
Enhanced Sequence Evolution Rates Correlate with Significant Rearrangements in Coccoid Mitochondrial Genomes



Abstract
1. Introduction
2. Materials and Methods
2.1. Specimens and Taxonomy
2.2. DNA and RNA Extraction
2.3. Mitogenome Assembly and Annotation
2.4. Sequence Alignment and Evolution Rate Analysis of Protein-Coding Genes
2.5. Phylogenetic Analysis
3. Results
3.1. General Characteristics of New Mitogenomes
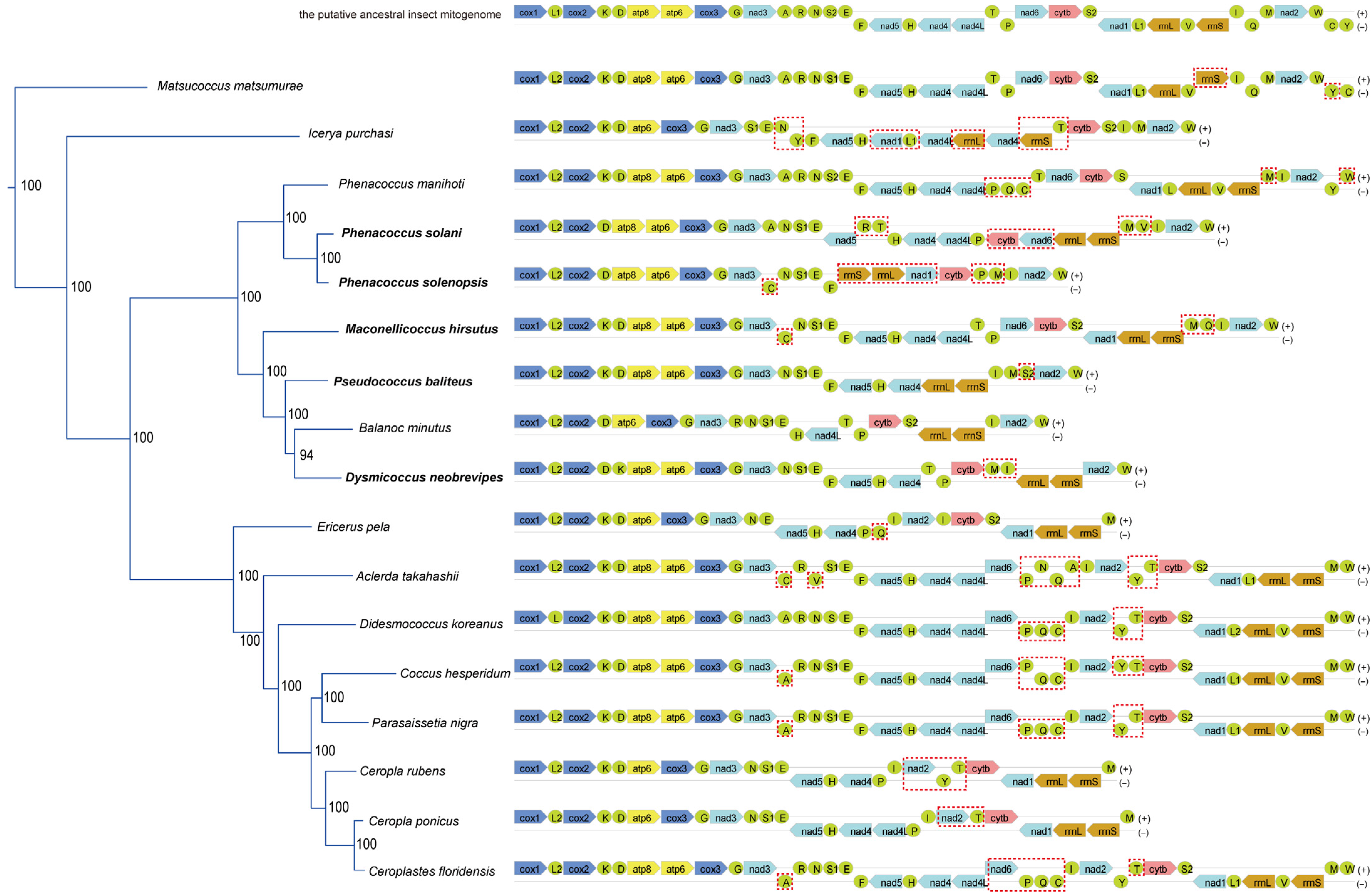
3.2. Rearrangement Patterns in Mitogenomes and Evolution Rate Analysis
3.3. Phylogenetic Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucl. Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria. Cold Spring Harb. Perspect. Biol. 2021, 13, a040543. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Schon, E.A. Mitochondria. J. Neurol. Neurosurg. Psychiat. 2003, 74, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.-J.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X.-X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genom. 2010, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.-J.; Li, Q.; van Achterberg, K.; Chen, X.-X. Two mitochondrial genomes from the families Bethylidae and Mutillidae: Independent rearrangement of protein-coding genes and higher-level phylogeny of the Hymenoptera. Mol. Phylogenet. Evol. 2014, 77, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Gibson, T.; Dowton, M. Evolutionary dynamics of the mitochondrial genome in the Evaniomorpha (Hymenoptera)—A group with an intermediate rate of gene rearrangement. Genome Biol Evol. 2014, 6, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Cameron, S.L.; Dowavic, J.I.; Austin, A.D.; Whiting, M.F. Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol. Biol. Evol. 2009, 26, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Austin, A.D. Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol. Biol. Evol. 1999, 16, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.-J.; Zhu, W.-C.; Rong, X.; Zhang, Y.-K.; Ding, X.-L.; Liu, J.; Chen, D.-S.; Du, Y.; Hong, X.-Y. The complete mitochondrial genomes of two rice planthoppers, Nilaparvata lugens and Laodelphax striatellus: Conserved genome rearrangement in Delphacidae and discovery of new characteristics of atp8 and tRNA genes. BMC Genom. 2013, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liang, A.-P. Complete mitochondrial genome of the small brown planthopper, Laodelphax striatellus (Delphacidae: Hemiptera), with a novel gene order. Zool. Sci. 2009, 26, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Huang, X.; Deng, J. Mitochondrial genomes of soft scales (Hemiptera: Coccidae): Features, structures and significance. BMC Genom. 2023, 24, 37. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-L.; Chen, Q.-D.; Chen, S.; Pu, D.-Q.; Chen, Z.-T.; Liu, Y.-Y.; Liu, X. The highly rearranged mitochondrial genomes of three economically important scale insects and the mitochondrial phylogeny of Coccoidea (Hemiptera: Sternorrhyncha). PeerJ 2020, 8, e9932. [Google Scholar] [CrossRef] [PubMed]
- Kômoto, N.; Yukuhiro, K.; Tomita, S. Novel gene rearrangements in the mitochondrial genome of a webspinner, Aposthonia japonica (Insecta: Embioptera). Genome 2012, 55, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Peng, L.; Vogler, A.P.; Morse, J.C.; Yang, L.; Sun, C.; Wang, B. Massive gene rearrangements of mitochondrial genomes and implications for the phylogeny of Trichoptera (Insecta). Syst. Entomol. 2023, 48, 278–295. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinfo 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinfo 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.Y.; Gao, F.; Jakovlić, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. Imeta 2023, 2, e87. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinfo 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucl. Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinfo 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Chernomor, O.; Von Haeseler, A.; Minh, B.Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Campbell, N.J.; Barker, S.C. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol. Biol. Evol. 2001, 18, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Barker, S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003, 20, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Johnson, K.P.; Sweet, A.D.; Yao, I.; Ferreira, R.L.; Cameron, S.L. Mitochondrial phylogenomics and genome rearrangements in the barklice (Insecta: Psocodea). Mol. Phylogenet. Evol. 2018, 119, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Wei, S.-J.; Wang, M. Mitochondrial genome rearrangements and phylogenomics of the Hymenoptera (Insecta) using an expanded taxon sample. Mitochondrial DNA A 2023, 34, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Weller, A.M.; Rödelsperger, C.; Eberhardt, G.; Molnar, R.I.; Sommer, R.J. Opposing forces of A/T-biased mutations and G/C-biased gene conversions shape the genome of the nematode Pristionchus pacificus. Genetics 2014, 196, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.L.; Claessens, A.; Otto, T.D.; Kekre, M.; Fairhurst, R.M.; Rayner, J.C.; Kwiatkowski, D. Extreme mutation bias and high AT content in Plasmodium falciparum. Nucl. Acids Res. 2017, 45, 1889–1901. [Google Scholar] [PubMed]
- Shao, R.; Dowton, M.; Murrell, A.; Barker, S.C. Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol. Biol. Evol. 2003, 20, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Gullan, P.J.; Kosztarab, M. Adaptations in scale insects. Annu. Rev. Entomol. 1997, 42, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Hardy, N.B.; Gullan, P.J.; Hodgson, C.J. A subfamily-level classification of mealybugs (Hemiptera: Pseudococcidae) based on integrated molecular and morphological data. Syst. Entomol. 2008, 33, 51–71. [Google Scholar] [CrossRef]
- Normark, B.B. The evolution of alternative genetic systems in insects. Annu. Rev. Entomol. 2003, 48, 397–423. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Kirkness, E.F.; Barker, S.C. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009, 19, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Ross, L. Genetic Conflict and Sex Allocation in Scale Insects. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2010. [Google Scholar]

{kind=link}
| Species Name | Data Type | Number of Clean Reads | Mitochondrial Contigs Matched Reads | Mitochondrial Contig Lengths | Mean Coverage | Maximum Coverage |
|---|---|---|---|---|---|---|
| D. neobrevipes | Genome | 84,116,612 | 80,697 | 14,118 | 776 | 3774 |
| M. hirsutus | Genome | 121,628,353 | 500,853 | 14,446 | 5089 | 28,787 |
| P. baliteus | Genome | 73,858,913 | 80,715 | 12,821 | 843 | 3039 |
| P. solani | Genome | 96,691,261 | 85,021 | 14,300 | 855 | 2916 |
| P. solenopsis | Transcriptome | 62,424,175 | 61,214 | 11,557 | 770 | 2924 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ji, J.; Xi, Y.; Song, N. Enhanced Sequence Evolution Rates Correlate with Significant Rearrangements in Coccoid Mitochondrial Genomes. Diversity 2025, 17, 515. https://doi.org/10.3390/d17080515
Zhang L, Ji J, Xi Y, Song N. Enhanced Sequence Evolution Rates Correlate with Significant Rearrangements in Coccoid Mitochondrial Genomes. Diversity. 2025; 17(8):515. https://doi.org/10.3390/d17080515
Chicago/Turabian StyleZhang, Lijuan, Junpeng Ji, Yuqiang Xi, and Nan Song. 2025. "Enhanced Sequence Evolution Rates Correlate with Significant Rearrangements in Coccoid Mitochondrial Genomes" Diversity 17, no. 8: 515. https://doi.org/10.3390/d17080515
APA StyleZhang, L., Ji, J., Xi, Y., & Song, N. (2025). Enhanced Sequence Evolution Rates Correlate with Significant Rearrangements in Coccoid Mitochondrial Genomes. Diversity, 17(8), 515. https://doi.org/10.3390/d17080515