
Rationally Designing Aptamer Sequences with Reduced Affinity for Controlled Sensor Performance

Abstract
:1. Introduction
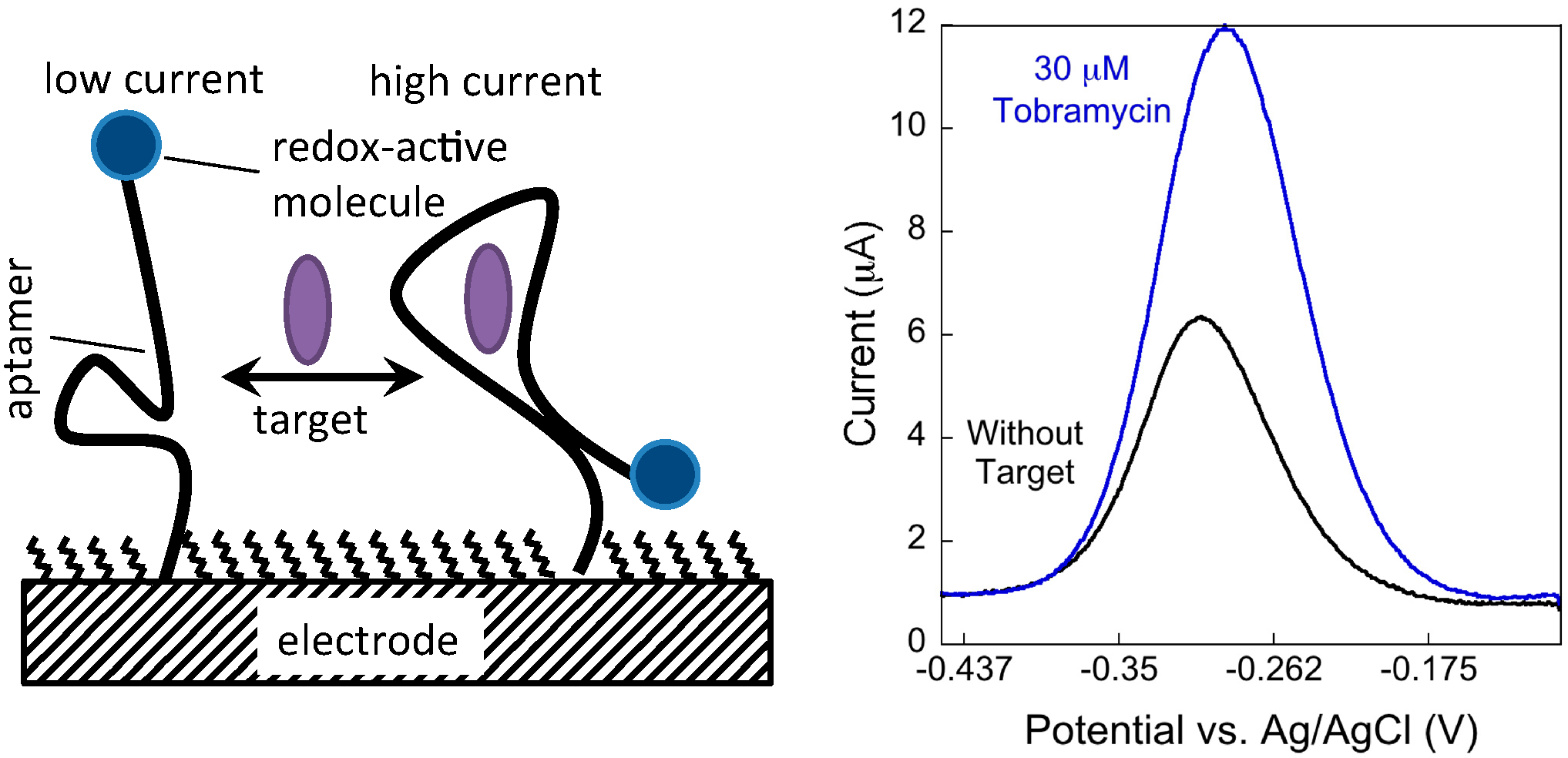
2. Experimental Section
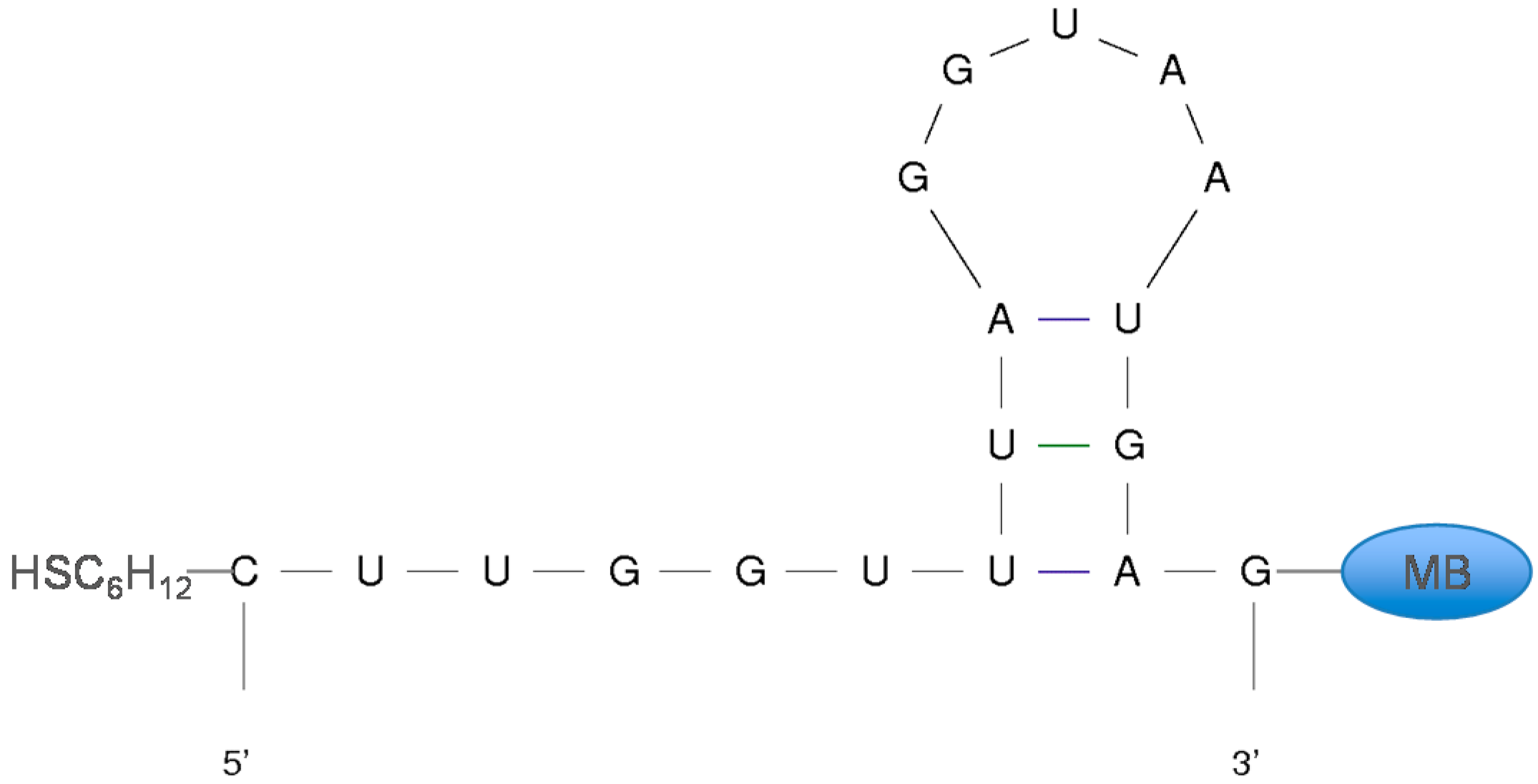
2.1. Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
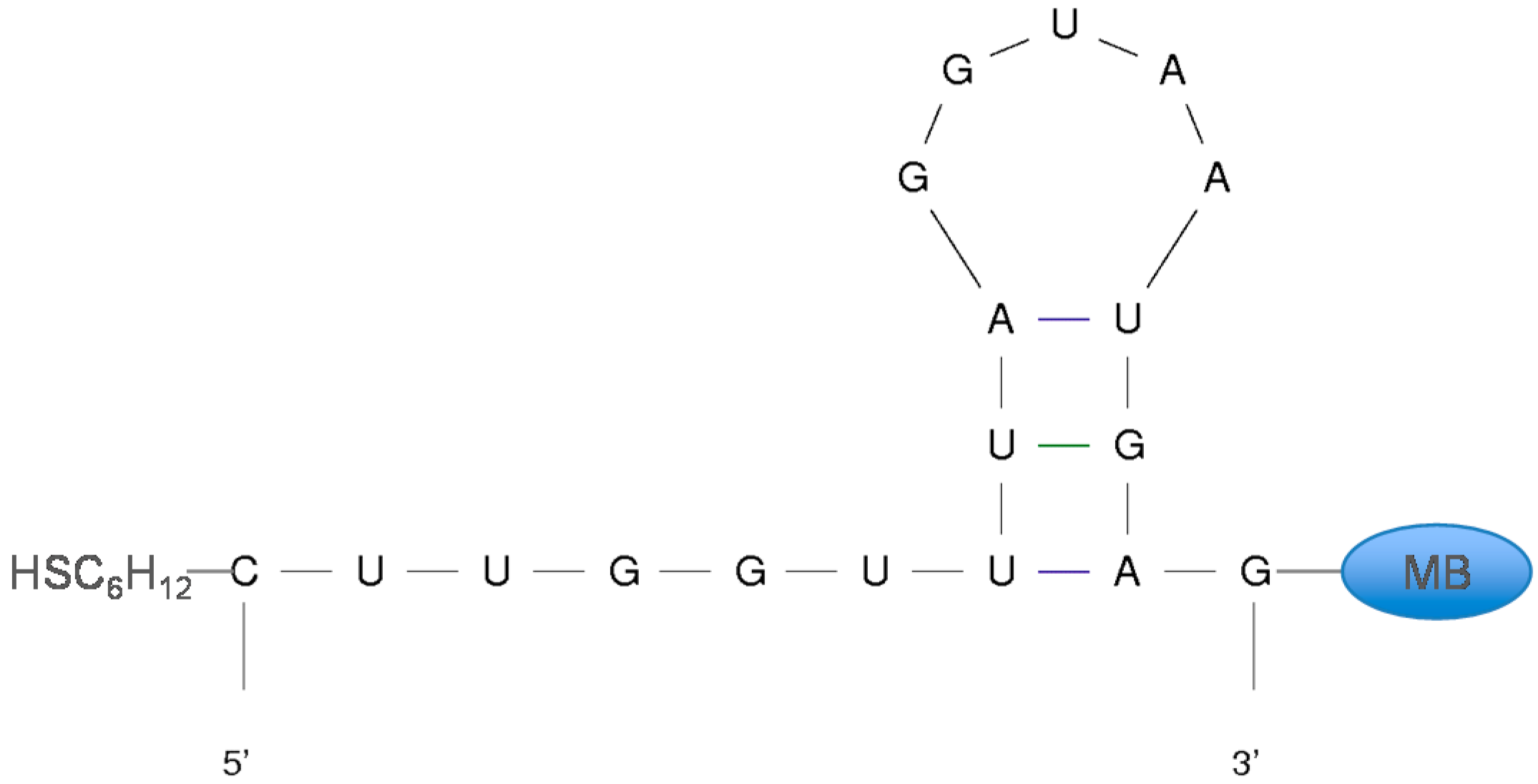
{kind=link}
| Sequence Name | Sequence |
|---|---|
| High-Gain Parent | 5'-HSC6H12-CUUGGUUUAGGUAAUGAG-MB-3' |
| 7UG | 5'-HSC6H12-CUUGGUGUAGGUAAUGAG-MB-3' |
| 7UC | 5'-HSC6H12-CUUGGUCUAGGUAAUGAG-MB-3' |
| 16GU | 5'-HSC6H12-CUUGGUUUAGGUAAUUAG-MB-3' |
| 3-UAC | 5'-HSC6H12-CUUGGUUUAGGUAAUGAGUAC-MB-3' |
2.2. Electrochemical Aptamer-Based (E-AB) Sensor Fabrication
2.3. Electrochemical Measurements

3. Results and Discussion
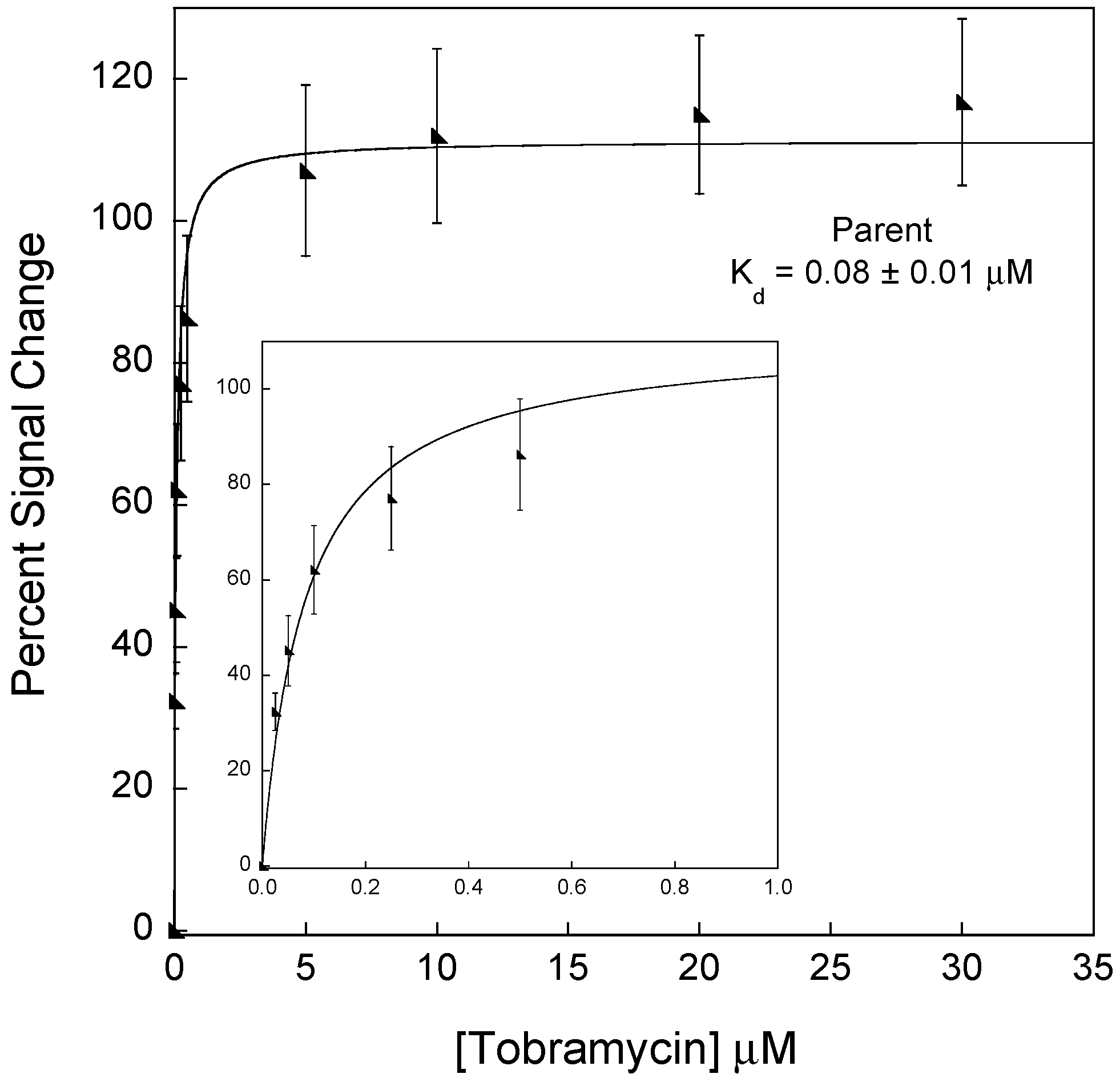
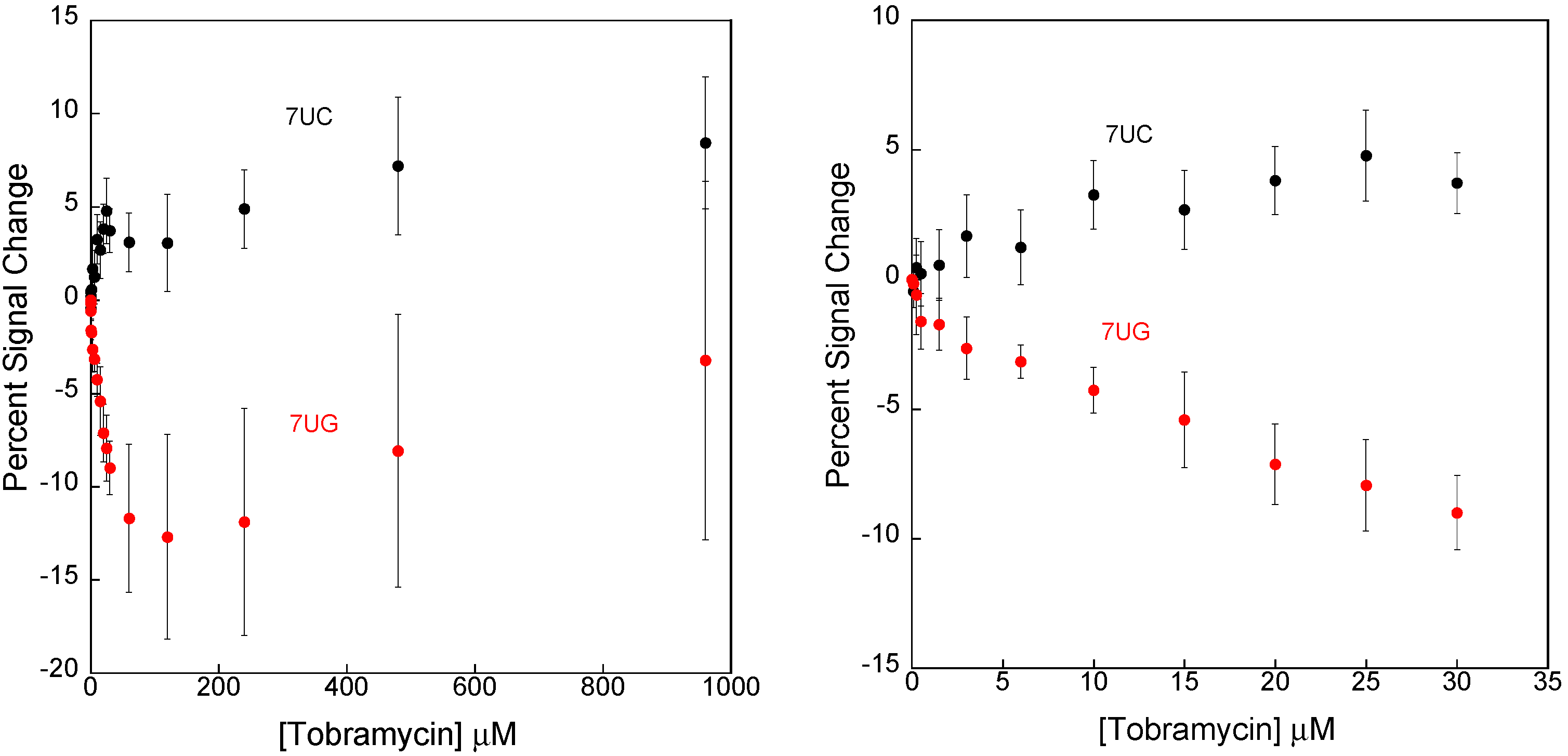
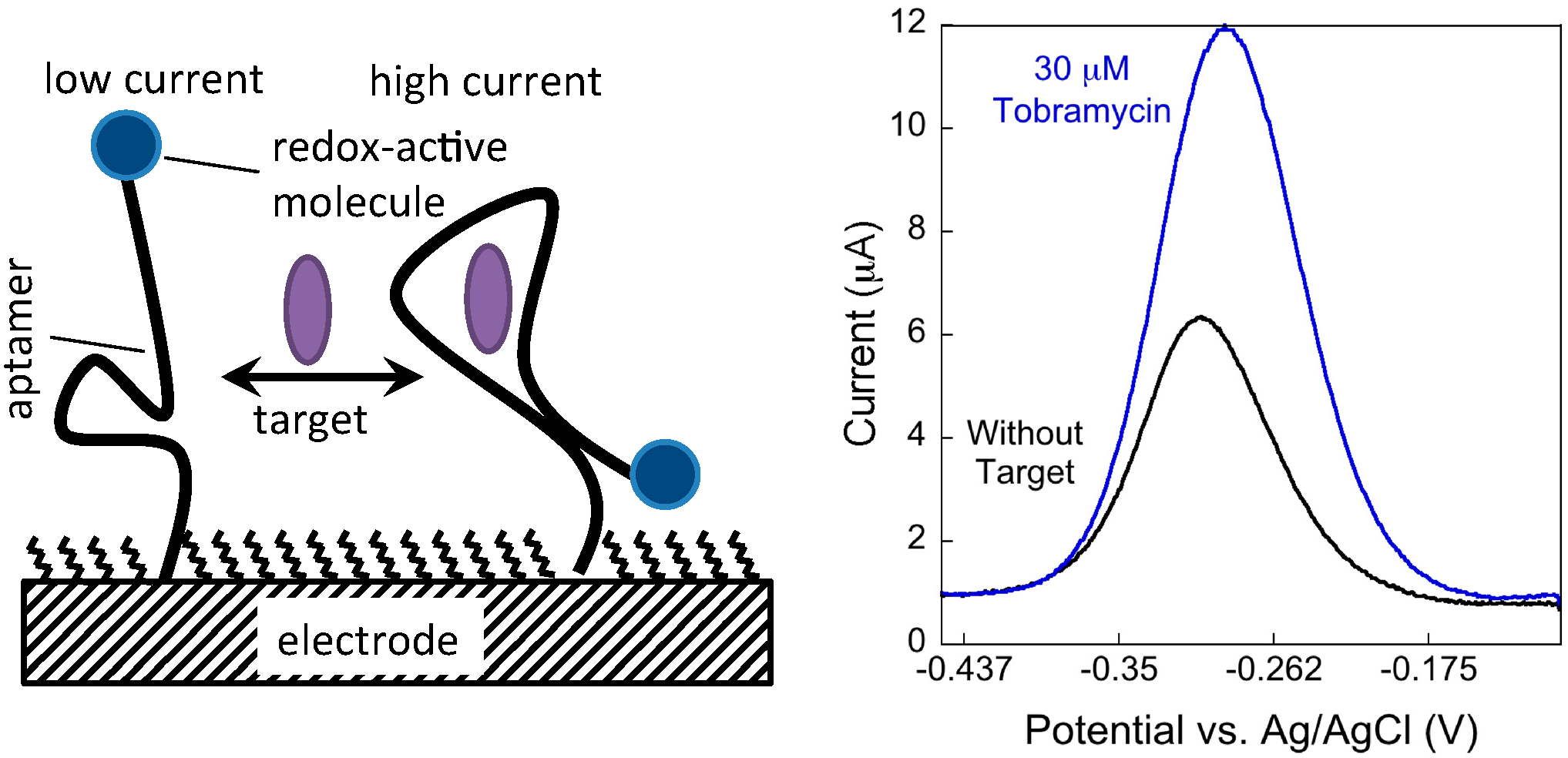
3.1. Disrupting the Aptamer–Target Interaction for Reduced Affinity Sensors
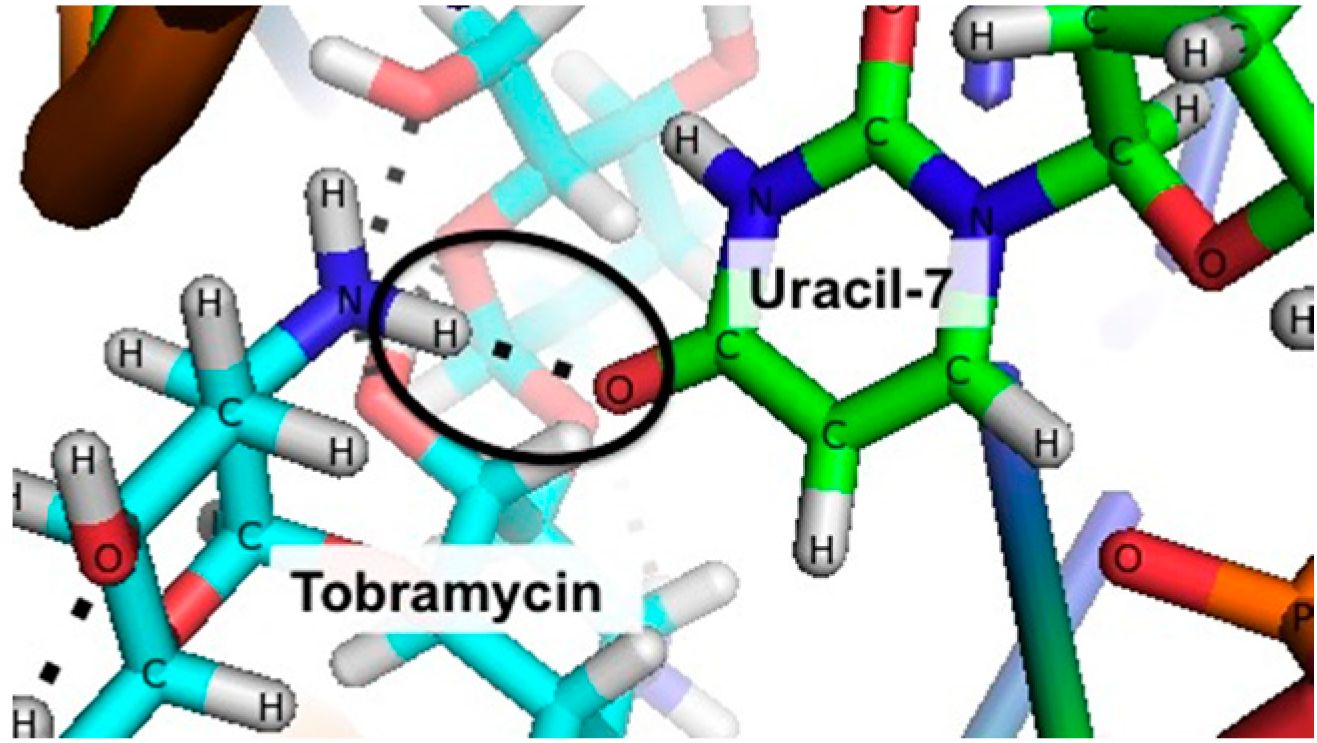
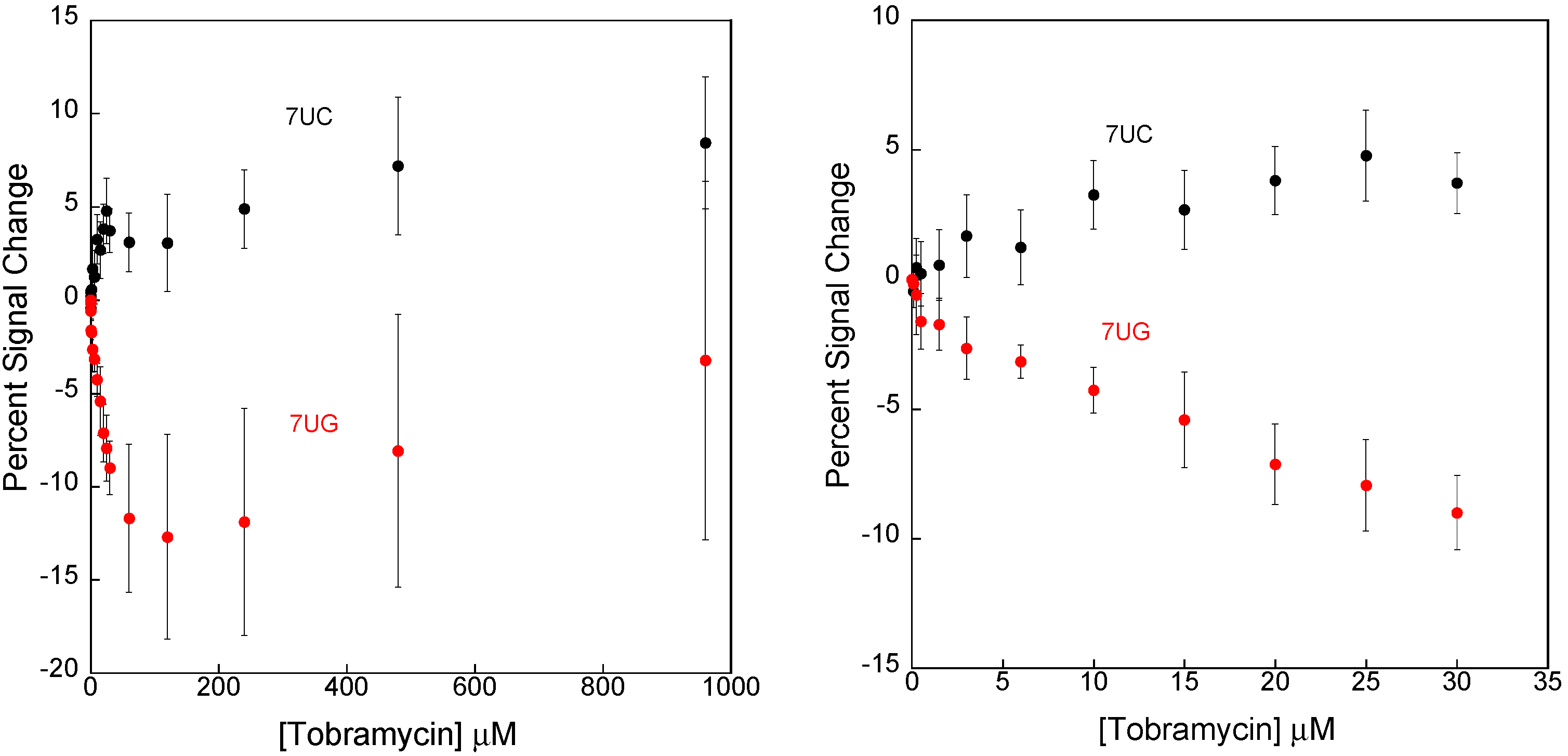
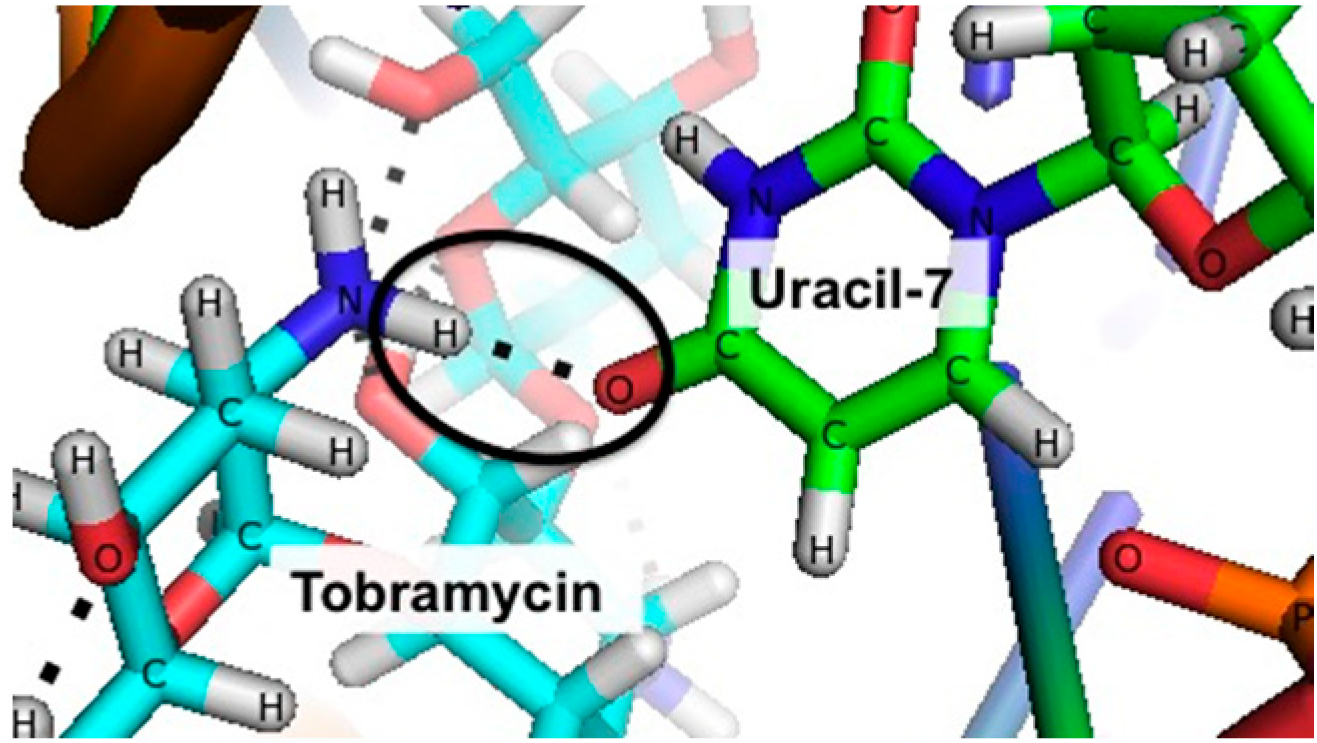
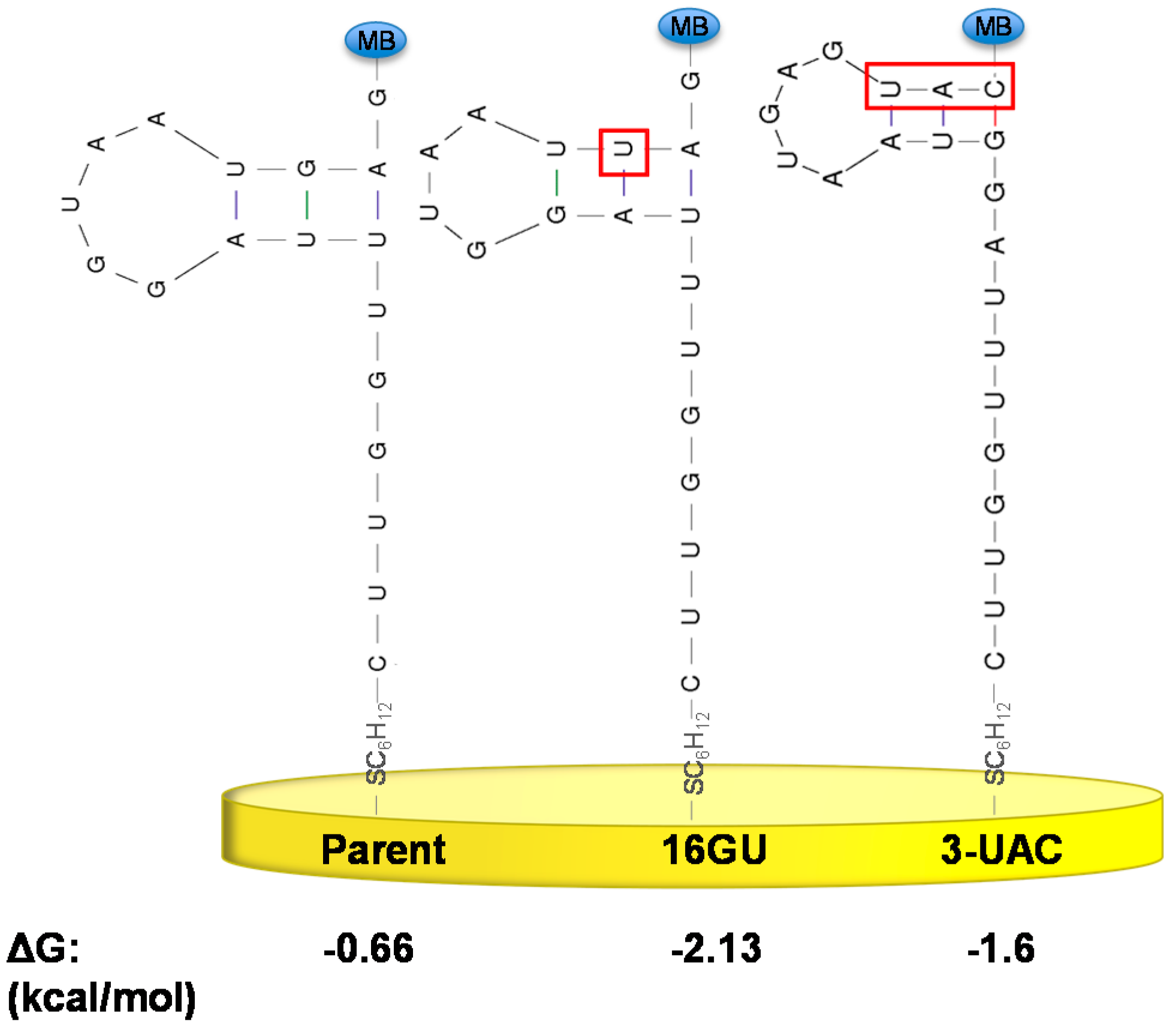
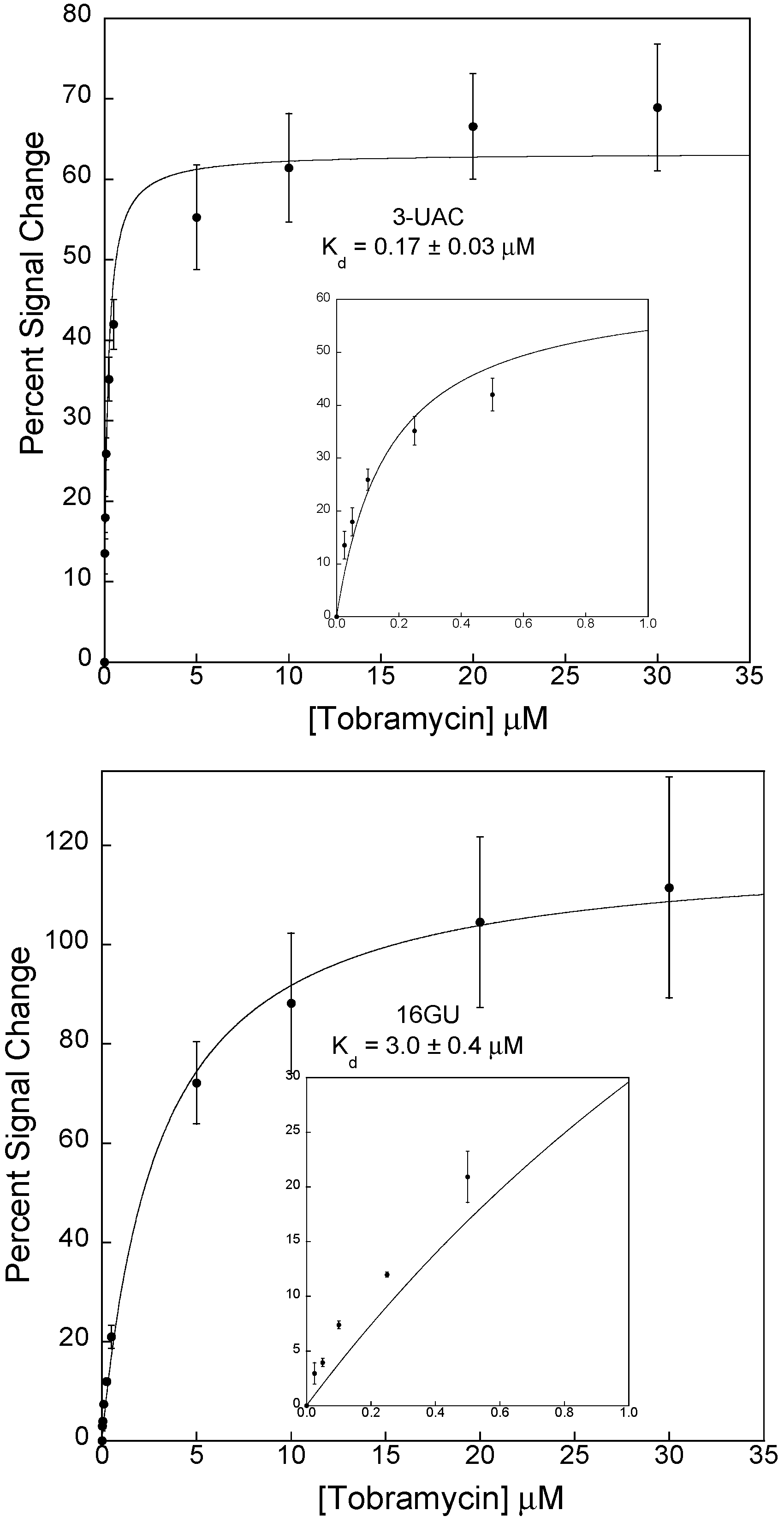
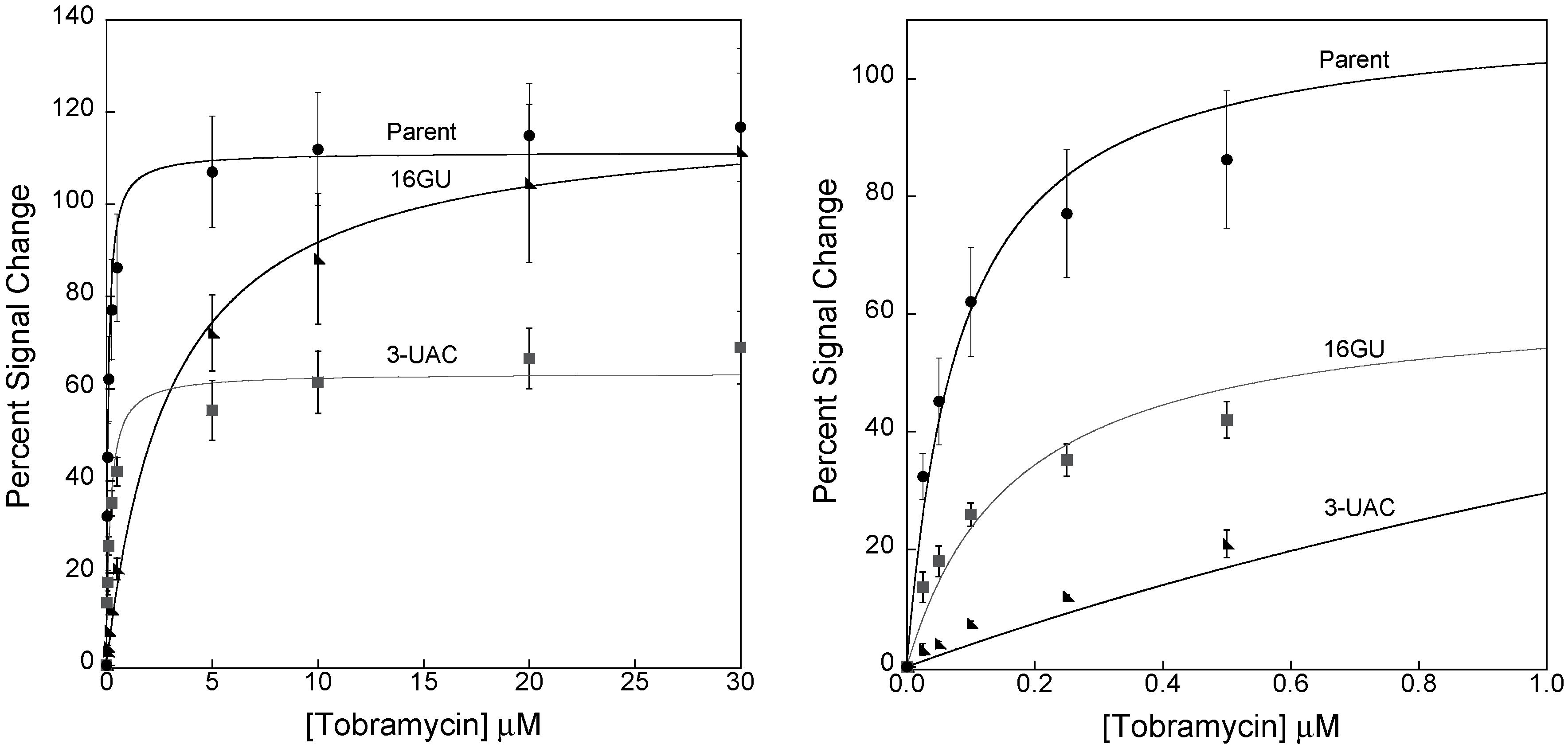
3.2. Stabilizing an Alternative Fold for Reduced Affinity Sensors
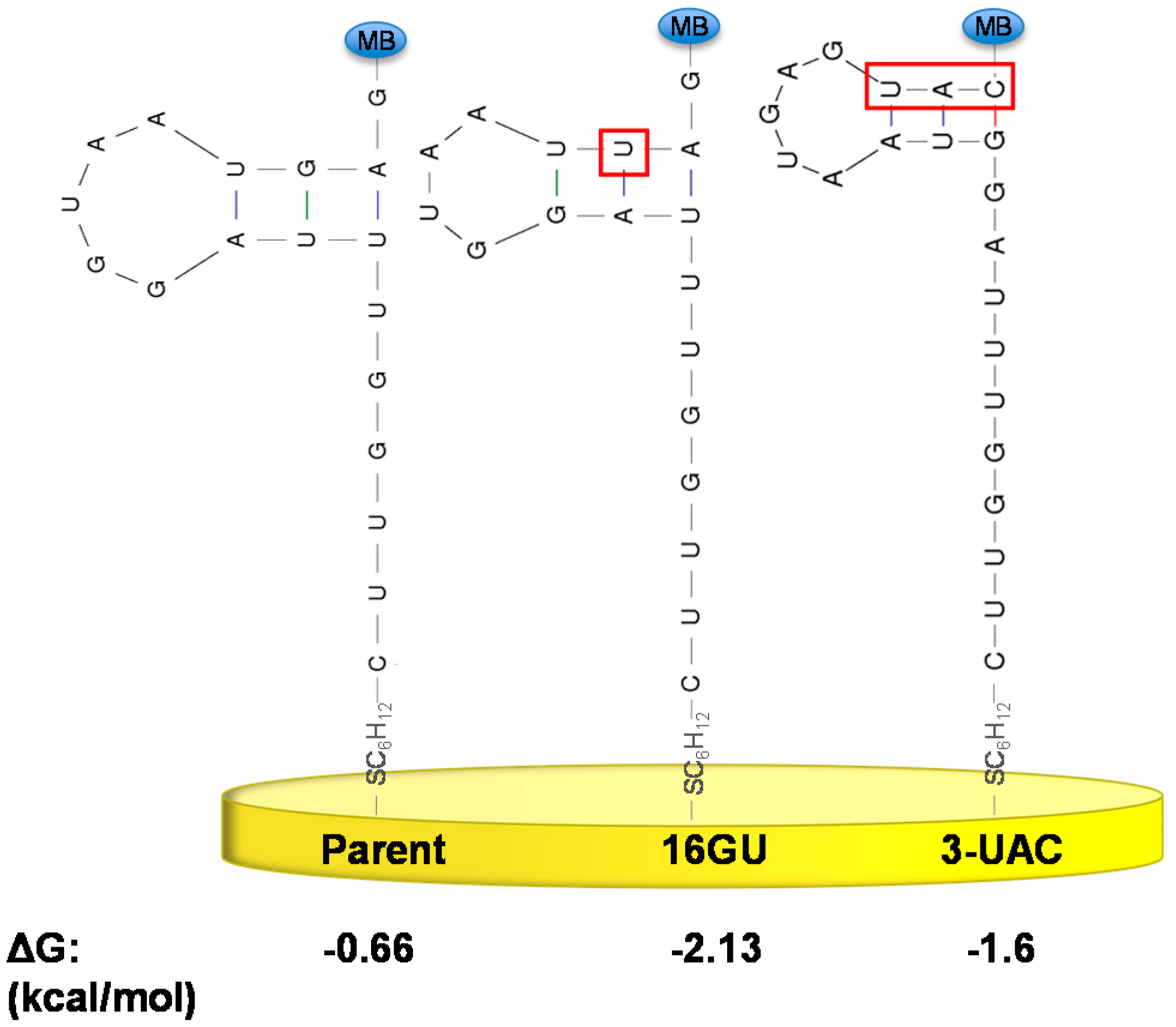
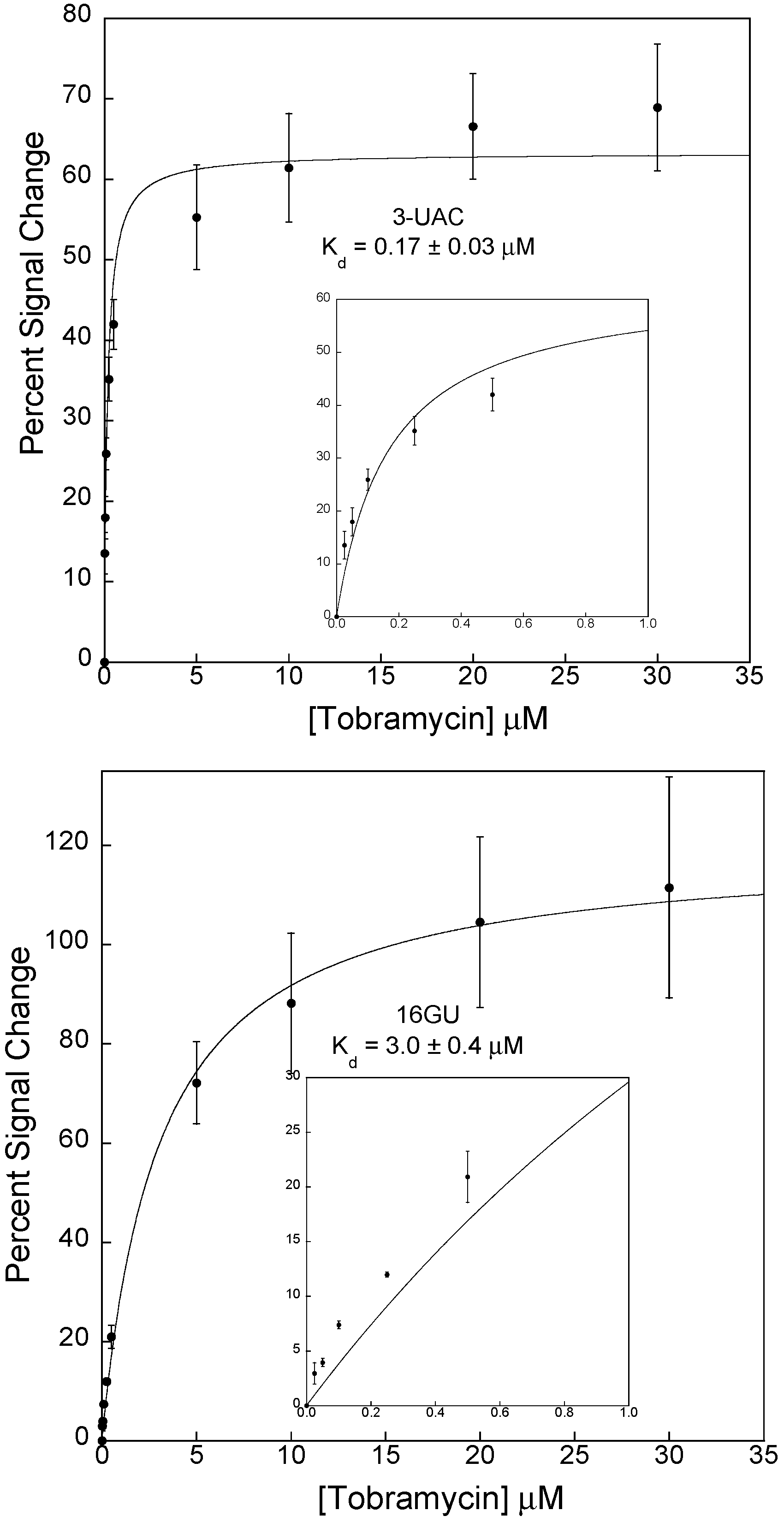
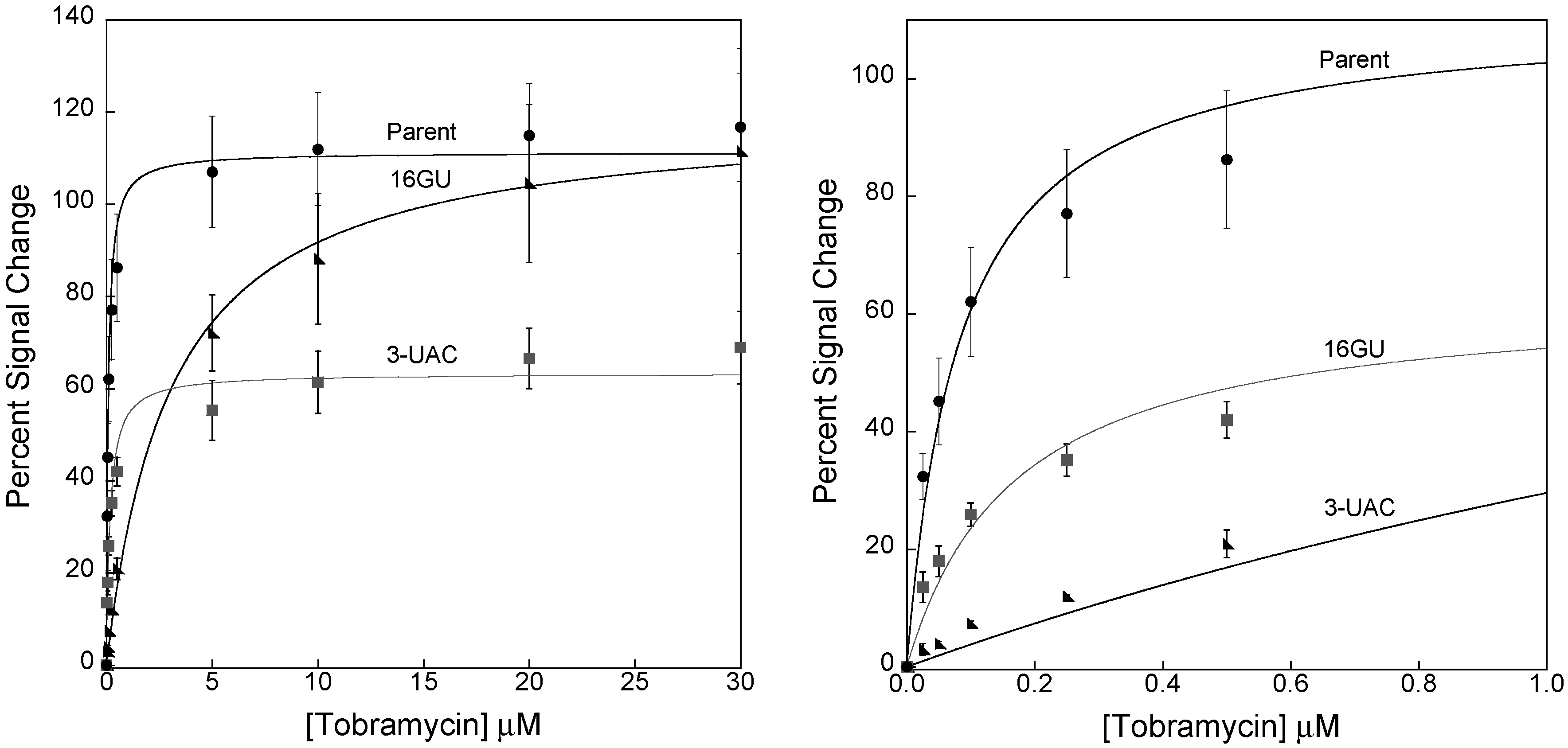
4. Conclusions
Acknowledgments
Author Contributions
Supplementary Materials
Conflicts of Interest
References
- Wilson, G.S.; Yibai, H. Enzyme-Based Biosensors for in Vivo Measurements. Chem. Rev. 2000, 100, 2693–2704. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, N.J.; Halsall, H.B.; Heineman, W.R. Electrochemical biosensors. Chem. Soc. Rev. 2010, 39, 1747–1763. [Google Scholar] [CrossRef] [PubMed]
- Yang, H. Enzyme-Based ultrasensitive electrochemical biosensors. Curr. Opin. Chem. Biol. 2012, 16, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, D.W.; Leblanc, G.; Meschievitz, M.E.; Cliffel, D.E. Electrochemical Sensors and Biosensors. Anal. Chem. 2012, 84, 685–707. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Morris, M.D.; Macazo, F.C.; Schoukroun-Barnes, L.R.; White, R.J. The Current and Future Role of Aptamers in Electroanalysis. J. Electrochem. Soc. 2014, 161, H301–H313. [Google Scholar] [CrossRef]
- Schoukroun-Barnes, L.R.; Wagan, S.; White, R.J. Enhancing the Analytical Performance of Electrochemical RNA Aptamer-Based Sensors for Sensitive Detection of Aminoglycoside Antibiotics. Anal. Chem. 2014, 86, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.A.; Miller, E.a; Plaxco, K.W. Reagentless measurement of aminoglycoside antibiotics in blood serum via an electrochemical, ribonucleic acid aptamer-based biosensor. Anal. Chem. 2010, 82, 7090–7095. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Anslyn, E.V. Signal amplification by allosteric catalysis. Angew. Chem. Int. Ed. 2006, 45, 1190–1196. [Google Scholar] [CrossRef]
- Liu, J.; Wagan, S.; Davila Morris, M.; Taylor, J.; White, R.J. Achieving Reproducible Performance of Electrochemical, Folding Aptamer-Based Sensors on Microelectrodes: Challenges and Prospects. Anal. Chem. 2014, 86, 11417–11424. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Claussen, J.C.; McLamore, E.S.; ul Haque, A.; Jaroch, D.; Diggs, A.R.; Calvo-Marzal, P.; Rickus, J.L.; Porterfield, D.M. A comparative study of enzyme immobilization strategies for multi-walled carbon nanotube glucose biosensors. Nanotechnology 2011, 22, 355502. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yan, X.; Zhao, J.; Liu, X.; Song, Y.; Luo, N.; Zhang, Y. Improved glucose electrochemical biosensor by appropriate immobilization of nano-ZnO. Colloids Surf. B. Biointerfaces 2011, 82, 168–72. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, M.P.; Reipa, V.; Holden, M.J.; Vilker, V.L. Improving the Cytochrome P450 Enzyme System for Electrode-Driven Biocatalysis of Styrene Epoxidation. Biotechnol. Prog. 2000, 16, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Interactions, H.À.G. Enhancing Binding Affinity by the Cooperativity between Host Conformation and Host–Guest Interactions. J. Am. Chem. Soc. 2011, 133, 8862–8865. [Google Scholar] [CrossRef] [PubMed]
- Schlehuber, S.; Skerra, A. Tuning ligand affinity, specificity, and folding stability of an engineered lipocalin variant—A so-called “anticalin ”—Using a molecular random approach. Biophys. Chem. 2002, 96, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Houk, K.N.; Leach, A.G.; Kim, S.P.; Zhang, X. Binding affinities of host-guest, protein-ligand, and protein-transition-state complexes. Angew. Chem. Int. Ed. 2003, 42, 4872–4897. [Google Scholar] [CrossRef]
- Hernandez, K.; Fernandez-Lafuente, R. Control of protein immobilization: Coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme Microb. Technol. 2011, 48, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Vallée-bélisle, A.; Ricci, F.; Plaxco, K.W. Engineering Biosensors with extended, narrowed, or arbitrarily edited dynamic range. J. Am. Chem. Soc. 2012, 20, 2876–2879. [Google Scholar] [CrossRef]
- Kang, D.; Vallée-Bélisle, A.; Porchetta, A.; Plaxco, K.W.; Ricci, F. Re-Engineering electrochemical biosensors to narrow or extend their useful dynamic range. Angew. Chem. Int. Ed. 2012, 51, 6717–6721. [Google Scholar] [CrossRef]
- Ricci, F.; Lai, R.Y.; Heeger, A.J.; Plaxco, K.W.; Sumner, J.J. Effect of molecular crowding on the response of an electrochemical DNA sensor. Langmuir 2007, 23, 6827–6834. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Zuo, X.; Yang, R.; Xia, F.; Plaxco, K.W.; White, R.J. Comparing the properties of electrochemical-based DNA sensors employing different redox tags. Anal. Chem. 2009, 81, 9109–9113. [Google Scholar] [CrossRef] [PubMed]
- Swensen, J.S.; Xiao, Y.; Ferguson, B.S.; Lubin, A.a; Lai, R.Y.; Heeger, A.J.; Plaxco, K.W.; Soh, H.T. Continuous, real-time monitoring of cocaine in undiluted blood serum via a microfluidic, electrochemical aptamer-based sensor. J. Am. Chem. Soc. 2009, 131, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, T.; Cheng, R.R.; White, R.J.; Makarov, D.E.; Plaxco, K.W. A Mechanistic Study of Electron Transfer from the Distal Termini of Electrode-Bound, Single-Stranded DNAs. J. Am. Chem. Soc. 2010, 132, 16120–16126. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Olsen, E.M.; Gothelf, K.V. An RNA aptamer-based electrochemical biosensor for detection of theophylline in serum. J. Am. Chem. Soc. 2008, 130, 4256–4258. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Gothelf, K.V. Effect of serum on an RNA aptamer-based electrochemical sensor for theophylline. Langmuir 2009, 25, 4279–4283. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Gothelf, K.V. Optimization of the Electrochemical RNA-Aptamer Based Biosensor for Theophylline by Using a Methylene Blue Redox Label. Electroanalysis 2009, 21, 1261–1266. [Google Scholar] [CrossRef]
- Ricci, F.; Zari, N.; Caprio, F.; Recine, S.; Amine, A.; Moscone, D.; Palleschi, G.; Plaxco, K.W. Surface chemistry effects on the performance of an electrochemical DNA sensor. Bioelectrochemistry 2009, 76, 208–213. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Plaxco, K.W. Exploiting Binding-Induced Changes in Probe Flexibility for the Optimization of Electrochemical Biosensors. Anal. Chem. 2010, 82, 73–76. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Plaxco, K.W. Engineering New Aptamer Geometries for Electrochemical Aptamer-Based Sensors. Proc. Soc. Photo Opt. Instrum. Eng. 2009, 7321. [Google Scholar] [CrossRef]
- White, R.J.; Rowe, A.A.; Plaxco, K.W. Re-Engineeing aptamers to support reagenless, self-reporting electrochemical sensors. Analyst 2010, 135, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Jarczewska, M.; Kékedy-Nagy, L.; Nielsen, J.S.; Campos, R.; Kjems, J.; Malinowska, E.; Ferapontova, E.E. Electroanalysis of pM-levels of urokinase plasminogen activator in serum by phosphorothioated RNA aptamer. Analyst 2015. [Google Scholar] [CrossRef]
- Farjami, E.; Campos, R.; Nielsen, J.S.; Gothelf, K.V.; Kjems, J.; Ferapontova, E.E. RNA aptamer-based electrochemical biosensor for selective and label-free analysis of dopamine. Anal. Chem. 2013, 85, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Patel, D. Solution sturucture of the tobramycin-RNA aptamer complex. Nat. Struct. Biol. 1998, 5, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Setia, U.; Gross, P.A. Administration of tobramycin and gentamicin by the intravenous route every 6 hr in patients with normal renal function. J. Infect. Dis. 1976, 134, S125–S129. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Phares, N.; Lubin, A.A.; Xiao, Y.; Plaxco, K.W. Optimization of Electrochemical Aptamer-Based Sensors via Optimization of Probe Packing Density and Surface Chemistry. Langmuir 2008, 24, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rando, R.R. Specific binding of aminoglycoside antibiotics to RNA. Chem. Biol. 1995, 2, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Bromberg, S. Molecular Driving Forces: Statistical Thermodynamics in Chemistry and Biology; Garland Science: New York, NY USA, 2003. [Google Scholar]
- Zuker, M.; Jacobson, A.B. Using reliability information to annotate RNA secondary structures. RNA 1998, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoukroun-Barnes, L.R.; White, R.J. Rationally Designing Aptamer Sequences with Reduced Affinity for Controlled Sensor Performance. Sensors 2015, 15, 7754-7767. https://doi.org/10.3390/s150407754
Schoukroun-Barnes LR, White RJ. Rationally Designing Aptamer Sequences with Reduced Affinity for Controlled Sensor Performance. Sensors. 2015; 15(4):7754-7767. https://doi.org/10.3390/s150407754
Chicago/Turabian StyleSchoukroun-Barnes, Lauren R., and Ryan J. White. 2015. "Rationally Designing Aptamer Sequences with Reduced Affinity for Controlled Sensor Performance" Sensors 15, no. 4: 7754-7767. https://doi.org/10.3390/s150407754
APA StyleSchoukroun-Barnes, L. R., & White, R. J. (2015). Rationally Designing Aptamer Sequences with Reduced Affinity for Controlled Sensor Performance. Sensors, 15(4), 7754-7767. https://doi.org/10.3390/s150407754