Nitrogen-Rich Multinuclear Ferrocenophanes as Multichannel Chemosensor Molecules for Transition and Heavy-Metal Cations

Abstract
: [m.n] Multinuclear ferrocenophanes prepared by aza-Wittig reaction of bisiminophosphoranes derived from 1,1′-diazidoferrocene and isophthaladelhyde or 2,5-diformylthiophene, behave as efficient electrochemical and chromogenic chemosensor molecules for Zn2+, Pb2+, and Hg2+ metal cations. Whereas the OSWV of receptor 3, bearing two m-phenylene units in the bridges, display one oxidation peak, receptor 4 incorporating two thiophene rings in the bridges, exhibits two well-separated oxidation peaks. In both receptors only the addition of Zn2+, Pb2+, and Hg2+ metal cations induced a remarkable anodic shift of ferrocene/ferrocenium redox couple. Likewise, in the absorption spectra of these receptors the low energy band is red-shifted by Δλ = 165 − 209 nm, and these changes promoted a significant color changes which could be used for the naked eye detection of these metal cations. The coordination modes for two representative cases were unveiled by DFT calculations that show an unsual coordination in the [42Pb]2+ complex with the Pb2+ cation in a distorted cubic N4S4 donor cage.1. Introduction
The development of simple and sensitive metal cation sensors continues to be a research area of considerable interest because of the important roles that these species play in biological, pathological or environmental processes. Among the various heavy metal cations, mercury and lead are especially hazardous because they are not biodegradable and can accumulate in the environment, which results in contaminated food and water, thus causing a variety of serious diseases such as neurological, metabolic, cognitive, kidney and motor disorders [1–6]. Consequently, the World Health Organization (WHO) and Environmental Protection Agency (EPA) have strictly defined their concentration limits in drinking water [7]. On the other hand, zinc is the second most abundant transition metal following iron and it plays well known roles in biological processes, the most important being as a structural cofactor in metalloproteins [8]. Thus, zinc metabolism disorders are closely associated with many severe neurological diseases such as Alzheimer's disease, amyotrophic lateral sclerosis and Parkinson's disease [9]. Therefore, during the last decades numerous efforts have been devoted to the development of abiotic receptors able to bind selectively cationic species with a concomitant change in one or more properties of the system, such as redox potentials, absorption or fluorescence spectra. In this context, the redox-active organometallic ferrocene scaffold has largely proved to be a simple and remarkably robust building block for the preparation of derivatives which have been considered as prototype chemosensor molecules displaying interesting electrochemical-sensing properties [10–16].
The advantage associated with the use of these functionalized ferrocene-containing ligands lies in the fact that, upon complexation with metal cations, they undergo significant perturbations of the ferrocene/ferrocinium redox couple and the values of the corresponding oxidation potential shifts are informative about the strength of the recognition event: the closer the binding site to the ferrocene unit, the higher oxidation potential shift. Despite the rich chemistry of ferrocene, [m.m]ferrocenophanes bridged by nitrogen-containing chains remain almost unexplored, and only the preparation and properties of some multinuclear nitrogen-rich [2.2]-, [3.3]-, and [4.4]ferrocenophanes have been reported [17–21].
In connection with our previous studies on the synthesis, structural characterization and properties of new families of azaferrocenophane ligands which incorporate binding sites in the bridge for the purpose of selective recognition and sensing of metal ions, we report herein the synthesis and sensing properties of two new highly preorganized tetraaza[7.7]ferrocenophane systems in which the two ferrocene units are linked to a thiophene or a benzene ring through two aldimine functions giving rise to the corresponding ferrocenophane framework. An interesting feature of the synthetic methodology used is that the starting 1,1′-bis(azido)ferrocene (1), has proved to be an excellent platform on which to build diazaferrocenophane frameworks. The combined effect of the binding capability of the aldimine moieties and the close proximity to the redox center makes this structural motif a likely candidate for displaying selective redox cation-sensing properties.
2. Experimental Section
2.1. General Information
Commercial starting materials were purchased to Aldrich (Madrid, Spain) and they were used without further purification. 1,1′-Bis(azido)ferrocene was prepared as described previously in the literature [17]. All reactions were carried out under N2 and using solvents which were dried by routine procedures. Melting points were determined on a Kofler hot-plate melting point apparatus and are uncorrected. 1H-NMR spectra were recorded on a Bruker AC 300 and 400. The following abbreviations for stating the multiplicity of the signals have been used; s (singlet), bs (broad singlet), d (doublet). Chemical shifts refer to signals of tetramethylsilane. High resolution electrospray (ESI) mass spectra were recorded on a Fisons AUTOSPEC 500 VG spectrometer. Cyclic Voltammetry (CV) and Osteryoung Square Wave Voltammetry (OSWV) techniques were performed with a conventional three-electrode configuration consisting of a carbon working and platinum auxiliary electrodes and a Ag/AgCl reference electrode. The experiments were carried out with a 10−4 M solution of sample in CH2Cl2 containing 0.1 M (n-C4H9)4NPF6 (TBAHP) as supporting electrolyte. All the potential values reported are relative to the decamethylferrocene (DMFc) couple at room temperature. Deoxygenation of the solutions was achieved by bubbling nitrogen for at least 10 min and the working electrode was cleaned after each run. The cyclic voltammograms were recorded with a scan rate of 0.1 Vs−1, while the OSWV were recorded at a scan rate of 100 mVs−1 with a pulse hight of 10 mV and a step time of 50 ms. Typically, receptor (10−4 M) was dissolved in the appropriate solvent (5 mL) and TBAHP (base electrolyte) (0.194 g) added. The guest under investigation was then added as a 2.5 × 10−2 M solution in CH3CN using a microsyringe whilst the electrochemical properties of the solution were monitored. DMFc was used as an external reference both for potential calibration and for reversibility criteria. UV-vis and emission spectra were recorded in the solvents and at the concentrations stated in the text and in the corresponding figure captions.
2.2. General Procedure for the Preparation of [7.7]ferrocenophanes 3, 4 and 5
To a solution of 1,1′-bis(azido)ferrocene (1, 0.1 g, 0.37 mmol) in dry THF (30 mL), Bu3P (0.31 mL, 1.2 mmol) was added. The resulting solution was stirred at room temperature and under nitrogen for 1.5 h. Then, the appropriate dialdehyde (0.37 mmol) was added and the reaction mixtures were refluxed for 12 h. On cooling, the resulting crude was crystallized from CH2Cl2/THF (1/5) to give the corresponding ferrocenophane.
2.2.1. Bis[1,3-phenylene-bis(methylimino)][7.7]ferrocenophane (3)
Red solid, 0.05 g, 21%; m.p. > 300 °C. 1H-NMR (400 MHz, CD2Cl2): δ 8.05 (s, 4H, = C–H), 7.56 (s, 2H, Ar–H2), 7.40 (d, 2H, J = 7.6 Hz, Ar–H5), 6.98 (m, 4H, Ar–H4), 4.62 (bs, 8H, Hα–Fc), 4.34 (bs, 8H, Hβ–Fc). HR-ESIMS m/z: calcd (C36H28N4Fe2, [M+ + 2]): 629.1080; found: 629.1092.
2.2.2. Bis[thiophene-2,5-diylbis(methylimino)][7.7]ferrocenophane (4)
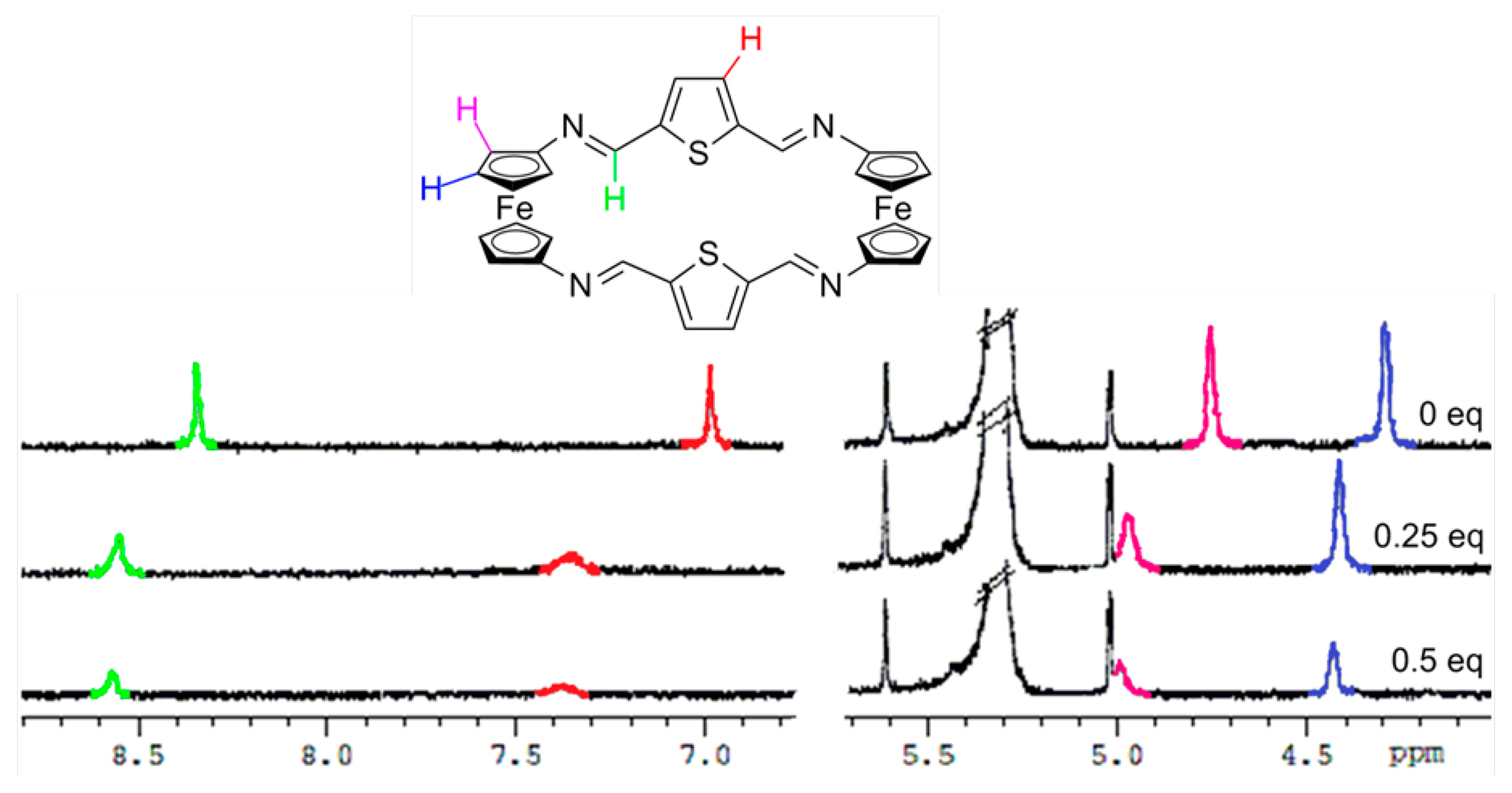
Purple solid, 0.085 g, 38%; m.p. > 300 °C. 1H-NMR (300 MHz, CD2Cl2): δ 8.33 (s, 4H, = C–H), 6.98 (s, 4H, thiophene), 4.74 (bs, 8H, Hα–Fc), 4.28 (bs, 8H, Hβ–Fc). HR-ESIMS m/z: calcd (C32H24N4Fe2 S2, [M+ + 2]): 641.0218; found: 641.0221.
2.2.3. Bis[1,10-phenanthroline-2,9-diylbis(methylimino)][7.7]ferrocenophane (5)
Pink solid, 0.110 g, 36%; m.p. > 300 °C. 1H-NMR (400 MHz, CD2Cl2): δ 8.63 (s, 4H, = C–H), 7.85 (d, 4H, J = 8.4 Hz), 7.70 (d, 4H, J = 8.4 Hz), 7.38 (s, 4H), 4.92 (bs, 4H), 4.82 (bs, 4H), 4.55 (bs, 4H), 4.43 (bs, 4H); HR-ESIMS m/z: calcd (C48H32N8Fe2, [M+ + 2]): 833.1536; found: 833.1529.
2.3. Computational Details
Quantum chemical calculations were performed with the ORCA electronic structure program package [22]. All geometry optimizations were run with tight convergence criteria using the B3LYP [23,24] functional together with the RIJCOSX algorithm [25] and the Ahlrichs' segmented def2-TZVP basis set [26,27], starting from preoptimized geometries obtained with the smaller def2-SVP basis set [28]. For Pb atoms the [SD(60,MDF)] effective core potential was used [29,30]. In all optimizations and energy evaluations, the latest Grimme's semiempirical atom-pair-wise correction (DFT-D3 methods), accounting for the major part of the contribution of dispersion forces to the energy, was included [31]. From these geometries obtained at the above mentioned level, all reported electronic data were obtained by means of single-point (SP) calculations using the same functional as well as the more polarized def2-TZVPP basis set. Reported energies are uncorrected for the zero-point vibrational term. The topological analysis of the electronic charge density, ρ(r), within Bader's Atoms-In-Molecules (AIM) methodology [32–34] was conducted using the AIM2000 software [35,36] and the wavefunctions (electron densities) generated with the Gaussian09 software package, [37] that were also used to perform the natural bond orbital (NBO) population analysis [38,39] with which Wiberg Bond Indices (WBI) [40] were obtained. Figures 6 and 7 were drawn with VMD [41].
3. Results and Discussion
3.1. Synthesis and Characterization of 3, 4 and 5
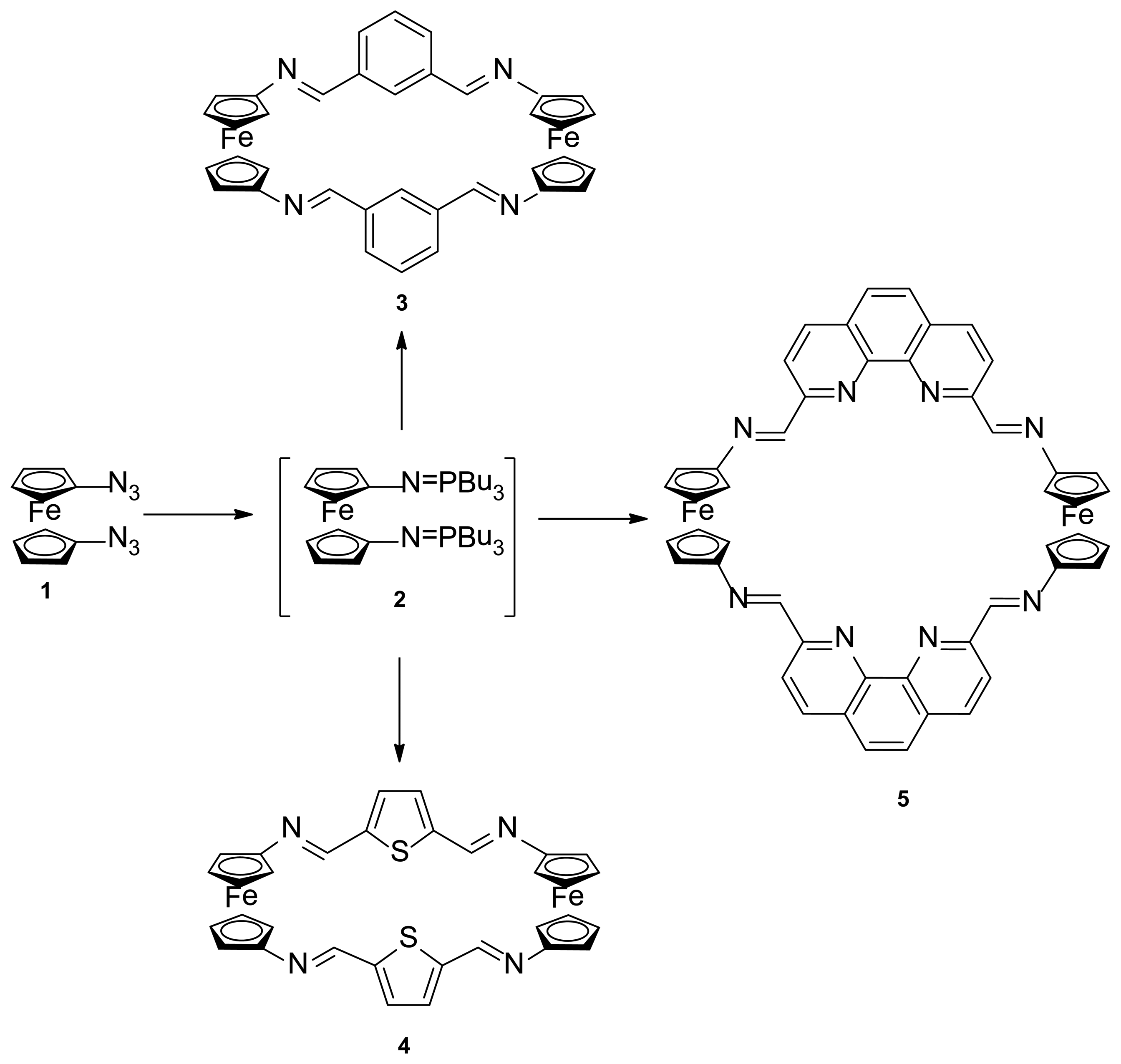
The synthesis of the targets ferrocenophane derivatives 3, 4 and 5 was carried out as depicted in Scheme 1, through a two-step procedure, starting from the previously reported 1,1′-bis(azido)ferrocene (1) [17], which in turn is available by the sequential treatment of ferrocene with n-BuLi followed by reaction with the strong azide-transfer agent 2,4,6-triisopropylbenzenesulfonyl azide (trisyl azide). Thus, compound 1 underwent Staudinger reaction with n-tributylphosphine under anhydrous conditions to give the not isolable bis-iminophosphorane 2 which, subsequently, underwent an aza-Wittig reaction with the appropriate dialdehyde in dry THF giving rise to the aforementioned ferrocenophanes 3, 4 and 5 in 21%, 38% and 36% yield, respectively.
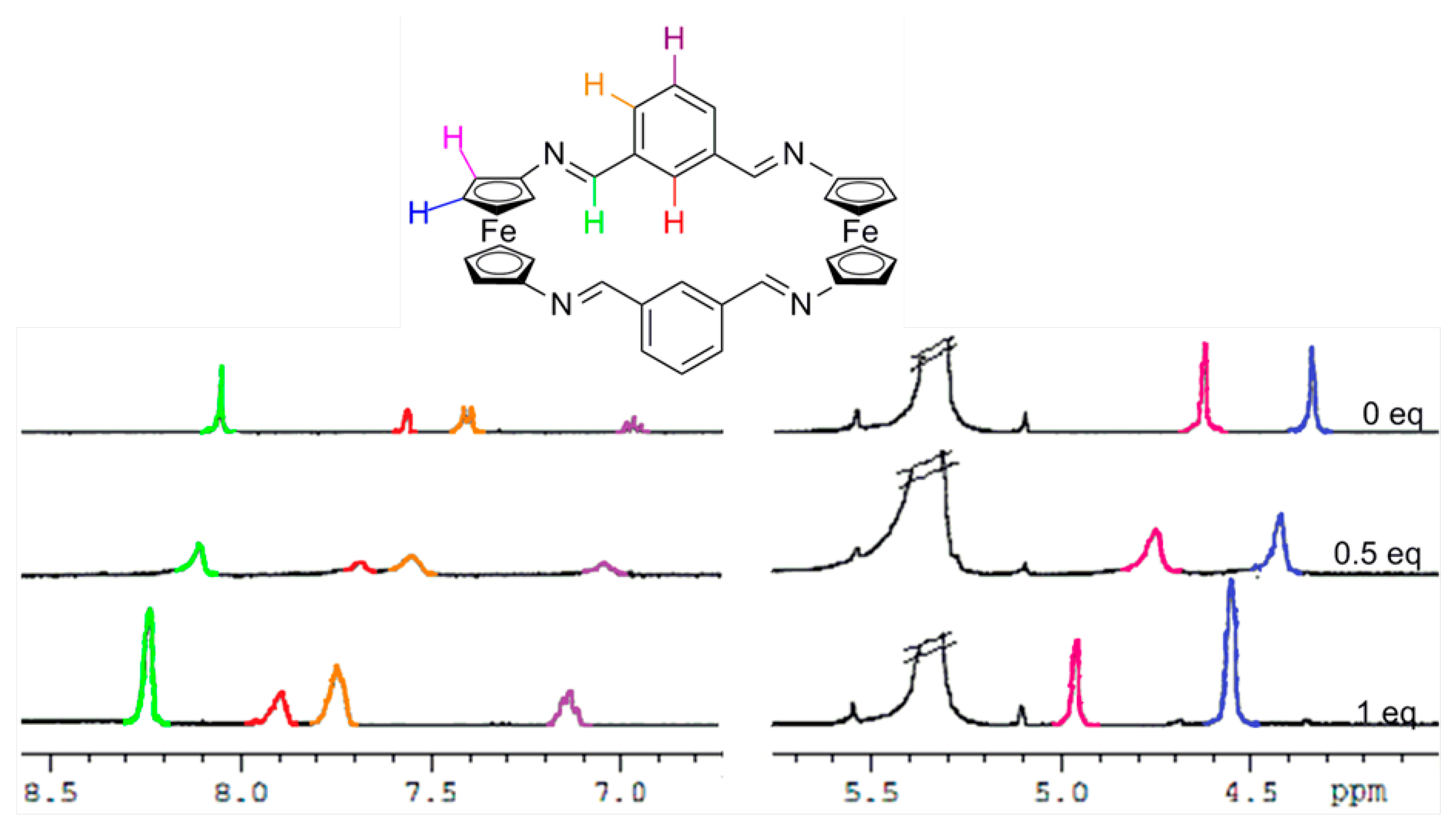
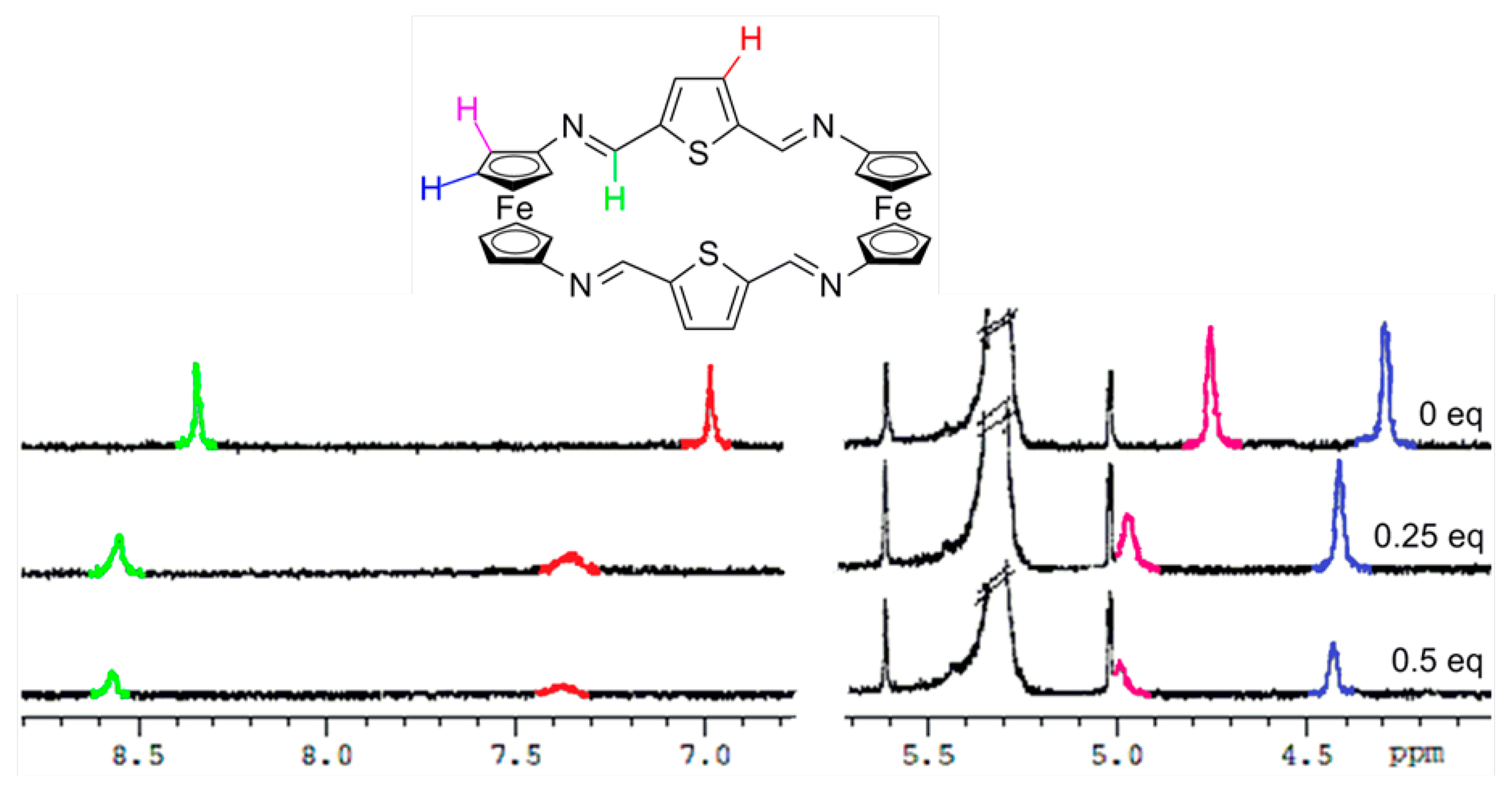
The structure of these compounds was elucidated using spectra 1H-NMR measurements as well as high resolution electrospray mass spectra (HR ESI-MS)]. In general, all the 1H NMR spectra of showed the presence of two pseudotriplets, integrating eight protons each, assigned to the Hα and Hβ protons within the symmetrically 1,1′-disubstituted ciclopentadienyl (Cp) rings present in the two equivalent ferrocene units. Additionally, one singlet, corresponding to the four iminic protons present in the bridges, together with the pattern of signals corresponding to the linked benzene, thiophene or 1,10-phenanthroline fragment was also observed (see ESI).
Reagents and conditions: (a) n-Bu3P, dry THF, 1.5 h, rt, N2; (b) isophthalaldehyde, dry THF, reflux 12 h; (c) 2,5-diformylthiophene, dry THF, reflux 12 h; (d) 2,9-diformyl-1,10-phenanthroline, dry THF, reflux 12 h.
3.2. Metal Cation Sensing Studies
An interesting common attribute of the new ferrocenophanes reported here is the presence of N-donor atoms on the bridges properly arranged with respect to the ferrocene redox-active moieties. Consequently, the receptors 3, 4 and 5 are good candidates to study their coordination behavior towards several metal cations, not only through electrochemical methods, [linear sweep voltammetry (LSV), cyclic voltammetry (CV), and Osteryoung square-wave voltammetry (OSWV)] [42] but also by UV-vis and 1H-NMR spectroscopic techniques, and quantum chemical calculations. However, it should be mentioned that although 1,10-phenanthroline is a classic chelating bidentate ligand whose nitrogen atoms are placed to act cooperatively in cation binding [43], the low solubility of receptor 5 in the common organic solvents, promoted that the recognition studies were only focused on the ferrocenophanes 3 and 4. We have found that all the iminoferrocenophane derivatives described in this paper are stable enough to undergo all the titration experiments without suffering any degradation. Only in the presence of traces of mineral acids does hydrolysis take place in a short period of time. To gain insight into the binding properties of both ligands, a set of alkali-metal ions (Li+, Na+ and K+), alkaline-earth metal ions (Mg2+ and Ca2+) and transition-metal metal ions (Ni2+, Cu2+, Zn2+, Cd2+, Hg2+, and Pb2+) as their triflate or perchlorate salts, were tested [44]. The titration experiments were further analyzed using the computer program SPECFIT [45].
3.2.1. Electrochemical Study
The reversibility and relative oxidation potential of the ferrocene/ferrocenium redox couple in receptors 3 and 4 were determined by cyclic voltammetry (CV) and Osteryoung square-wave voltammetry (OSWV) in solutions of CH2Cl2 (c = 1 × 10−4 M) containing 0.1 M [(n-Bu)4N]PF6 as supporting electrolyte.
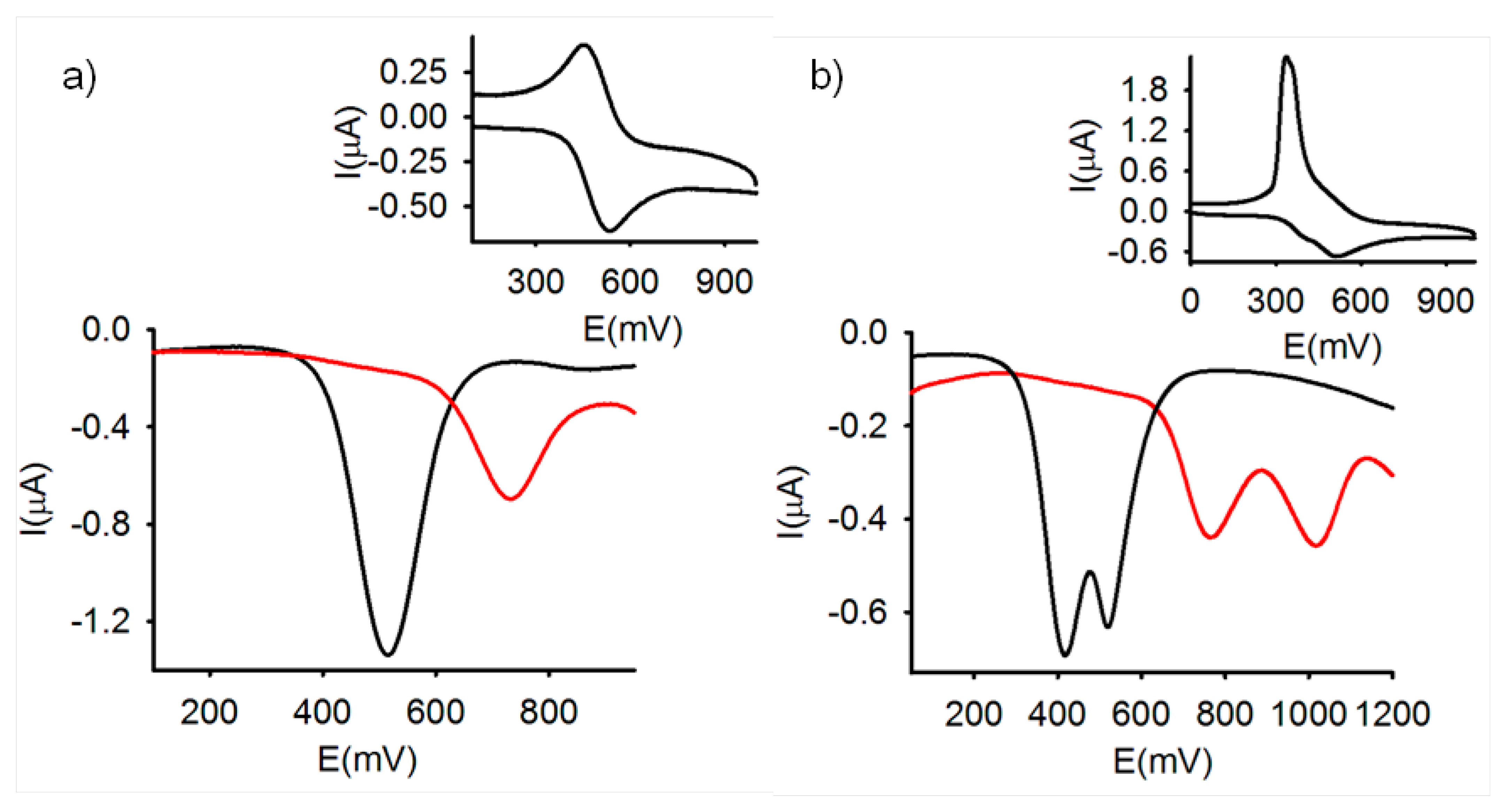
As expected, the CV of the free receptor 3 in CH2Cl2 (c = 1 × 10−4 M), showed a reversible two-electron oxidation wave at E1/2 = 517 mV vs the decamethylferrocene (DMFc) redox couple indicating that the two metal centers are electronically decoupled. Similarly, OSWV also exhibits one oxidation peak at the same potential vs DMFc (Figure 1). By contrast the ferrocenophane 4 bearing the two redox centers separated by two 2,5-iminomethyl disubstituted thiophene bridges gave rise, under similar electrochemical conditions, to two successive one-electron oxidations at E11/2 = 420 and E21/2 = 530 mV, respectively (Figure 1), indicating the existence of an electronic interaction between the two organoiron centers, through such organic bridge.
It is worth mentioning that optically induced intramolecular electron transfer processes have been previously reported in electrochemically active ferrocenyl-thiophene derivatives [46–50]. Moreover, it has also been reported that the intramolecular electron transfer phenomena can be monitored by the study of the intervalence charge-transfer bands (IVCT) occurring in the electrochemically oxidized state of this type of π-bridged systems [51–55]. However, the detection of this IVCT bands through spectroelectrochemical experiments followed by UV-vis-near IR spectrocopy was unsuccessful due to the insolubility of this compound in the common organic solvents. Nevertheless, it has also been shown that the magnitude of the separation ΔE1/2 between the first and second redox events gives an indication of the interaction through the bridge between the two Fe sites [56]. In the present work this value has been calculated by the Richardson-Taube method [57] (E11/2 = 420 mV, E21/2 = 530 mV, cf ferrocene 570 mV vs. DMFc). From the separation |E21/2 − E11/2|= 110 mV a comproportionation constant Kc = 74 was calculated, which indicates that the monocation [4]+ could be an example of a slightly delocalized mixed-valence species.
The electrochemical changes upon addition of the above mentioned set of metal cations to the receptors 3 and 4 were also investigated by using OSWV. In the case of 3, these experiments demonstrated that only the addition of Zn2+, Hg2+ and Pb2+ induced the appearance, in the OSWV, of a new oxidation peak at a remarkably more positive potential: E1/2 = 635 mV (ΔE1/2 = 118 mV) for Zn2+, E1/2 = 729 mV (ΔE1/2 = 212 mV) for Pb2+, and E1/2 = 745 mV (ΔE1/2 = 228 mV) for Hg2+ (Figures 1a, S1 and S2). Interestingly, while addition Zn2+, Pb2+ and Hg2+ metal cations to 3, promotes the formation of the corresponding complexes, addition of Cu2+ induces the oxidation of the ferrocene moieties present in the free receptor. Thus, LSV studies carried out upon addition of Cu2+ to the CH2Cl2 electrochemical solutions of receptor 3, showed a significant shift of the sigmoidal voltammetric wave toward cathodic currents, indicating that this metal cation promotes the oxidation of the free receptors. By contrast, the same experiments carried out upon addition of Zn2+, Hg2+ and Pb2+ metal cations, revealed a shift of the linear sweep voltammogram toward more positive potentials, which is in agreement with the complexation process previously observed by CV and OSWV (Figure S3).
On the other hand, the results obtained on the stepwise addition of substoichiometric amounts of the metal cations to receptor 4, under the same electrochemical conditions, also revealed that only the addition of Zn2+, Hg2+ and Pb2+ metal cations promoted the appearance of two new waves anodically shifted (E11/2 = 640 mV and E21/2 = 954 mV for Zn2+, E11/2 = 780 mV and E21/2 = 1040 mV for Pb2+, and E11/2 = 780 mV and E21/2 = 805 mV for Hg2+) while the addition of the other metal cations tested had no effect on its CV or OSWV even when they were added in a large excess (Figures 1b and S4).
3.2.2. UV-vis Absorption Study
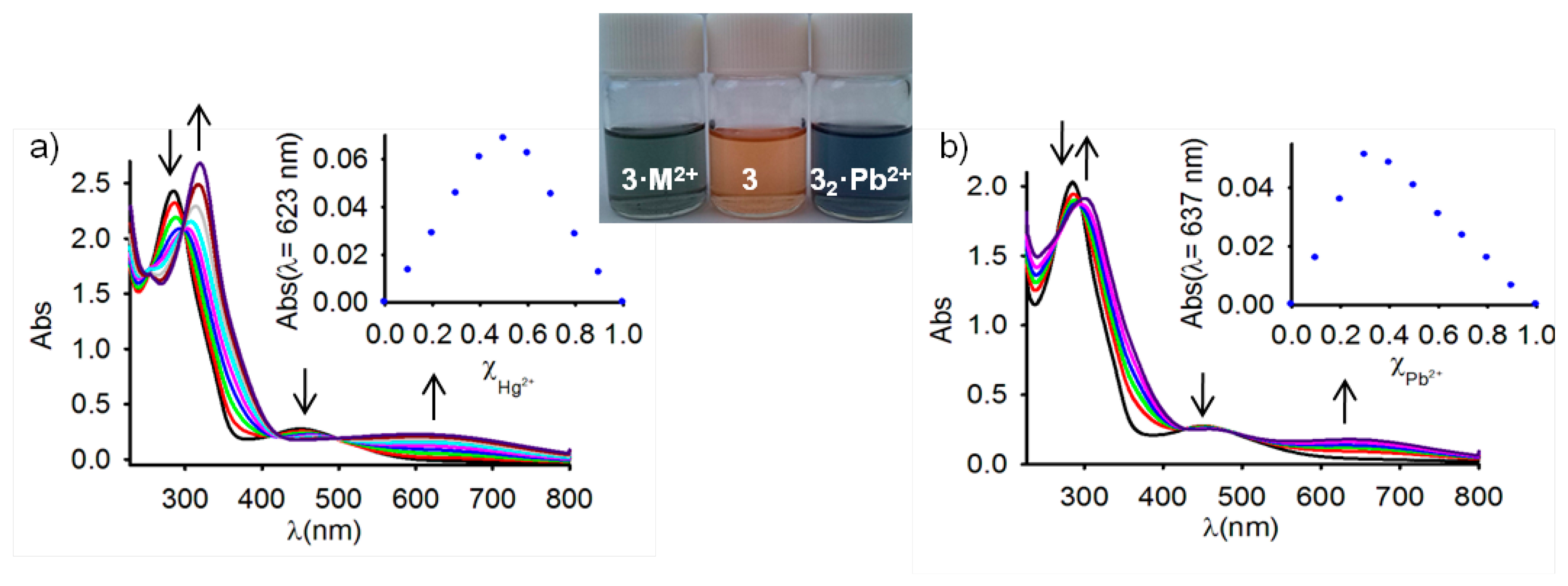
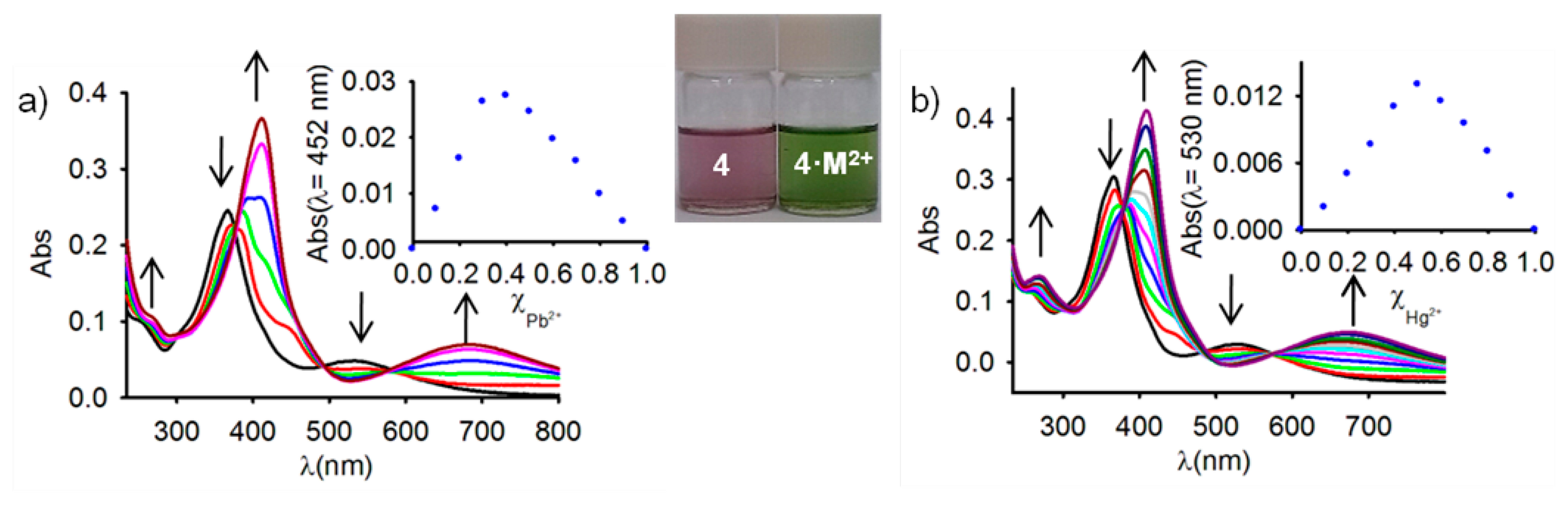
Because studies on ferrocene-based ligands have clearly shown that their characteristic low-energy bands in the absorption spectra are perturbed on complexation [58–60], the cation binding properties of the ferrocenophanes 3 and 4 were also examined using UV-vis spectroscopic measurements. The absorption properties of both free receptors were first determined in CH2Cl2 and are summarized in Table 1. In both cases, a characteristic absorption band, present in most of the ferrocenyl derivatives, is detected at λ = 450 and λ = 532 nm. This weak absorption is produced either by two nearly degenerate transitions: a Fe(II) d-d transition [61,62] or by a metal-ligand charge transfer (MLCT) process (dπ − π*). This assignment is in agreement with the latest theoretical treatment (model III) reported by Barlow et al. [63]. The optical detection capability of these receptors toward the above mentioned set of metal cations was carried out by the progressive addition of the corresponding metal cations into a solution of them in CH2Cl2. These titration experiments confirmed the electrochemical results previously shown in the sense that only the presence of Zn2+, Hg2+ and Pb2+ metal cations displayed modifications of the UV-vis spectrum of the free receptors, as a consequence of its coordination to those metal cations, while no significant spectral changes were observed upon addition of the other metal cations tested. The changes observed during such coordination processes are similar in both cases (Figures 2, 3, S5 and S7 and Table 1). In particular, the lower energy band in 3 shifts from λ = 450 nm to higher wavelengths (λ = 642 nm for Zn2+, λ = 615 nm for Hg2+, and λ = 659 nm for Pb2+), a similar trend being also observed for the case of receptor 4, in which the lower energy band appearing at λ = 532 nm is also red shifted to λ = 675 nm, λ = 680 nm, and λ = 685 nm, upon addition of Zn2+, Hg2+, and Pb2+, respectively. These progressive changes in the spectra revealed clear isosbestic points, indicating the presence of only two species in equilibrium in the solution, namely, the free receptor and the corresponding metal complex. As shown in Figures 2 and 3, such changes also promoted a significant colour changes in the solutions of the free receptors which could be used for the naked eye detection of these metal cations. Furthermore, Job's plots and titration profiles obtained on the basis of the changes in the absorption spectra upon addition of these metal cations also indicated 1:1 stoichiometries for the complexes between both receptors and Zn2+ and Hg2+, and 1:2 stoichiometries for the complexes formed by Pb2+ metal cations. Moreover, the calculated association constant [45] and detection limits [64] (Figures S6 and S8) are collected in Table 1.
3.2.3. 1H-NMR Study
In order to gain further understanding on the recognition processes, 1H-NMR titration experiments in CD2Cl2 were carried out. As shown in Figures 4, S9 and S10, and in Table 2, the most significant changes observed upon the gradual addition of Zn2+, Hg2+ and Pb2+ metal cations to receptor 3 are the clear downfield shifts promoted in the protons present within both the ferrocene and the bridge units. Similarly, the two single peaks assigned for the four iminic protons, the four thiophene protons as well as the eight Hα and eight Hβ protons within the 1,1'-disubstituted ferrocene units present in 4 were also downfield shifted (Figure 5, S11 and S12 and Table 2). However, it is worth mentioning that, in both receptors, the ferrocenyl protons were distinctly downfield shifted upon complexation, indicating the different electron deshielding effect promoted by the metal cations bound in the proximity of such ferrocene moieties. Moreover, the strong thiophilic affinity by Hg2+ is also clearly evidenced in receptor 4 where the downfield shifts promoted upon complexation are significantly higher than in the species [3·Hg2+]. Moreover, the observed downfield shifts of the thiophene protons in 4 upon recognition of Hg2+, in comparison to other metal cations, also indicates the cooperative role that the sulfur atom, present in this rigid structural motif, should play during the recognition process. In addition, the variation of 1H NMR spectra upon titration with Pb2+ show that both receptors 3 and 4 experienced saturation of the change in chemical shift when 0.5 equiv of Pb2+ was added, while in the cases of Zn2+, and Hg2+ such saturation was achieved when 1 equiv of these metal cations was added.
3.2.4. Computational Study
Computational calculations were carried out to find out the binding mode of the Zn2+ and Hg2+ metal cations with receptor 4. QC calculations show that the most stable geometry for ligand 4 (see the SI) features a parallel alignment of the two (Z,Z)-2,5-thienyl-bisiminoyl bridges at typical π-stacking distance (ca. 3.62 Å at thiophene α positions). Four main conformers of 4 were compared at a preliminary B3LYP-D3/def2-SVP level of theory. The most stable arc-shaped Z,Z-bridges allow the thiophene units to be far away (i.e., no stacked) in the “antiparallel” arrangement 4ZZ−anti that was found to be 3.73 kcal/mol less stable. This energy difference could be used as a rough estimation of the magnitude for the π-stacking interaction in 4 [65], that is additionally evidenced by one bond critical point (BCP) between both S atoms (dS⋯S = 3.669 Å; WBI = 0.002; ρ(r) = 0.69 × 10−2 e/ao3), as well as two other BCPs between both formal thienyl diene moieties (average dC⋯C = 3.609 Å; ΣWBI = 0.010; Σρ(r) = 0.91 × 10−2 e/ao3) and one BCP between each C = N pair (WBI = 0.008; ρ(r) = 0.41 × 10−2 e/ao3).
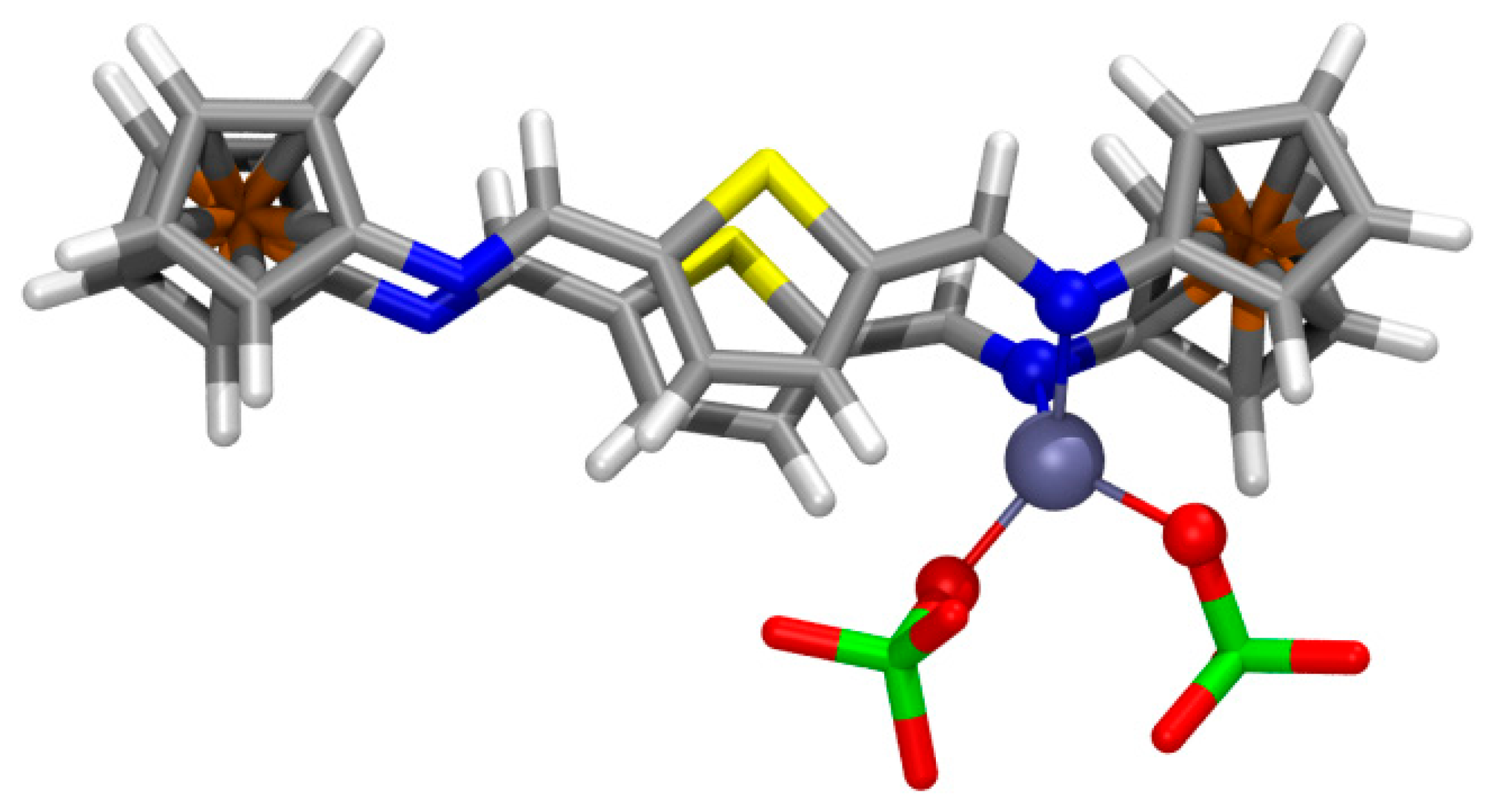
Among several different structural possibilities computed for the 1:1 complex of 4 with Zn(ClO4)2, the most stable geometry (Figure 6) displays the ligand with E,E-configured bridges and the metal cation chelated by two imine N atoms (dZn-N = 2.115, 2.143 Å; WBI = 0.186, 0.182; ρ(r) = 6.79 × 10−2, 6.43 × 10−2 e/ao3; angle N-Zn-N: 89.1°). The parallel alignment of the bridges is significantly distorted by elongating the S⋯S distance (dS⋯S = 4.170 Å; ρ(r) = 0.29 × 10−2 e/ao3) thus allowing the approach of both chelating imine N atoms (dN⋯N = 2.987 Å) [66]. The E-configuration at the imine groups allows the formation of two moderately strong hydrogen bonds (HB) of one perchlorate unit with thienyl H atoms (dO⋯H = 2.009, 2.204 Å; WBI = 0.015, 0.012; ρ(r) = 2.42 × 10−2, 1.60 × 10−2 e/ao3), whereas two other weaker HB are formed between the other perchlorate unit and ferrocenyl H atoms (dO⋯H = 2.184, 2.287 Å; WBI = 0.005, 0.009; ρ(r) = 1.68 × 10−2, 1.27 × 10−2 e/ao3).
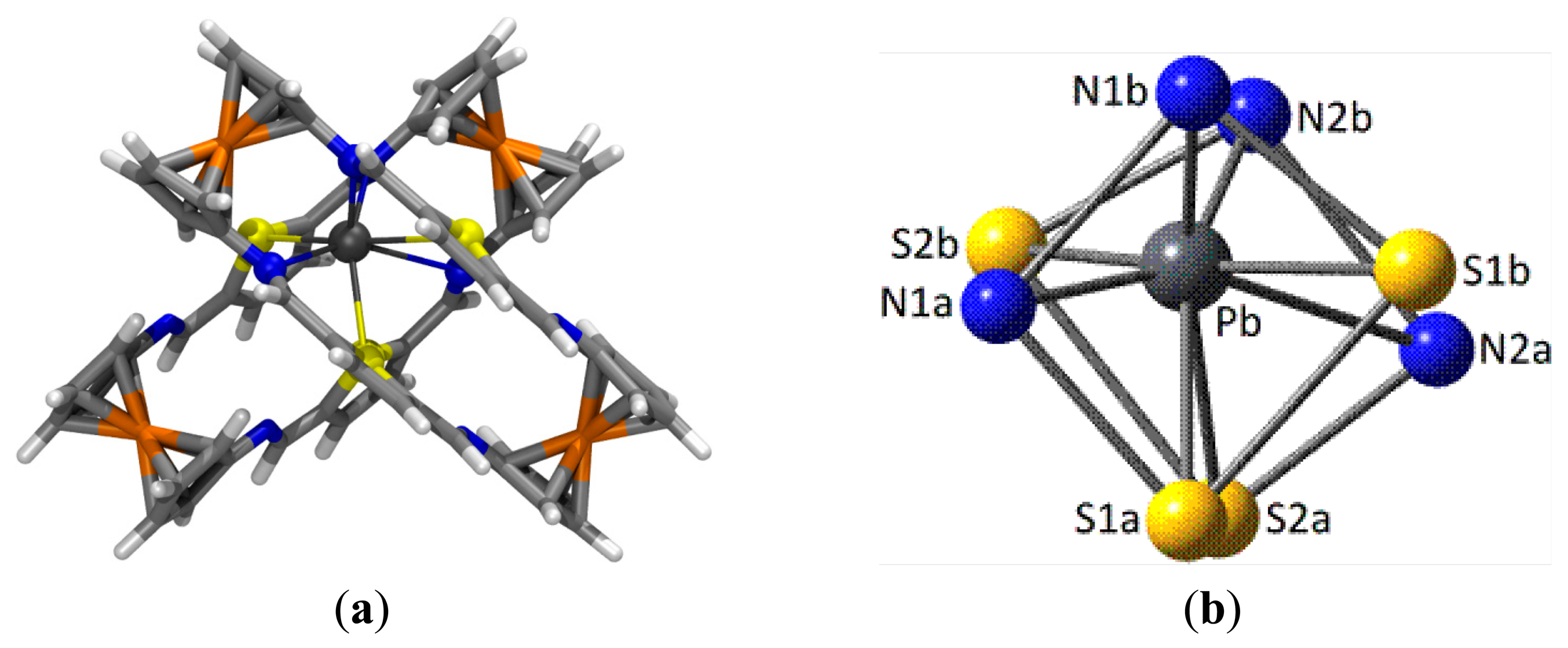
In the case of the complex with Pb2+ cations and taking into account the experimentally obtained 2:1 ligand to metal stoichiometry, the most stable geometry for the 42·Pb2+ complex (Figure 7a) shows an unusual distorted cubic 8-coordination around the metal cation, made up by four N and four S atoms. To some extent, the highly distorted coordination sphere arises from the fact that two ligands (labeled as “1” and “2”) in their most stable parallel Z,Z-arrangement 4, must approach each other using a four donor atoms S2N2 set each, consisting of one S and one N atoms belonging to one bridge (labeled as “a”) and the corresponding atoms in the other bridge (“b”). As a result of the geometrical constraints in the ligands, all donor atoms approach leading to one S⋯S and one N⋯N (homo)pairings and two S⋯N (hetero)pairings (Figure 7b).
The central Pb2+ cation lies very much closer to the N1b⋯N2b (dPb-N1b = 2.611 Å, WBI = 0.126; ρ(r) = 4.19 × 10−2 e/ao3; dPb-N2b = 2.733 Å, WBI = 0.117; ρ(r) = 3.34 × 10−2 e/ao3) and the N1a⋯S2b pairs (dPb-N1a = 2.899 Å, WBI = 0.115; ρ(r) = 2.41 × 10−2 e/ao3; dPb-S2b = 3.082 Å, WBI = 0.161; ρ(r) = 2.52 × 10−2 e/ao3). The other three S donor atoms bind the metal with moderate strength (dPb-S1b = 3.113 Å, WBI = 0.153; ρ(r) = 2.33x10−2 e/ao3; dPb-S1a = 3.310 Å, WBI = 0.129; ρ(r) = 1.52 × 10−2 e/ao3; dPb-S2a = 3.417 Å, WBI = 0.124; ρ(r) = 1.27x10−2 e/ao3), whereas the interaction with the remaining N2a atom is remarkably long (dPb-N2a = 3.533 Å, WBI = 0.070; ρ(r) = 0.72 × 10−2 e/ao3). Very likely some long contacts (especially that with N2a) within the distorted cubic coordination is a consequence of the need for a hemidirected environment around the metal allowing some room to accommodate the stereochemical electron pair [67,68], characteristic of Pb(II) and other species. In addition, the structure of the 42·Pb2+ complex is further stabilized by secondary interactions of HB or T-stacking nature between the two ligand units.
4. Conclusions
The synthesis and electrochemical, optical, and ion sensing properties of [m.n]ferrocenophanes with bridges decorated with aldimines as cation-binding sites and an aromatic or heteroaromatic (thiophene) spacer, are presented. Both receptors act as efficient redox/chromogenic chemosensor molecules for Zn2+, Pb2+, and Hg2+ cations in CH3-CN, whereas Cu2+ cations induced oxidation of the ferrocenyl end groups, which is confirmed by linear sweep voltammetry (LSV) data. The absorption spectra changes are accompanied by a color change which suggests the potential for “naked eye” detection. It is worth mentioning that from the calculated Kas and detection limits no pronounced differences in terms of selectivity and sensitivity of the highly preorganized ligands 3 and 4 toward Zn2+, Pb2+, and Hg2+ cations were observed, probably due to the fact that the ferrocenophane hole is not involved in the coordination mode. DFT calculations reveal an unusual distorted cubic coordination for the Pb2+ cation in the [42Pb]2+ complex in a N4S4 donor cage.
Acknowledgments
We gratefully acknowledge the financial support from MICINN-Spain, Project CTQ2011/27175, FEDER.
Author Contributions
The work presented in this paper is a collaborative research by all the authors. Antonia Sola performed the experiments; Arturo Espinosa carried out and wrote the computational study and Alberto Tárraga and Pedro Molina defined the research line, analyzed the data and wrote the rest of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, A.; Siegel, H.; Siegel, R.K.O. Metal Ions in Life Sciences. Neurodegenerative Diseases and Metal Ions; Wiley: New York, NY, USA, 2006; Volume 1. [Google Scholar]
- Nordberg, G.F.; Fowler, B.A.; Nordberg, M.; Friberg, L. Handbook on the Toxicology of Metals, 3rd ed.; Elsevier: New York, NY, USA, 2007. [Google Scholar]
- de Vries, W.; Roemkens, P.F.A.M.; Schütze, G. Critical soil concentrations of cadmium, lead and mercury in view of health effects on humans and animals. Rev. Environ. Contam. Toxicol. 2007, 191, 91–130. [Google Scholar]
- Nolan, E.M.; Lippard, S.J. Tools and tactics for the optical detection of mercuric ion. Chem. Rev. 2008, 108, 3443–3480. [Google Scholar]
- Kim, H.N.; Ren, W.X.; Kim, J.S.; Yoon, J. Fluorescent and colorimetric sensors for detection of lead, cadmium, and mercury ions. Chem. Soc. Rev. 2012, 41, 3210–3244. [Google Scholar]
- Charlet, L.; Chapron, Y.; Faller, P.; Kirsch, R.; Stone, A.T.; Baveye, P.C. Neurodegenerative diseases and exposure to the environmental metals Mn, Pb and Hg. Coord. Chem. Rev. 2012, 256, 2147–2163. [Google Scholar]
- World Health Organization. Guidelines for Drinking-Water Quality, 3rd ed.; WHO: Geneva, Switzerland, 2004; Volume 1, p. 188. [Google Scholar]
- de Silva, J.J.R.F.; Williams, R.J.O. Zinc: Lewis Acid Catalysis and Regulation. In The Biological Chemistry of Elements: The Inorganic Chemistry of Life, 2nd ed.; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Jiang, P.; Guo, Z. Fluorescent detection of zinc in biological systems: Recent development of the design of chemosensors and biosensors. Coord. Chem. Rev. 2004, 248, 205–229. [Google Scholar]
- Molina, P.; Tárraga, A.; Caballero, A. Ferrocene-based small molecules for multichannel molecular recognition of cations and anions. Eur. J. Inorg. Chem. 2008, 3401–3417. [Google Scholar]
- Molina, P.; Tárraga, A.; Alfonso, M. Preparation of nitrogen-substituted ferrocene derivatives by aza-Wittig methodologies. Eur. J. Org. Chem. 2011, 4505–4518. [Google Scholar]
- Beer, P.D. Redox-responsive macrocyclic receptor molecules containing transition-metal redox centers. Chem. Soc. Rev. 1989, 18, 409–450. [Google Scholar]
- Beer, P.D.; Gale, P.A.; Chen, G.Z. Mechanisms of electrochemical recognition of cations, anions and neutral guest species by redox-active receptor molecules. Coord. Chem. Rev. 1999, 185, 3–36. [Google Scholar]
- Beer, P.D.; Cadman, J. Electrochemical and optical sensing of anions by transition metal based receptors. Coord. Chem. Rev. 2000, 205, 131–155. [Google Scholar]
- Beer, P.D.; Hayes, R.J. Transition metal and organometallic anion complexation agents. Coord. Chem. Rev. 2003, 240, 167–189. [Google Scholar]
- Beer, P.D.; Bayly, S.R. Anion sensing by metal-based receptors. Top. Curr. Chem. 2005, 255, 125–162. [Google Scholar]
- Tárraga, A.; Otón, F.; Espinosa, A.; Velasco, M.D.; Molina, P.; Evans, D.J. Synthesis and properties of a new class of nitrogen-rich multinuclear [m.n]ferrocenophanes. Chem. Commun. 2004, 4, 458–459. [Google Scholar]
- Caballero, A.; Lloveras, V.; Tárraga, A.; Espinosa, A.; Velasco, M.D.; Vidal-Gancedo, J.; Rovira, C.; Wurst, K.; Molina, P.; Veciana, J. An electroactive nitrogen-rich [4.4]ferrocenophane displaying redox-switchable behavior: selective sensing, complexation, and decomplexation of Mg2+ ions. Angew. Chem. Int. Ed. Engl. 2005, 44, 1977–1981. [Google Scholar]
- Otón, F.; Tárraga, A.; Molina, P. A bis-guanidine-based multisignaling sensor molecule that display redox-ratiometric behavior or fluorescence enhancement in the presence of anions and cations. Org. Lett. 2006, 8, 2107–2110. [Google Scholar]
- Otón, F.; Espinosa, A.; Tárraga, A.; Ramírez de Arellano, C.; Molina, P. [3.3]Ferrocenophanes with guanidine bridging units as multisignaling receptor moleculares for selective recognition of anions, cations, and aminoacids. Chem. Eur. J. 2007, 13, 5742–5752. [Google Scholar]
- Otón, F.; Ratera, I.; Espinosa, A.; Tárraga, A.; Veciana, J.; Molina, P. Conformationally modulated intramolecular electron transfer process in a diaza[2.2]ferrocenophane. Inorg. Chem. 2010, 49, 3183–3191. [Google Scholar]
- Neese, F. The ORCA program system. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadrupole zeta valence quality for H to Rn: design and assessment of accuracy. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms lithium to krypton. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac-Hartree-Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar]
- ECP Parameters for Pb [SD(60,MDF)] have been Obtained from the Pseudopotential Library of the Stuttgart/Cologne Group. Available online: http://www.theochem.uni-stuttgart.de/pseudopotentials/ (accessed on 9 September 2013).
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules; Matta, C.F., Boyd, R.J., Eds.; Wiley-VCH: New York, NY, USA, 2007; pp. 1–34. [Google Scholar]
- König, F.B.; Schönbohm, J.; Bayles, D. AIM2000-a program to analyze and visualize atoms in molecules. J. Comp. Chem. 2001, 22, 545–559. [Google Scholar]
- Biegler-König, F.; Schönbohm, J. Update of the AIM2000-Program for atoms in molecules. J. Comp. Chem. 2002, 23, 1489–1494. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc: Wallingford, CT, USA, 2009. [Google Scholar]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar]
- Wiberg, K.B. Application of the Pople-Santry-Segal complete neglect of differential overlap method to the cyclopropyl-carbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar]
- Serr, B.R.; Andersen, K.A.; Elliot, C.M.; Anderson, O.P. A triply-bridged dinuclear tris(pipyridine)iron(II) complex: Synthesis and electrochemical and structural studies. Inorg. Chem. 1988, 27, 4499–4504. [Google Scholar]
- Bencini, A.; Lippolis, V. 1,10-Phenanthroline: A versatile building block for the construction of ligands for various purposes. Coord. Chem. Rev. 2010, 254, 2096–2180. [Google Scholar]
- Li+, K+, Mg2+, Ni2+, Cd2+ and Pb2+ were added as perchlorate salts (Warning! Perchlorate salts are hazardous because of the possibility of explosion; only small amounts of this material should be handled and with great caution), while Na+, Ca2+, Cu2+, Zn2+ and Hg2+ were added as triflate salts.
- Specfit/32 Global Analysis System, 1994−2004 Spectrum Software Associates (SpecSoft@compuserve.com, acquired from Biologic SA ( www.bio-logic.info) in January 2005.
- Zu, Y.B.; Wolff, M.O. Electropolymerization of oligothienylferrocene complexes: Spectroscopic and electrochemical characterization. Chem. Mater. 1999, 11, 2995–3001. [Google Scholar]
- Zu, Y.B.; Millet, D.B.; Wolf, M.O.; Rettig, S.J. Models for conjugated metal acetylide polymers: Ruthenium oligothienylacetylide complexes. Organometallics 1999, 18, 1930–1938. [Google Scholar]
- Zu, Y.B.; Wolf, M.O. Charge transfer and delocalization in conjugated (ferrocenylethynyl) oligothiophene complexes. J. Am. Chem. Soc. 2000, 122, 10121–10125. [Google Scholar]
- Justin, T.K.R.; Lin, J.T.; Wen, Y.S. Biferrocenes with heteroaromatic spacers: synthesis, structure, and electrochemistry. Organometallics 2000, 19, 1008–1012. [Google Scholar]
- Chawdhury, N.; Long, N.L.; Mahon, M.F.; Ooi, L.; Raithby, P.R.; Rooke, S.; White, A.J.P.; Williams, D.J.; Younus, M. Synthesis and characterization of aromatic ethynyl-bridged ferrocenes. J. Organomet. Chem. 2004, 689, 840–847. [Google Scholar]
- Creutz, C. Mixed valence complexes of d5-d6 metal centers. Prog. Inorg. Chem 1983, 30, 1–73. [Google Scholar]
- Crutchley, R.J. Intervalence charge transfer and electron exchange studies of dinuclear ruthenium complexes. Adv. Inorg. Chem. 1994, 41, 273–325. [Google Scholar]
- Barlow, S.; O'Hare, D. Metal-metal interactions in linked metallocenes. Chem. Rev. 1997, 97, 637–669. [Google Scholar]
- Lambert, C.; Nöll, G.; Schelter, J. Bridge-mediated hopping or superexchange electron-transfer processes in bis (triarylamine) systems. Nat. Mater. 2002, 1, 69–73. [Google Scholar]
- Low, P.J.; Paterson, M.A.J.; Puchmann, H.; Goeta, A.E.; Howard, J.A.K.; Lambert, C.; Cherryman, J.C.; Tackley, D.R.; Leeming, S.; Brown, B. Crystal, molecular and electronic structure of N,N′-diphenyl-N,N′-bis(2,4-dimethylphenyl)-(1,1′-biphenyl)-4,4′-diamine and the corresponding radical cation. Chem. Eur. J. 2004, 10, 83–91. [Google Scholar]
- Flanagan, J. B.; Margell, S.; Bard, A. J.; Anson, F. C. Electron transfer to and from molecules containing multiple, noninteracting redox centers. Electrochemical oxidation of poly(vinylferrocene). J. Am. Chem. Soc. 1978, 100, 4248–4253. [Google Scholar]
- Richardson, D.E.; Taube, H. Determination of E2°-E1° in multistep charge transfer by stationary-electrode pulse and cyclic voltammetry: application to binuclear ruthenium ammines. Inorg. Chem. 1981, 20, 1278–1285. [Google Scholar]
- Marder, S.R.; Perry, J.W.; Tiemann, B.G. Organometallic salts with large second-armonic generation powder efficiencies: (E)-1-ferrocenyl-2-(1-meyhyl-4-pyridiniumyl)ethylene salts. Organometallics 1991, 10, 1896–1901. [Google Scholar]
- Coe, B.J.; Jones, C.J.; McCleverty, J.A.; Bloor, D.; Cross, G.J. An assessment of second harmonic generation by donor acceptor molecules containing stilbenyl or diarylazo bridges between ferrocenyl donor and nitro acceptor groups. J. Organomet. Chem. 1994, 464, 225–232. [Google Scholar]
- Müller, T.J.; Netz, A.; Ansorge, M. Synthesis and NLO properties of chromium carbonyl arene complexes with conjugated side chains: The amphoteric nature of chromium carbonyl complexation in push-pull chromophores. Organometallics 1999, 18, 5066–5074. [Google Scholar]
- Sohn, Y.S.; Hendrickson, D.N.; Gray, H.B. Electronic structure of metallocenes. J. Am. Chem. Soc. 1971, 93, 3603–3612. [Google Scholar]
- Geoffroy, G.L.; Wrighton, M.S. Organometallic Photochemistry; Academic Press: New York, NY, USA, 1979. [Google Scholar]
- Barlow, S.; Bunting, H.E.; Ringham, C.; Green, J.C.; Bublitz, G.U.; Boxer, S.G.; Perry, J.W.; Marder, S.R. Studies of the electronic structure of metallocene-based second-order nonlinear optical dyes. J. Am. Chem. Soc. 1999, 121, 3715–3723. [Google Scholar]
- Shortreed, M.; Kopelman, R.; Kuhn, M.; Hoyland, B. Fluorescent fiber-optic calcium sensor for physiological measurements. Anal. Chem. 1996, 68, 1414–1418. [Google Scholar]
- A parallel arrangement of two E,E-bridges led to an 4EE located 2.90 kcal/mol above 4 in the B3LYP-D3/def2-SVP potential energy surface.
- Compare with the non-chelating imine N atoms in the same complex: dN⋯N = 3.438 Å.
- Shimoni-Livny, L.; Glusker, J.P.; Bock, C.W. Lone pair functionality in divalent lead comopounds. Inorg. Chem. 1998, 37, 1853–1867. [Google Scholar]
- Otón, F.; González, M.C.; Espinosa, A.; Tárraga, A.; Molina, P. Synthesis, structural characterization, and sensing properties of clickable unsymmetrical 1,1′-disubstituted ferrocene-triazole derivatives. Organometallic 2012, 31, 2085–2096. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | UV-vis λmax (10−3 ε) a | IP b | Kas | Dlim c |
|---|---|---|---|---|
| 3 | 285(20.27), 450(2.73) | - | - | - |
| 3·Zn2+ | 312(23.03), 469(2.27), 642(2.23) | 497, 419, 298, 245 | 1.44 × 106 e | 7.1 × 10−6 |
| 3·Hg2+ | 320(26.81), 615(2.26) | 496, 419, 298, 251 | 4.58 × 105 e | 1.4 × 10−5 |
| 3·Pb2+ | 296(18.69), 469(2.53), 659(1.54) | 484, 427, 296, 263 | 1.26 × 108 d | 6.7 × 10−6 |
| 4 | 250(5.06), 367(12.27), 532(2.41) | - | - | - |
| 4·Zn2+ | 269(5.99), 392(17.71), 408(18.12), 675(1.97) | 570, 489, 375 | 2.86 × 10−6 e | 1.5 × 10−6 |
| 4·Hg2+ | 268(7.04), 408(20.68), 680(2.47) | 574, 497, 377 | 2.17 × 106 e | 1.8 × 10−5 |
| 4·Pb2+ | 272(6.12), 411(21.00), 685(3.86) | 588, 489, 376, 298 | 5.89 × 109 d | 6.7 × 10−6 |
aε in·dm3·mol−1·cm−1;bisosbestic points in nm;cdetection limits in M;din M−2;ein M−1.
| Comp. | ΔδCH=N | ΔδH2 | ΔδH4 | ΔδH5 | ΔδCH thiop | ΔδHα | ΔδHβ |
|---|---|---|---|---|---|---|---|
| 3·Zn2+ | 0.20 | 0.34 | 0.33 | 0.19 | - | 0.32 | 0.22 |
| 3·Hg2+ | 0.35 | 0.52 | 0.43 | 0.24 | - | 0.44 | 0.27 |
| 3·Pb2+ | 0.12 | 0.23 | 0.25 | 0.13 | - | 0.22 | 0.15 |
| 4·Zn2+ | 0.32 | - | - | - | 0.56 | 0.33 | 0.15 |
| 4·Hg2+ | 1.12 | - | - | - | 1.21 | 0.92 | 0.38 |
| 4·Pb2+ | 0.21 | - | - | - | 0.37 | 0.22 | 0.12 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sola, A.; Espinosa, A.; Tárraga, A.; Molina, P. Nitrogen-Rich Multinuclear Ferrocenophanes as Multichannel Chemosensor Molecules for Transition and Heavy-Metal Cations. Sensors 2014, 14, 14339-14355. https://doi.org/10.3390/s140814339
Sola A, Espinosa A, Tárraga A, Molina P. Nitrogen-Rich Multinuclear Ferrocenophanes as Multichannel Chemosensor Molecules for Transition and Heavy-Metal Cations. Sensors. 2014; 14(8):14339-14355. https://doi.org/10.3390/s140814339
Chicago/Turabian StyleSola, Antonia, Arturo Espinosa, Alberto Tárraga, and Pedro Molina. 2014. "Nitrogen-Rich Multinuclear Ferrocenophanes as Multichannel Chemosensor Molecules for Transition and Heavy-Metal Cations" Sensors 14, no. 8: 14339-14355. https://doi.org/10.3390/s140814339
APA StyleSola, A., Espinosa, A., Tárraga, A., & Molina, P. (2014). Nitrogen-Rich Multinuclear Ferrocenophanes as Multichannel Chemosensor Molecules for Transition and Heavy-Metal Cations. Sensors, 14(8), 14339-14355. https://doi.org/10.3390/s140814339