Macchesney, J.B. Optical fiber chemical sensors utilizing dye-doped silicone polymer claddings. Polym. Eng. Sci. 1989, 29, 1215–1218. [Google Scholar] [CrossRef]
- Makhloufi, C.; Roizard, D.; Favre, E. Reverse selective NH3/CO2 permeation in fluorinated polymers using membrane gas separation. J. Membr. Sci. 2013, 441, 63–72. [Google Scholar] [CrossRef]
- Gadre, S.R.; Yeole, S.D.; Sahu, N. Quantum Chemical Investigations on Molecular Clusters. Chem. Rev. 2014, 114, 12132–12173. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.C.; Friess, K.; Drioli, E. Organic vapour transport in glassy perfluoropolymer membranes: A simple semi-quantitative approach to analyze clustering phenomena by time lag measurements. J. Membr. Sci. 2011, 367, 141–151. [Google Scholar] [CrossRef]
- Ricci, A.; Rosi, M. Gas-Phase Chemistry of NHxCly+. 1. Structure, Stability, and Reactivity of Protonated Monochloramine. J. Phys. Chem. A 1998, 102, 10189–10194. [Google Scholar] [CrossRef]
- Ricci, A.; Rosi, M. Gas Phase Chemistry of NHxCly+ Ions. II. Structure, Stability and Reactivity of Protonated Dichloramine. J. Phys. Chem. A 2000, 104, 5617–5624. [Google Scholar] [CrossRef]
- Burgess, D.R.F. Thermochemical Data. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; NIST: Gaithersburg, MD, USA, 2018; Volume 20899. [Google Scholar]
- Latappy, H.; Lemaire, J.; Heninger, M.; Louarn, E.; Bauchard, E.; Mestdagh, H. Protonated 1,4-difluorobenzene C6H5F2+: A promising precursor for proton-transfer chemical ionization. Int. J. Mass Spectrom. 2016, 405, 13–23. [Google Scholar] [CrossRef]
- Dehon, C.; Lemaire, J.; Heninger, M.; Chaput, A.; Mestdagh, H. Chemical ionization using CF3+: Efficient detection of small alkanes and fluorocarbons. Int. J. Mass Spectrom. 2011, 299, 113–119. [Google Scholar] [CrossRef]
- Grandinetti, F.; Occhiucci, G.; Crestoni, M.E.; Fornarini, S.; Speranza, M. Ionic Lewis superacids in the gas phase. Part 2. Reactions of gaseous CF+3 with oxygen bases. Int. J. Mass Spectrom. Ion Process. 1993, 127, 123–135. [Google Scholar] [CrossRef]
- Hunter, E.P.L.; Lias, S.G. Evaluated gas phase basicities and proton affinities of molecules: An update. J. Phys. Chem. Ref. Data 2009, 27, 413. [Google Scholar] [CrossRef]
- Francisco, J.S.; Sander, S.P. Protonated hydrochlorous acid (HOClH+): Molecular structure, vibrational frequencies, and proton affinity. J. Chem. Phys. 1995, 102, 9615. [Google Scholar] [CrossRef]
- Provin, C.; Fujii, T. Reaction–diffusion phenomena in a PDMS matrix can modify its topography. Lab Chip 2011, 11, 2948–2954. [Google Scholar] [CrossRef]
- Rodier, J.; Legube, B.; Merlet, N. L’analyse de L’eau, 9th ed.; Dunod: Paris, France, 2009; ISBN 978-2-10-007246-0. [Google Scholar]
- Mauclaire, G.; Lemaire, J.; Boissel, P.; Bellec, G.; Heninger, M. MICRA: A compact permanent magnet Fourier transform ion cyclotron resonance mass spectrometer. Eur. J. Mass Spectrom. 2004, 10, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, J.; Thomas, S.; Lopes, A.; Louarn, E.; Mestdagh, H.; Latappy, H.; Leprovost, J.; Heninger, M. Compact FTICR Mass Spectrometry for Real Time Monitoring of Volatile Organic Compounds. Sensors 2018, 18, 1415. [Google Scholar] [CrossRef] [PubMed]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Process. 1998, 173, 191–241. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neutral Precursor | Ionic Precursor | PA [29] (kJ·mol−1) | Product 1 | Product 2 |
|---|---|---|---|---|
| H2O | H3O+ | 691 | NH3Cl+ | NH4+ |
| F2C6H4 | F2C6H5+ | 718.7 | NH3Cl+ | NH4+ |
| (CH3)3C6H3 | (CH3)3C6H4+ | 853.6 | - | NH4+ |
| CF4 | CF3+ | - | CF2NHCl+ | CF2NH2+ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louarn, E.; Asri-Idlibi, A.M.; Leprovost, J.; Héninger, M.; Mestdagh, H. Evidence of Reactivity in the Membrane for the Unstable Monochloramine during MIMS Analysis. Sensors 2018, 18, 4252. https://doi.org/10.3390/s18124252
Louarn E, Asri-Idlibi AM, Leprovost J, Héninger M, Mestdagh H. Evidence of Reactivity in the Membrane for the Unstable Monochloramine during MIMS Analysis. Sensors. 2018; 18(12):4252. https://doi.org/10.3390/s18124252
Chicago/Turabian StyleLouarn, Essyllt, Abdoul. Monem Asri-Idlibi, Julien Leprovost, Michel Héninger, and Hélène Mestdagh. 2018. "Evidence of Reactivity in the Membrane for the Unstable Monochloramine during MIMS Analysis" Sensors 18, no. 12: 4252. https://doi.org/10.3390/s18124252
APA StyleLouarn, E., Asri-Idlibi, A. M., Leprovost, J., Héninger, M., & Mestdagh, H. (2018). Evidence of Reactivity in the Membrane for the Unstable Monochloramine during MIMS Analysis. Sensors, 18(12), 4252. https://doi.org/10.3390/s18124252