
Bench-Top Fabrication of an All-PDMS Microfluidic Electrochemical Cell Sensor Integrating Micro/Nanostructured Electrodes


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
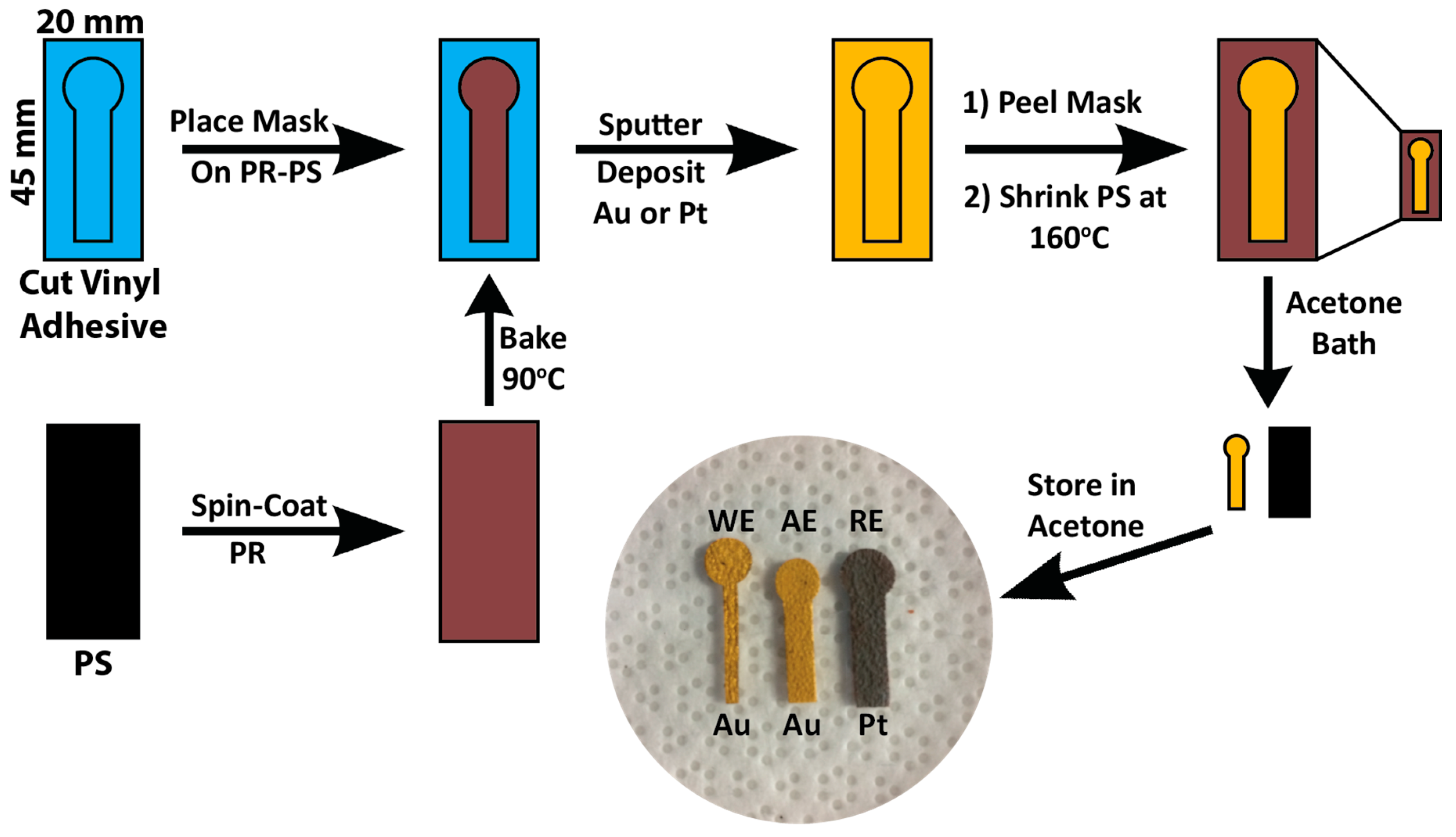
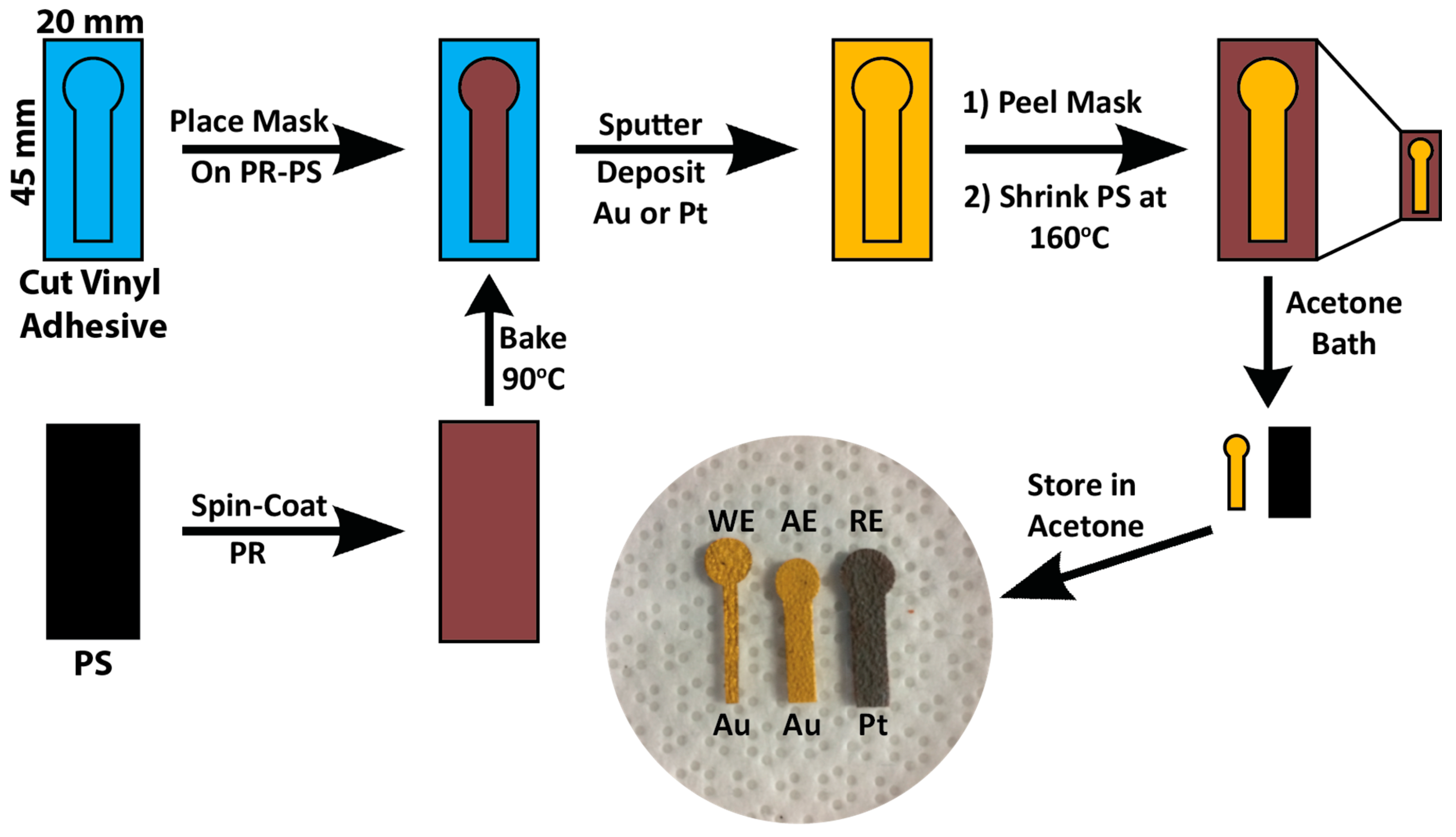
2.1. Electrode Fabrication
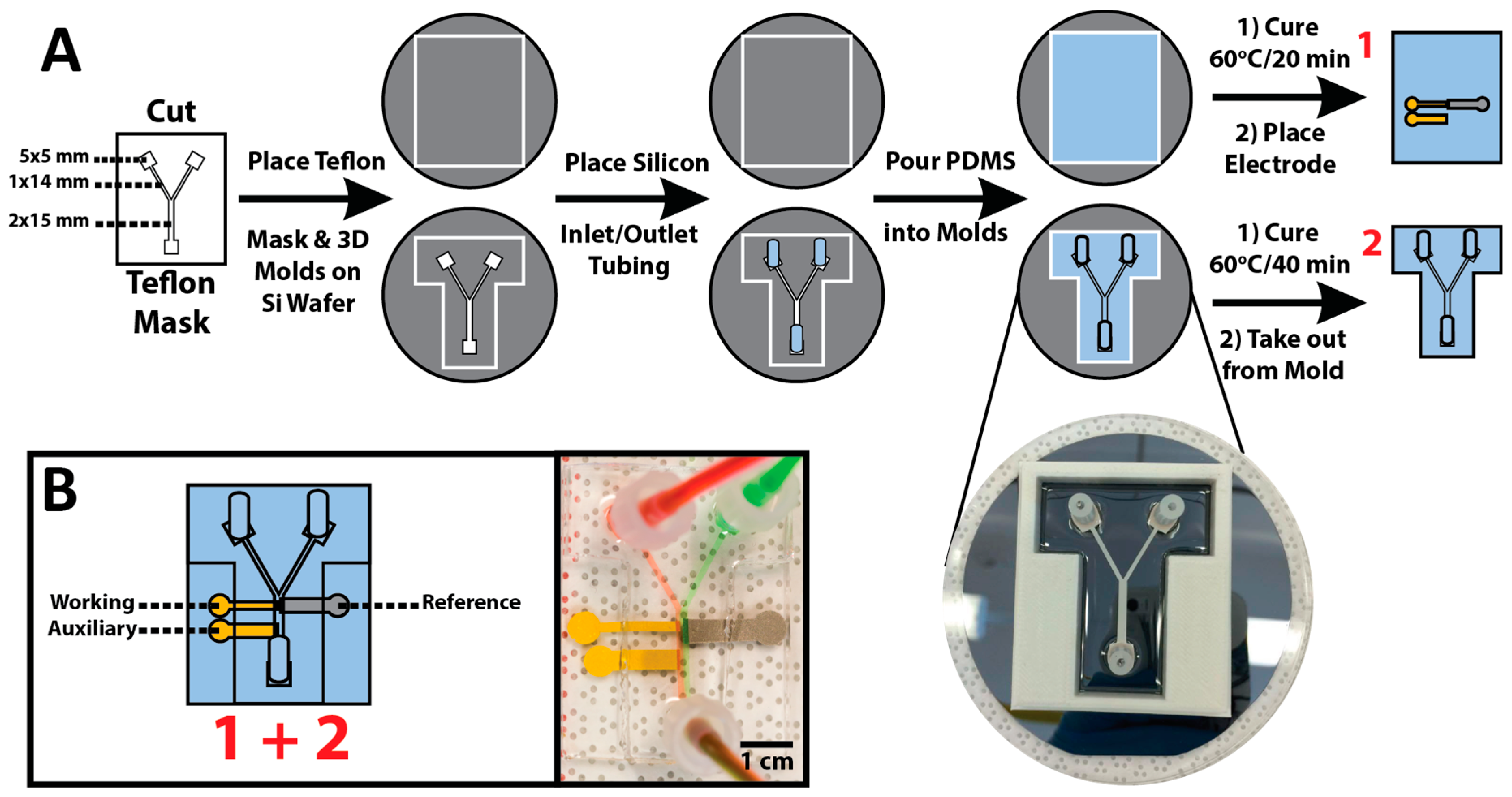
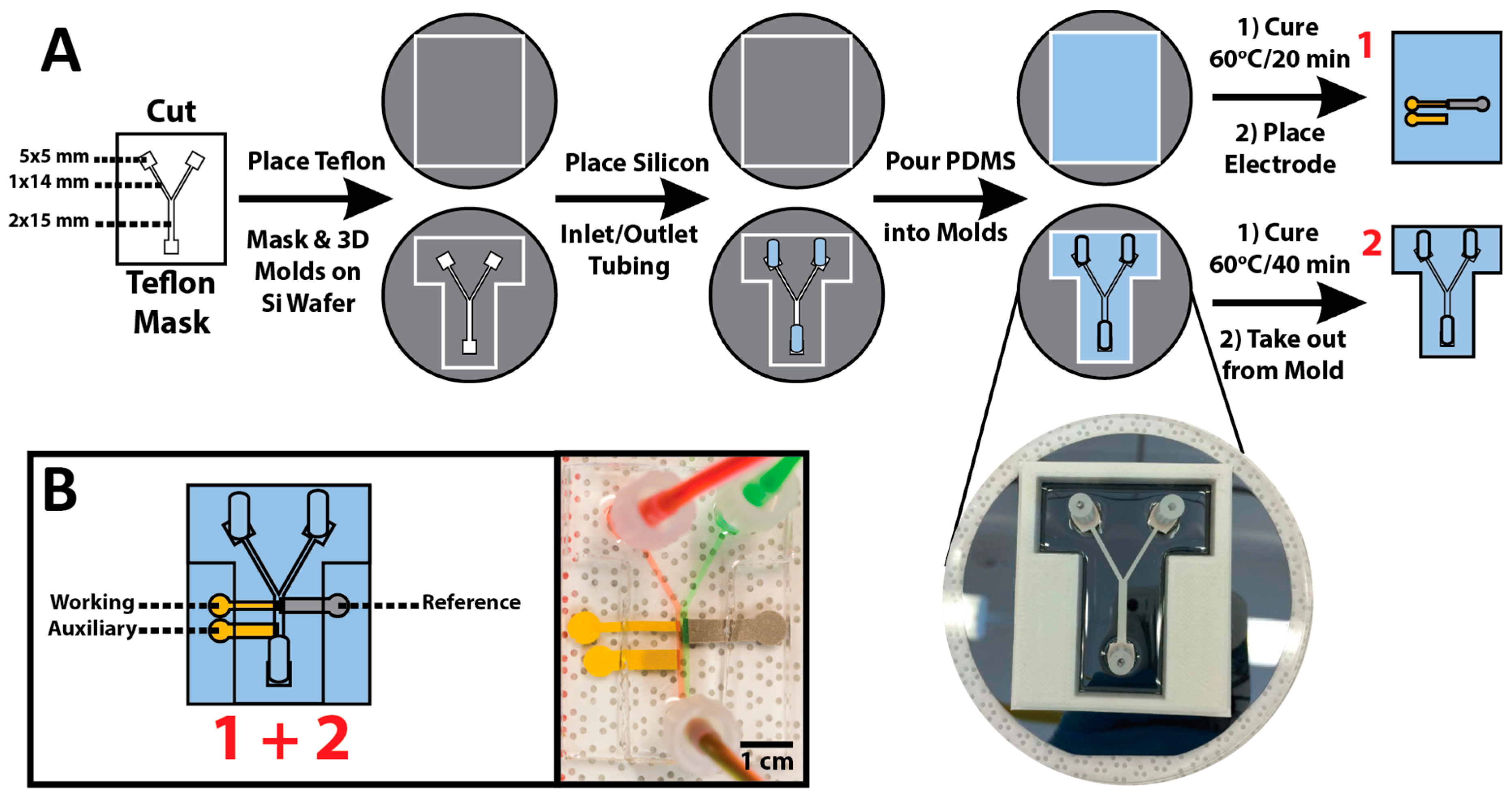
2.2. All PDMS μF Device Fabrication
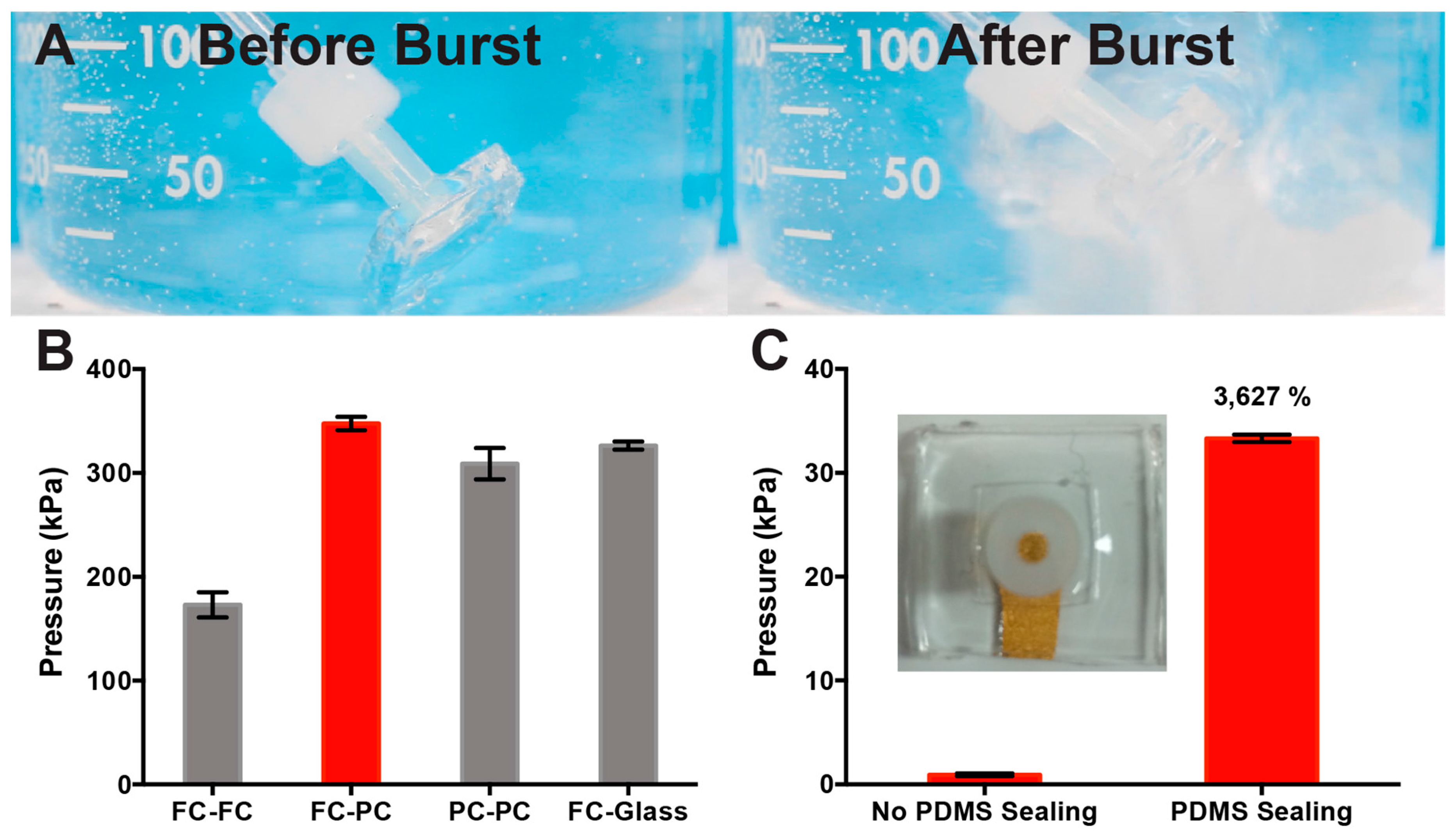
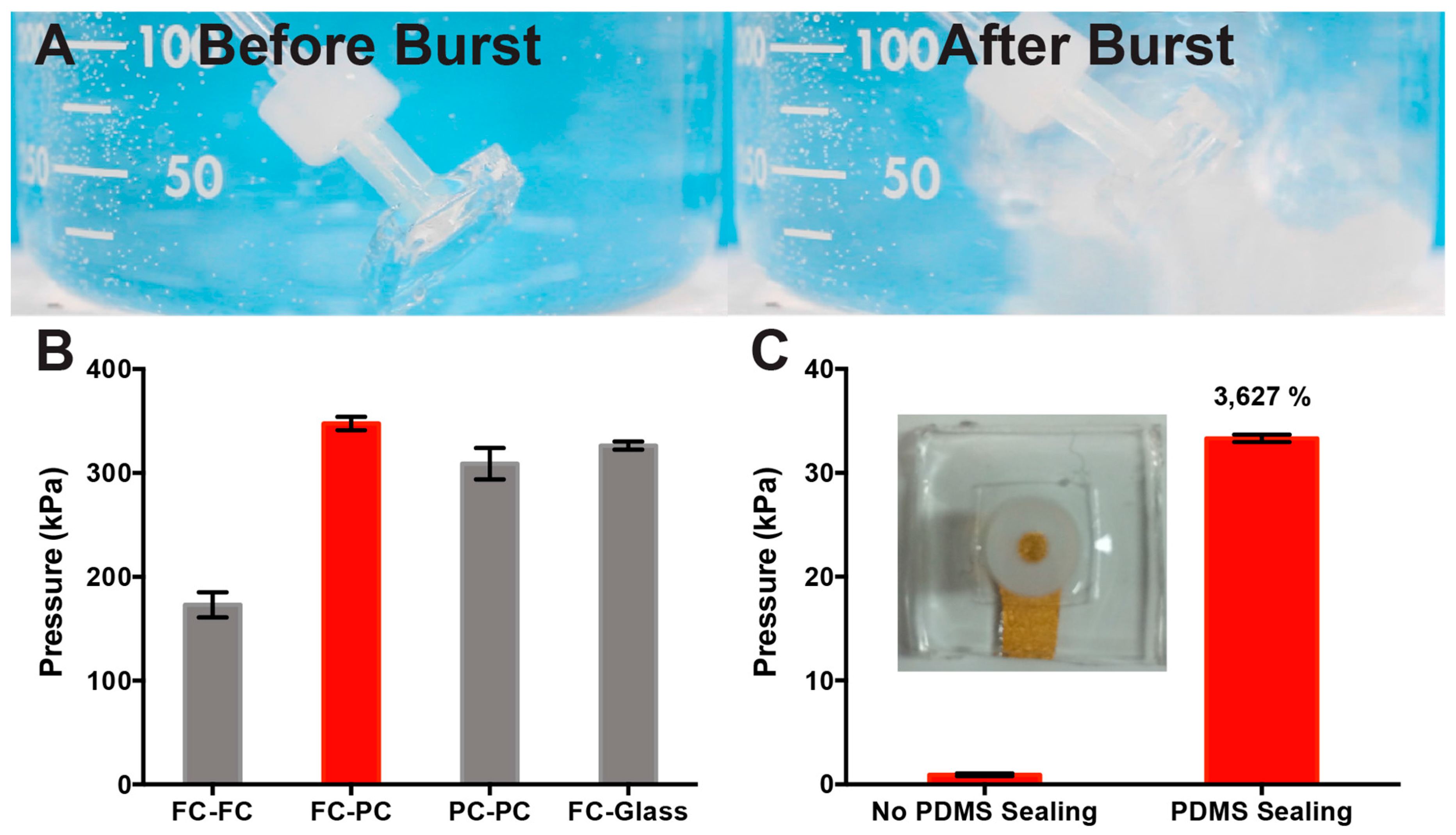
2.3. Dead-End Pressure Test of PDMS Bond Strength
2.4. Murine 3T3 Fibroblast Cell Culture
2.5. CFSE Fibroblast Cell Staining
2.6. Cell Viability Assay
2.7. Murine 3T3 Fibroblast Cell Sensing
2.8. Fluorescent Microscopy Image Acquisition
3. Results and Discussion
3.1. Optimization of PDMS Device Bond Strength for μF Device Fabrication
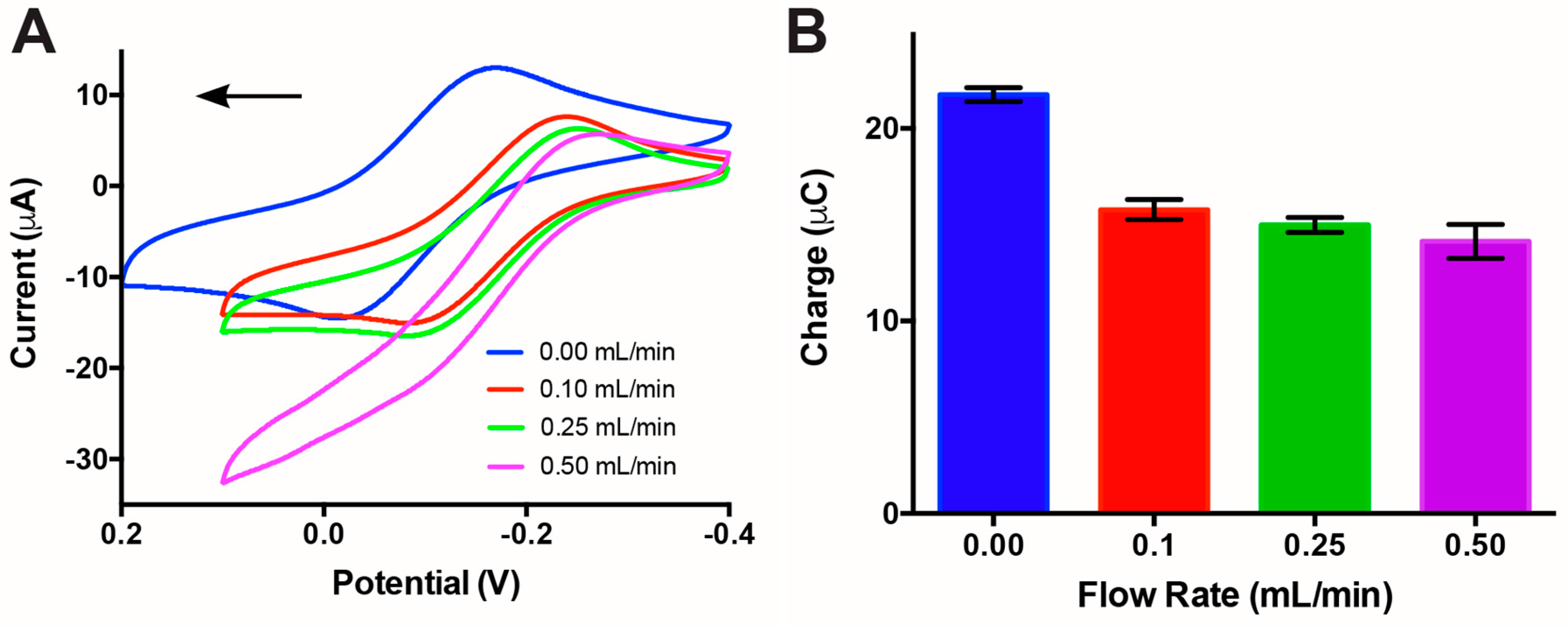
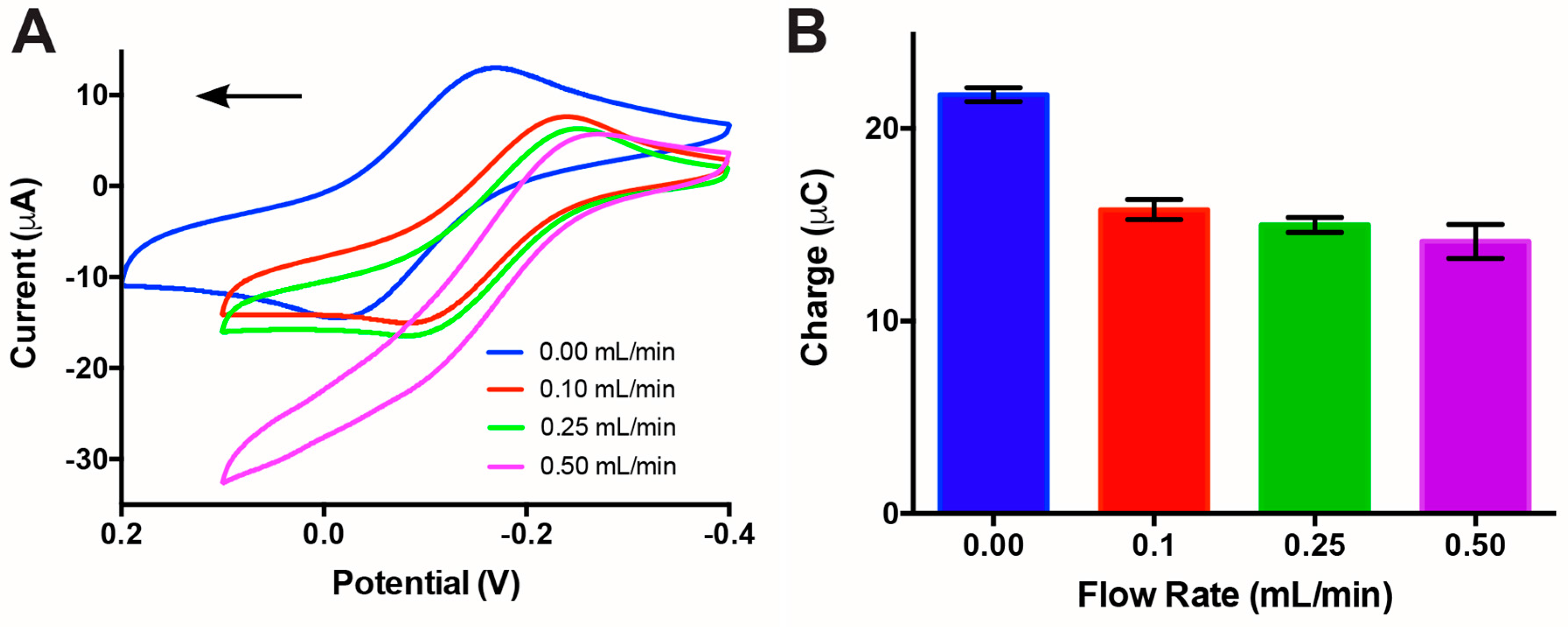
3.2. Impact of Flow Rate on Electrochemical Sensing
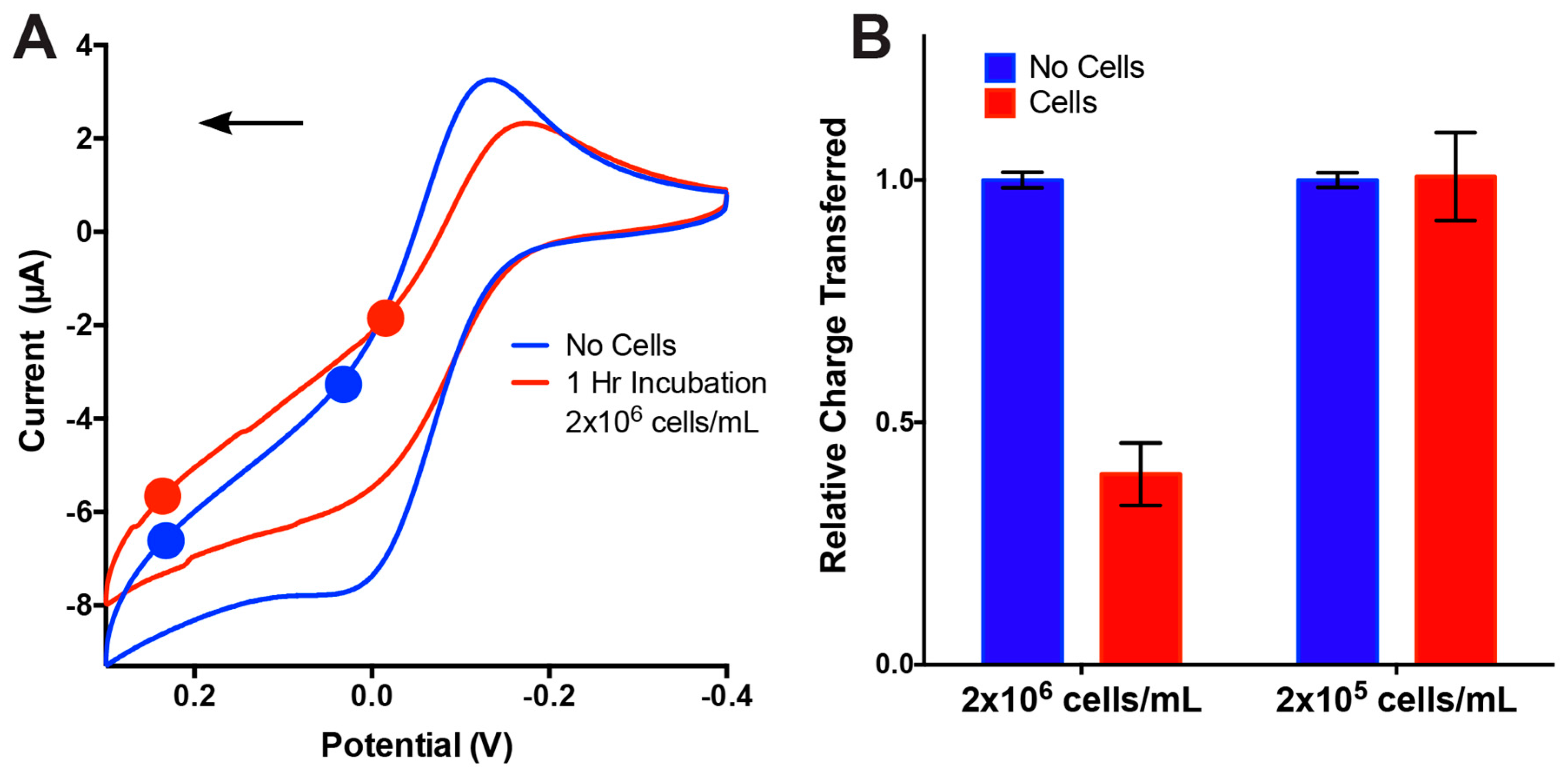
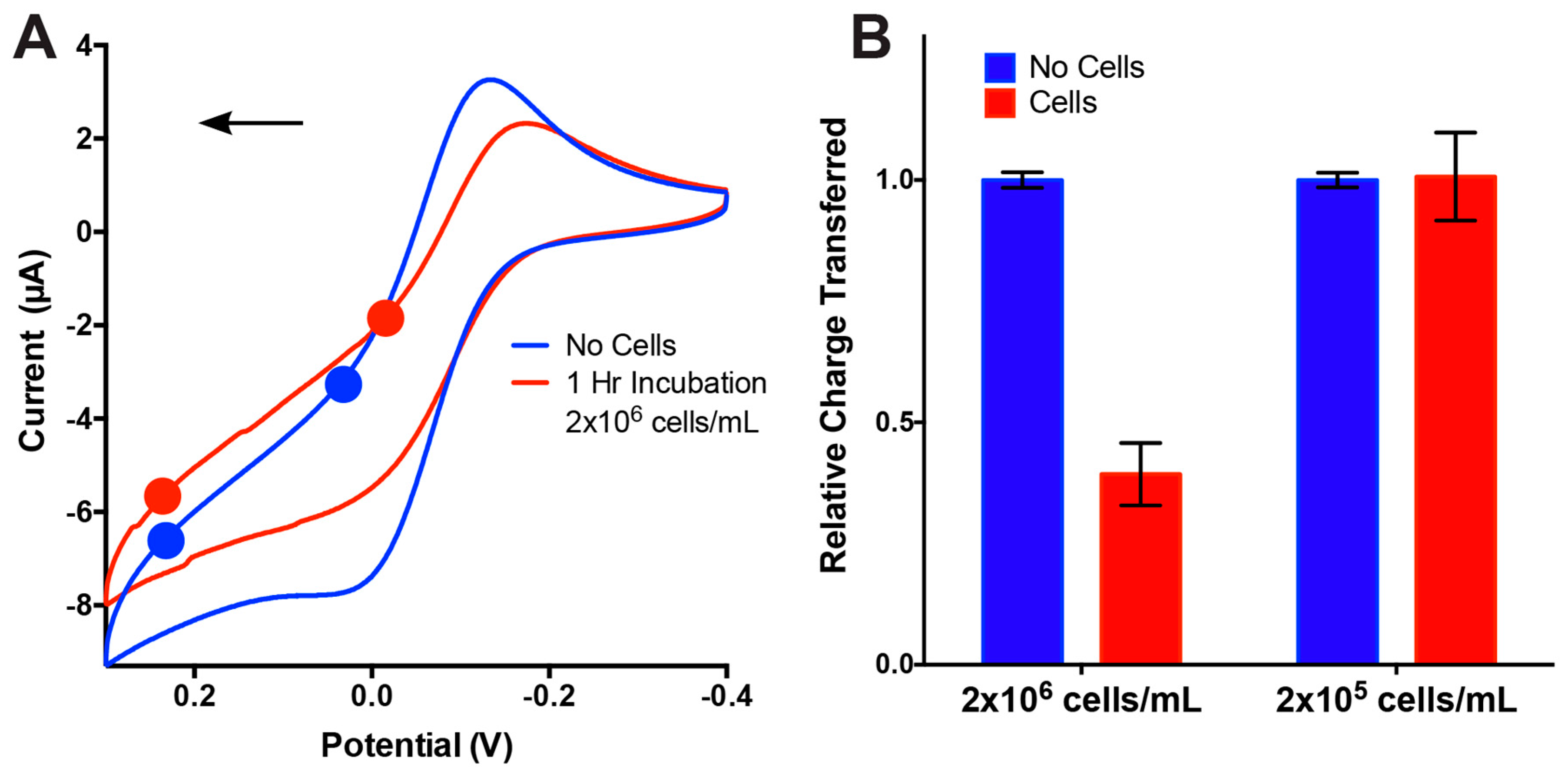
3.3. On-Chip Cell Detection
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cordell, G.A. Sustainable medicines and global health care. Planta Med. 2011, 77, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.D.; Linder, V.; Sia, S.K.; Daar, A.S.; Thorsteinsdottir, H.; Martin, D.K. Lab-on-a-chip devices for global health: Past studies and future opportunities. Lab Chip 2007, 7, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Yager, P.; Domingo, G.J.; Gerdes, J. Point-of-care diagnostics for global health. Annu. Rev. Biomed. Eng. 2008, 10, 107–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wright, G.; Yang, Y. Materials and techniques for electrochemical biosensor design and construction. Biosens. Bioelectron. 2000, 15, 273–282. [Google Scholar] [CrossRef]
- Dungchai, W.; Chailapakul, O.; Henry, C.S. Use of multiple colorimetric indicators for paper-based microfluidic devices. Anal. Chim. Acta 2010, 674, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Chabinyc, M.L.; Chiu, D.T.; McDonald, J.C.; Stroock, A.D.; Christian, J.F.; Karger, A.M.; Whitesides, G.M. An integrated fluorescence detection system in poly (dimethylsiloxane) for microfluidic applications. Anal. Chem. 2001, 73, 4491–4498. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Nijhuis, C.A.; Gong, J.; Chen, X.; Kumachev, A.; Martinez, A.W.; Narovlyansky, M.; Whitesides, G.M. Electrochemical sensing in paper-based microfluidic devices. Lab Chip 2010, 10, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Kwakye, S.; Goral, V.N.; Baeumner, A.J. Electrochemical microfluidic biosensor for nucleic acid detection with integrated minipotentiostat. Biosens. Bioelectron. 2006, 21, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Dhahi, T.H.S.; Bin Hashim, U.D.A.; Ahmed, N.M.; Mat Taib, A. A review on the electrochemical sensors and biosensors composed of nanogaps as sensing material. J. Optoelectron. Adv. Mater. 2010, 12, 1857–1862. [Google Scholar]
- Bhattacharyya, A.; Klapperich, C.M. Design and testing of a disposable microfluidic chemiluminescent immunoassay for disease biomarkers in human serum samples. Biomed. Microdevices 2007, 9, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Khine, M.; Ionescu-Zanetti, C.; Blatz, A.; Wang, L.-P.; Lee, L.P. Single-cell electroporation arrays with real-time monitoring and feedback control. Lab Chip 2007, 7, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ziaie, B.; Baldi, A.; Lei, M.; Gu, Y.; Siegel, R.A. Hard and soft micromachining for BioMEMS: Review of techniques and examples of applications in microfluidics and drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Nguyen, D.; Chen, A.; Lew, V.; Khine, M. Unconventional low-cost fabrication and patterning techniques for point of care diagnostics. Ann. Biomed. Eng. 2011, 39, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Waits, C.M.; Morgan, B.; Kastantin, M.; Ghodssi, R. Microfabrication of 3D silicon MEMS structures using gray-scale lithography and deep reactive ion etching. Sens. Actuators A Phys. 2005, 119, 245–253. [Google Scholar] [CrossRef]
- Sonney, S.; Shek, N.; Moran-Mirabal, J.M. Rapid bench-top fabrication of poly(dimethylsiloxane)/polystyrene microfluidic devices incorporating high-surface-area sensing electrodes. Biomicrofluidics 2015, 9, 026501. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, P.S.; Manz, A. Lab-on-a-chip: Microfluidics in drug discovery. Nat. Rev. Drug Discov. 2006, 5, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Eddings, M.A.; Johnson, M.A.; Gale, B.K. Determining the optimal PDMS–PDMS bonding technique for microfluidic devices. J. Micromech. Microeng. 2008, 18, 67001. [Google Scholar] [CrossRef]
- Faustino, V.; Catarino, S.O.; Lima, R.; Minas, G. Biomedical microfluidic devices by using low-cost fabrication techniques: A review. J. Biomech. 2016, 49, 2280–2292. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Sung, K.E.; Burns, M.A. Microstencils for the patterning of nontraditional materials. Langmuir 2006, 22, 5392–5397. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Moran-Mirabal, J. Highly bendable and stretchable electrodes based on micro/nanostructured gold films for flexible sensors and electronics. Adv. Electron. Mater. 2016, 2, 1500345. [Google Scholar] [CrossRef]
- Behl, M.; Lendlein, A. Shape-memory polymers. Mater. Today 2007, 10, 20–28. [Google Scholar] [CrossRef]
- Liu, C.; Qin, H.; Mather, P.T. Review of progress in shape-memory polymers. J. Mater. Chem. 2007, 17, 1543. [Google Scholar] [CrossRef]
- Cummins, G.; Desmulliez, M.P.Y. Inkjet printing of conductive materials: A review. Circuit World 2012, 38, 193–213. [Google Scholar] [CrossRef]
- Derby, B. Inkjet Printing of Functional and Structural Materials: Fluid Property Requirements, Feature Stability, and Resolution. Annu. Rev. Mater. Res. 2010, 40, 395–414. [Google Scholar] [CrossRef]
- Carrilho, E.; Martinez, A.W.; Whitesides, G.M. Understanding wax printing: A simple micropatterning process for paper-based microfluidics. Anal. Chem. 2009, 81, 7091–7095. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Phillips, S.T.; Whitesides, G.M.; Carrilho, E. Diagnostics for the developing world: Microfluidic paper-based analytical devices. Anal. Chem. 2010, 82, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Dungchai, W.; Chailapakul, O.; Henry, C.S. Electrochemical detection for paper-based microfluidics. Anal. Chem. 2009, 81, 5821–5826. [Google Scholar] [CrossRef] [PubMed]
- Kitson, P.J.; Rosnes, M.H.; Sans, V.; Dragone, V.; Cronin, L. Configurable 3D-Printed millifluidic and microfluidic “lab on a chip” reactionware devices. Lab Chip 2012, 12, 3267. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.C.; Erkal, J.L.; Lockwood, S.Y.; Chen, C.; Spence, D.M. Evaluation of 3D printing and its potential impact on biotechnology and the chemical sciences. Anal. Chem. 2014, 86, 3240–3253. [Google Scholar] [CrossRef] [PubMed]
- Gabardo, C.M.; Zhu, Y.; Soleymani, L.; Moran-Mirabal, J.M. Bench-top fabrication of hierarchically structured high-surface-area electrodes. Adv. Funct. Mater. 2013, 23, 3030–3039. [Google Scholar] [CrossRef]
- Fu, C.C.; Grimes, A.; Long, M.; Ferri, C.G.L.; Rich, B.D.; Ghosh, S.; Ghosh, S.; Lee, L.P.; Gopinathan, A.; Khine, M. Tunable nanowrinkles on shape memory polymer sheets. Adv. Mater. 2009, 21, 4472–4476. [Google Scholar] [CrossRef]
- Pegan, J.D.; Ho, A.Y.; Bachman, M.; Khine, M. Flexible shrink-induced high surface area electrodes for electrochemiluminescent sensing. Lab Chip 2013, 13, 4205–4209. [Google Scholar] [CrossRef] [PubMed]
- Gabardo, C.M.; Yang, J.; Smith, N.J.; Adams-McGavin, R.C.; Soleymani, L. Programmable Wrinkling of Self-Assembled Nanoparticle Films on Shape Memory Polymers. ACS Nano 2016, 10, 8829–8836. [Google Scholar] [CrossRef] [PubMed]
- Bowden, N.; Brittain, S.; Evans, A.G.; Hutchinson, J.W.; Whitesides, G.M. Spontaneous formation of ordered structures in thin films of metals supported on an elastomeric polymer. Nature 1998, 393, 146–149. [Google Scholar]
- Lacour, S.P.; Wagner, S.; Huang, Z.; Suo, Z. Stretchable gold conductors on elastomeric substrates. Appl. Phys. Lett. 2003, 82, 2404–2406. [Google Scholar] [CrossRef]
- Meng, Q.; Hu, J. A review of shape memory polymer composites and blends. Compos. Part A Appl. Sci. Manuf. 2009, 40, 1661–1672. [Google Scholar] [CrossRef]
- Woo, S.M.; Gabardo, C.M.; Soleymani, L. Prototyping of wrinkled nano-/microstructured electrodes for electrochemical DNA detection. Anal. Chem. 2014, 86, 12341–12347. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jayatissa, A.H. Detection of organic vapors by graphene films functionalized with metallic nanoparticles. J. Appl. Phys. 2012, 112. [Google Scholar] [CrossRef]
- Steuwe, C.; Kaminski, C.F.; Baumberg, J.J.; Mahajan, S. Surface enhanced coherent anti-stokes Raman scattering on nanostructured gold surfaces. Nano Lett. 2011, 11, 5339–5343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lang, X.; Hirata, A.; Chen, M. Wrinkled nanoporous gold films with ultrahigh surface-enhanced raman scattering enhancement. ACS Nano 2011, 5, 4407–4413. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Koo, L.Y.; Wang, W.M.; Lauffenburger, D.A.; Griffith, L.G.; Jensen, K.F. Microfluidic Shear Device for Quantitative Analysis of Cell Adhesion. Anal. Chem. 2004, 76, 5257–5264. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saem, S.; Zhu, Y.; Luu, H.; Moran-Mirabal, J. Bench-Top Fabrication of an All-PDMS Microfluidic Electrochemical Cell Sensor Integrating Micro/Nanostructured Electrodes. Sensors 2017, 17, 732. https://doi.org/10.3390/s17040732
Saem S, Zhu Y, Luu H, Moran-Mirabal J. Bench-Top Fabrication of an All-PDMS Microfluidic Electrochemical Cell Sensor Integrating Micro/Nanostructured Electrodes. Sensors. 2017; 17(4):732. https://doi.org/10.3390/s17040732
Chicago/Turabian StyleSaem, Sokunthearath, Yujie Zhu, Helen Luu, and Jose Moran-Mirabal. 2017. "Bench-Top Fabrication of an All-PDMS Microfluidic Electrochemical Cell Sensor Integrating Micro/Nanostructured Electrodes" Sensors 17, no. 4: 732. https://doi.org/10.3390/s17040732
APA StyleSaem, S., Zhu, Y., Luu, H., & Moran-Mirabal, J. (2017). Bench-Top Fabrication of an All-PDMS Microfluidic Electrochemical Cell Sensor Integrating Micro/Nanostructured Electrodes. Sensors, 17(4), 732. https://doi.org/10.3390/s17040732