
Reconstruction of Pleistocene Evolutionary History of the Root Vole Alexandromys oeconomus (Cricetidae, Rodentia) in Northern Asia

 ,
,  , , , and
, , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Isolation and Mitochondrial Cytochrome B Amplification
2.2. Phylogenetic Analyses
2.3. Divergence Dating
2.4. Demographic Analyses
3. Results
3.1. Sequence Analysis
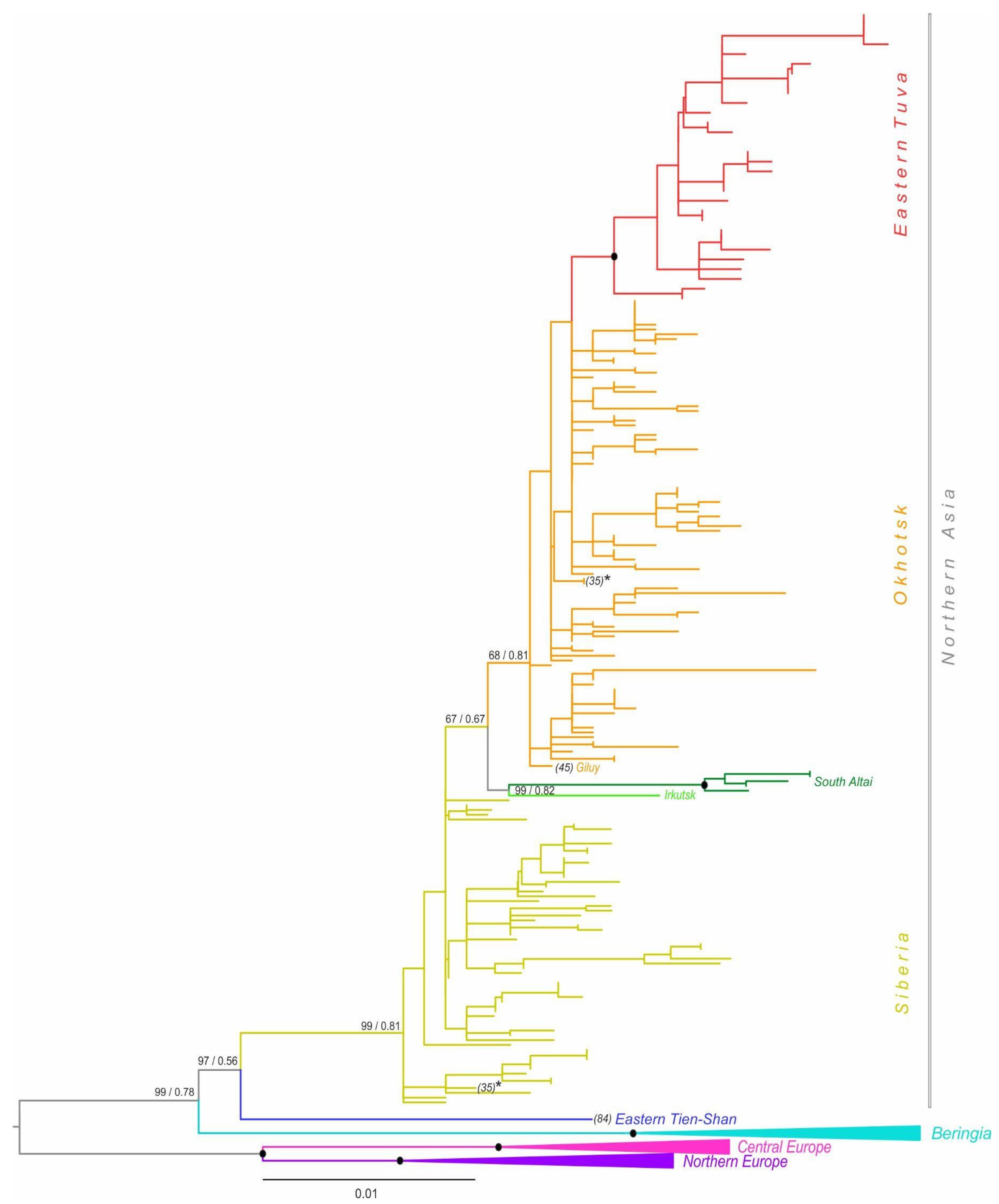
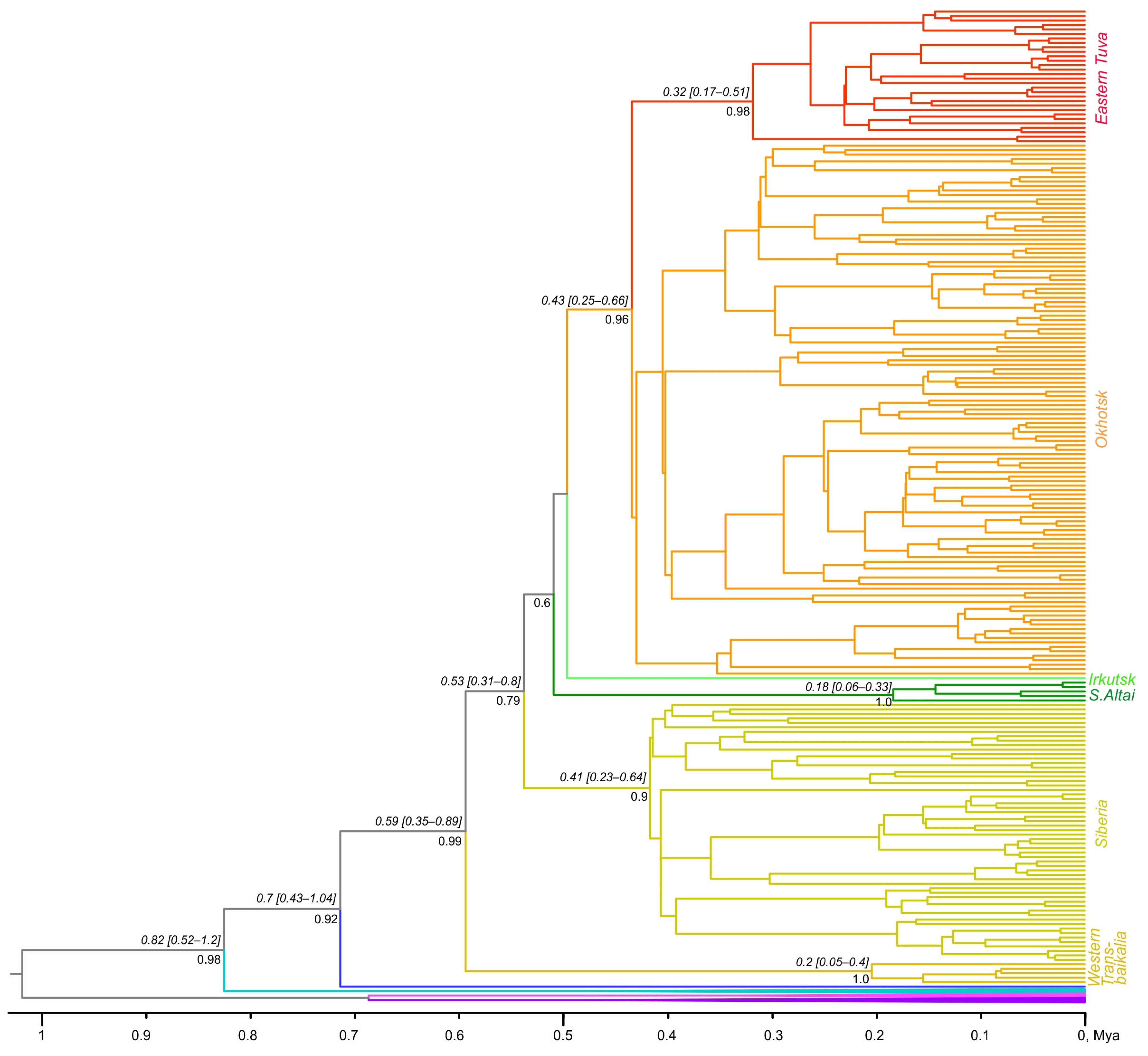
3.2. Phylogenetic Analyses and Divergence Time
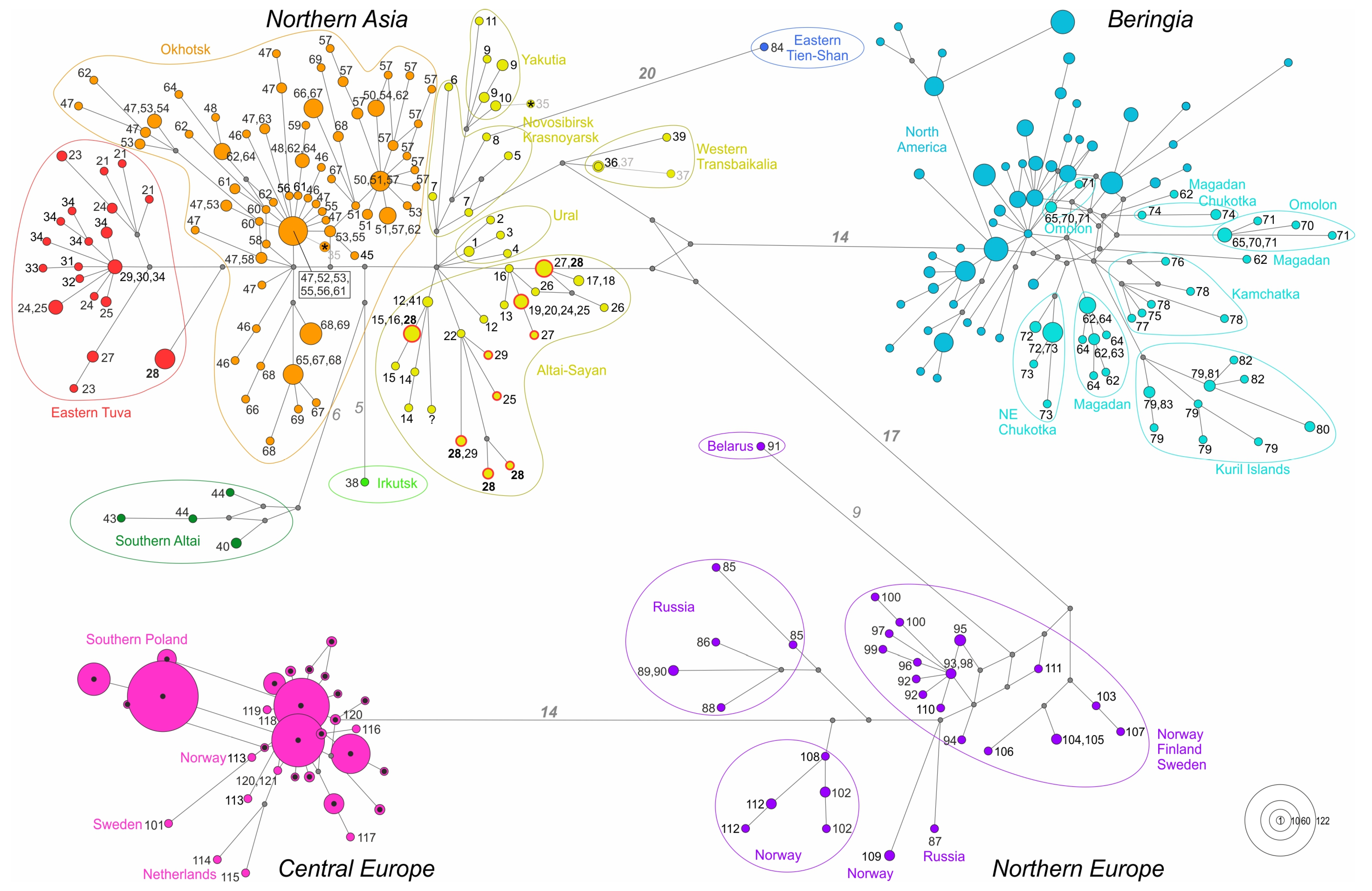
3.3. Demographic Analyses
4. Discussion
4.1. The Early Divergence of the Root Vole
4.2. Evolutionary History of A. oeconomus in Northern Asia
4.3. Comparative Phylogeography of Wide-Ranging Small Rodent Species in Northern Palearctic
4.4. Genetic Diversity and Taxonomic Structure
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, S.; Cai, B. Micromammalian Fossils from Danangou of Yuxian, Hebei. In Contributions to the XIII INQUA; Beijing Scientific and Technological Publishing House: Beijing, China, 1991; pp. 100–131. [Google Scholar]
- Erbaeva, M.A. The Late Cenozoic Faunistic Complexes of Transbaikalia with Special Reference to the Micromammalia. In Quartärpaläontologie Band 6; Kahlke, H.D., Ed.; De Gruyter: Berlin, Germany, 1986; pp. 25–28. ISBN 978-3-11-265256-5. [Google Scholar]
- Markova, A.K. The Sequence of Early Pleistocene Small-Mammal Faunas from the South Russian Plain. Quartarpalaontologie 1990, 8, 131–151. [Google Scholar]
- Janossy, D. Pleistocene Vertebrate Faunas of Hungary. In Developments in Palaeontology and Stratigraphy; Elsevier: Amsterdam, The Netherlands, 1986; Volume 8. [Google Scholar]
- Chaline, J. Arvicolid Data (Arvicolidae, Rodentia) and Evolutionary Concepts. Evol. Biol. 1987, 21, 237–310. [Google Scholar]
- Nadachowski, A. Origin and History of the Present Rodent Fauna in Poland Based on Fossil Evidence. Acta Theriol. 1989, 34, 37–53. [Google Scholar] [CrossRef]
- Kordos, L. The Evolution of Upper Pleistocene Voles in Central Europe. In International Symposium Evolution, Phylogeny and Biostratigraphy of Arvicolids (Rodentia, Mammalia); Fejfar, O., Heinrich, W.-D., Eds.; Geological Survey: Prague, Czech Republic, 1990; pp. 275–284. [Google Scholar]
- Rausch, R.L. A Review of the Distribution of Holarctic Recent Mammals. In Pacific Basin Biogeography; Gressett, J.L., Ed.; Bishop Museum Press: Honolulu, HI, USA, 1963; pp. 29–44. [Google Scholar]
- MacPherson, A.H. The Origin of Diversity in Mammals of the Canadian Arctic Tundra. Syst. Zool. 1965, 14, 153–173. [Google Scholar] [CrossRef]
- Jopling, A.V.; Irving, W.N.; Beebe, B.F. Stratigraphic, Sedimentological and Faunal Evidence for the Occurrence of Presangamonian Artifacts in Northern Yukon. Arctic 1981, 34, 3–33. [Google Scholar] [CrossRef]
- MacDonal, S.O.; Cook, J.A. Recent Mammals of Alaska; University of Alaska Press: Fairbanks, AK, USA, 2009; Volume 91, ISBN 978-1-60223-047-7. [Google Scholar]
- Lance, E.W.; Cook, J.A. Biogeography of Tundra Voles (Microtus oeconomus) of Beringia and the Southern Coast of Alaska. J. Mammal. 1998, 79, 53–65. [Google Scholar] [CrossRef]
- Brunhoff, C.; Galbreath, K.E.; Fedorov, V.B.; Cook, J.A.; Jaarola, M. Holarctic Phylogeography of the Root Vole (Microtus oeconomus): Implications for Late Quaternary Biogeography of High Latitudes. Mol. Ecol. 2003, 12, 957–968. [Google Scholar] [CrossRef]
- Galbreath, K.E.; Cook, J.A. Genetic Consequences of Pleistocene Glaciations for the Tundra Vole (Microtus oeconomus) in Beringia. Mol. Ecol. 2004, 13, 135–148. [Google Scholar] [CrossRef]
- Brunhoff, C.; Yoccoz, N.G.; Ims, R.A.; Jaarola, M. Glacial Survival or Late Glacial Colonization? Phylogeography of the Root Vole (Microtus oeconomus) in North-west Norway. J. Biogeogr. 2006, 33, 2136–2144. [Google Scholar] [CrossRef]
- Iwasa, M.A.; Kostenko, V.A.; Frisman, L.V.; Kartavtseva, I.V. Phylogeography of the Root Vole Microtus oeconomus in Russian Far East: A Special Reference to Comparison between Holarctic and Palaearctic Voles. Mammal Study 2009, 34, 123–130. [Google Scholar] [CrossRef]
- Jancewicz, E.; Falkowska, E.; Ratkiewicz, M. mtDNA Evidence for a Local Northern Latitude Pleistocene Refugium for the Root Vole (Microtus oeconomus, Arvicolinae, Rodentia) from Eastern Poland. J. Zool. Syst. Evol. Res. 2015, 53, 331–339. [Google Scholar] [CrossRef]
- Pereverzeva, V.V.; Dokuchaev, N.E.; Primak, A.A.; Dubinin, E.A. Variability of the Gene Cytochrome b mtDNA of the Tundra Vole (Alexandromys oeconomus Pallas, 1776) in North-Eastern Asia and Alaska. Usp. Sovrem. Biol. 2023, 143, 149–164. [Google Scholar] [CrossRef]
- Pereverzeva, V.V.; Dokuchaev, N.E.; Primak, A.A.; Dubinin, E.A.; Kiselev, S.V. Variability of the Cytochrome b mtDNA Gene of the Root Vole (Alexandromys oeconomus Pallas, 1776) from the Northern Coast of the Sea of Okhotsk. Usp. Sovrem. Biol. 2022, 142, 90–104. [Google Scholar]
- Bannikova, A.A.; Lebedev, V.S.; Lissovsky, A.A.; Matrosova, V.; Abramson, N.I.; Obolenskaya, E.V.; Tesakov, A.S. Molecular Phylogeny and Evolution of the Asian Lineage of Vole Genus Microtus (Rodentia: Arvicolinae) Inferred from Mitochondrial Cytochrome b Sequence: Molecular Phylogeny of Asian Microtus. Biol. J. Linn. Soc. 2010, 99, 595–613. [Google Scholar] [CrossRef]
- Lissovsky, A.A.; Petrova, T.V.; Yatsentyuk, S.P.; Golenishchev, F.N.; Putincev, N.I.; Kartavtseva, I.V.; Sheremetyeva, I.N.; Abramson, N.I. Multilocus Phylogeny and Taxonomy of East Asian Voles Alexandromys (Rodentia, Arvicolinae). Zool. Scr. 2018, 47, 9–20. [Google Scholar] [CrossRef]
- Wang, X.; Liang, D.; Wang, X.; Tang, M.; Liu, Y.; Liu, S.; Zhang, P. Phylogenomics Reveals the Evolution, Biogeography, and Diversification History of Voles in the Hengduan Mountains. Commun. Biol. 2022, 5, 1124. [Google Scholar] [CrossRef]
- Abramson, N.I.; Rodchenkova, E.N.; Kostygov, A.Y. Genetic Variation and Phylogeography of the Bank Vole (Clethrionomys glareolus, Arvicolinae, Rodentia) in Russia with Special Reference to the Introgression of the mtDNA of a Closely Related Species, Red-Backed Vole (Cl. Rutilus). Russ. J. Genet. 2009, 45, 533–545. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Heled, J.; Bouckaert, R.R. Looking for Trees in the Forest: Summary Tree from Posterior Samples. BMC Evol. Biol. 2013, 13, 221. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the Cytochromeb Gene of Mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef]
- Linzey, A.V.; Shar, S.; Lkhagvasuren, D.; Juškaitis, R.; Sheftel, B.; Meinig, H.; Amori, G.; Henttonen, H. Microtus oeconomus (Errata Version Published in 2017). The IUCN Red List of Threatened Species 2016: e.T13451A115113894. 2016. Available online: https://dx.doi.org/10.2305/IUCN.UK.2016-3.RLTS.T13451A22347188.en (accessed on 17 July 2025).
- Bowen, D.Q.; Richmond, G.M.; Fullerton, D.S.; Sibrava, V.; Fulton, R.J.; Velichko, A.A. Correlation of Quaternary Glaciations in the Northern Hemisphere. Quat. Sci. Rev. 1986, 5, 509–510. [Google Scholar] [CrossRef]
- Zakrzewski, R.J. The Fossil Record. In Biology of New World Microtus; Tamarin, R.H., Ed.; American Society of Mammalogists: Washington, DC, USA, 1985. [Google Scholar]
- Herman, J.S.; McDevitt, A.D.; Kawałko, A.; Jaarola, M.; Wójcik, J.M.; Searle, J.B. Land-Bridge Calibration of Molecular Clocks and the Post-Glacial Colonization of Scandinavia by the Eurasian Field Vole Microtus agrestis. PLoS ONE 2014, 9, e103949. [Google Scholar] [CrossRef] [PubMed]
- Haring, E.; Sheremetyeva, I.N.; Kryukov, A.P. Phylogeny of Palearctic Vole Species (Genus Microtus, Rodentia) Based on Mitochondrial Sequences. Mamm. Biol. 2011, 76, 258–267. [Google Scholar] [CrossRef]
- Avenarius, I.G.; Muratova, M.V.; Spasskaya, I.I. Paleogeography of Northern Eurasia in the Late Pleistocene-Holocene and Geographical Forecast; Nauka: Moscow, Russia, 1978. [Google Scholar]
- Grunert, J.; Lehmkuhl, F.; Walther, M. Paleoclimatic Evolution of the Uvs Nuur Basin and Adjacent Areas (Western Mongolia). Quat. Int. 2000, 65–66, 171–192. [Google Scholar] [CrossRef]
- Obruchev, S.V. The eastern part of the sayan-tuva highland in the quaternary period. Izv Union Geogr. Soc. 1953, 85, 533–547. [Google Scholar]
- Krivonogov, S.K.; Sheinkman, V.S.; Mistruykov, A.A. Stages in the Development of the Darhad Dammed Lake (Northern Mongolia) during the Late Pleistocene and Holocene. Quat. Int. 2005, 136, 83–94. [Google Scholar] [CrossRef]
- Gillespie, A.R.; Burke, R.M.; Komatsu, G.; Bayasgalan, A. Late Pleistocene Glaciers in Darhad Basin, Northern Mongolia. Quat. Res. 2008, 69, 169–187. [Google Scholar] [CrossRef]
- Namzalov, B.B. The most important nodes of biodiversity and phytogeographic phenomena of mountain steppes of Southern Siberia. Arid Ecosyst. 2021, 27, 24–36. [Google Scholar] [CrossRef]
- McGaughran, A.; Liggins, L.; Marske, K.A.; Dawson, M.N.; Schiebelhut, L.M.; Lavery, S.D.; Knowles, L.L.; Moritz, C.; Riginos, C. Comparative Phylogeography in the Genomic Age: Opportunities and Challenges. J. Biogeogr. 2022, 49, 2130–2144. [Google Scholar] [CrossRef]
- Abramson, N.I.; Petrova, T.V.; Dokuchaev, N.E.; Obolenskaya, E.V.; Lissovsky, A.A. Phylogeography of the Gray Red-Backed Vole Craseomys Rufocanus (Rodentia: Cricetidae) across the Distribution Range Inferred from Nonrecombining Molecular Markers. Russ. J. Theriol. 2012, 11, 137–156. [Google Scholar] [CrossRef]
- Kohli, B.A.; Fedorov, V.B.; Waltari, E.; Cook, J.A. Phylogeography of a Holarctic Rodent (Myodes rutilus): Testing High-latitude Biogeographical Hypotheses and the Dynamics of Range Shifts. J. Biogeogr. 2015, 42, 377–389. [Google Scholar] [CrossRef]
- Fedorov, V.B.; Goropashnaya, A.V.; Boeskorov, G.G.; Cook, J.A. Comparative Phylogeography and Demographic History of the Wood Lemming (Myopus schisticolor): Implications for Late Quaternary History of the Taiga Species in Eurasia. Mol. Ecol. 2008, 17, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.V.; Kuksin, A.N.; Putintsev, N.I.; Lopatina, N.V.; Lissovsky, A.A. Phylogeography of Lasiopodomys gregalis (Cricetidae, Rodentia) in the Southern Part of Its Geographic Range. Russ. J. Theriol. 2023, 22, 102–119. [Google Scholar] [CrossRef]
- Petrova, T.V.; Genelt-Yanovskiy, E.A.; Lissovsky, A.A.; Chash, U.-M.G.; Masharsky, A.E.; Abramson, N.I. Signatures of Genetic Isolation of the Three Lineages of the Narrow-Headed Vole Lasiopodomys gregalis (Cricetidae, Rodentia) in a Mosaic Steppe Landscape of South Siberia. Mamm. Biol. 2021, 101, 275–285. [Google Scholar] [CrossRef]
- Gromov, I.M.; Erbaeva, M.A. Mammals of the Fauna of Russia and Adjacent Regions. Lagomorphs and Rodents; Zoological Institute RAS: Saint Petersburg, Russia, 1995. [Google Scholar]
- Ognev, S.I. Zveri SSSR I Prilezhashhih Stran (The Mammals of the USSR and Adjacent Countries). Vol VII. Gryzuny (Rodentia); Publ. Acad. Nauk USSR: Leningrad, Russia, 1950. [Google Scholar]
- Gromov, I.M.; Polyakov, I.Y. Voles (Microtinae); Brill: Leiden, The Netherlands, 1992; Volume 3. [Google Scholar]
- Bobrinsky, N.A.; Kuznetsov, B.A.; Kuzyakin, A.P. Check-List of the Mammals of the USSR; Prosveshchenie: Moscow, Russia, 1965. [Google Scholar]
- Vorontsov, N.N.; Lyapunova, E.A.; Boeskorov, G.G.; Revin, Y.V. Stability of the root vole (Microtus oeconomus) karyotype in the central part of its range and the history of formation of the species present range. Zool. Zhurnal 1986, 65, 1705–1715. [Google Scholar]
- Pavlinov, I.Y.; Rossolimo, O.L. Systematics of the USSR Mammals; Moscow State University Press: Moscow, Russia, 1987. [Google Scholar]
- Kostenko, V.A.; Allenova, T.V. Intraspecies differentiation of the root vole in the Far East and distributional history of its subspecific form. In Theriological Studies of the Southern Far East; Kostenko, V.A., Ed.; Far East Div. Russian Acad. Sci.: Vladivostok, Russia, 1989; pp. 4–25. [Google Scholar]
- Abramson, N.I.; Tikhonova, E.P. Reevaluation of Taxonomic Structure of the Root Vole (Microtus oeconomus Pallas, 1776, Rodentia, Arvicolidae) from the Territory of the Former USSR Based on Evidence of Craniometric and Molecular Data. Russ. J. Theriol. 2005, 4, 63–73. [Google Scholar] [CrossRef]
- Lissovsky, A.A.; Obolenskaya, E.V. The structure of craniometrical diversity of grey voles Microtus subgenus Alexandromys. Proc. Zool. Inst. RAS 2011, 315, 461–477. [Google Scholar] [CrossRef]
- Pozdnyakov, A.A.; Litvinov, Y.N. Ecogeographic interpretation of morphotypical variability of the molars in Microtus oeconomus (Rodentia, Arvicolidae). Zool. J. 1994, 73, 151–157. [Google Scholar]
- Kryštufek, B.; Shenbrot, G. Voles and Lemmings (Arvicolinae) of the Palaearctic Region; University of Maribor, University Press: Maribor, Slovenia, 2022; ISBN 978-961-286-611-2. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Northern Europe | Central Europe | Beringia | Northern Asia | |
|---|---|---|---|---|
| Northern Europe | 0.0089 | |||
| Central Europe | 0.0240 | 0.0049 | ||
| Beringia | 0.0392 | 0.0395 | 0.0080 | |
| Northern Asia | 0.0343 | 0.0397 | 0.0296 | 0.0097 |
| Lineage/Region Within N. Asian Lineage | n | H | Hd (SD) | Pi% (SD) | Fs | R2 | D |
|---|---|---|---|---|---|---|---|
| Northern Europe | 36 | 29 | 0.987 (0.010) | 0.889 (0.061) | −17.018 | 0.0726 | −1.19686 |
| Central Europe | 33 | 31 | 0.996 (0.009) * | 0.493 (0.050) | −34.098 | 0.0528 | −1.76647 |
| Beringia | 204 | 83 | 0.980 (0.003) | 0.795 (0.031) | −69.256 | 0.0316 | −1.92808 |
| Northern Asia | 250 | 126 | 0.987 (0.002) | 0.968 (0.028) | −32.694 | 0.0311 | −1.90935 |
| Siberia (except Altai and Transbaikalia) | 19 | 14 | 0.965 (0.028) | 0.589 (0.045) | −5.279 | 0.0760 | −1.26709 |
| Okhotsk (locs. 46–65) ** | 144 | 63 | 0.969 (0.006) | 0.612 (0.024) | −54.443 | 0.0391 | −1.73805 |
| Ural-Krasnoyarsk (locs. 1–8) | 10 | 9 | 0.978 (0.054) | 0.487 (0.067) | −4.086 | 0.0865 | −1.29682 |
| Yakutia (locs. 9–11) | 9 | 5 | 0.861 (0.087) | 0.287 (0.063) | −0.306 | 0.1609 | −0.59820 |
| Altai–Sayan (locs. 12–34) | 77 | 41 | 0.971 (0.008) | 1.033 (0.030) | n/a | n/a | n/a |
| Altai–Sayan (Siberian haplogroup) | 37 | 22 | 0.949 (0.020) | 0.630 (0.058) | −8.871 | 0.0655 | −1.47954 |
| Eastern Tuva | 40 | 19 | 0.935 (0.022) | 0.544 (0.037) | −5.779 | 0.0741 | −1.08068 |
| Eastern Tuva (except Torekhol) | 32 | 18 | 0.954 (0.018) | 0.428 (0.038) | −8.490 | 0.0638 | −1.45114 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, T.V.; Lissovsky, A.A.; Bodrov, S.Y.; Kuular, A.V.; Putintsev, N.I.; Bariushaa, M.; Abramson, N.I. Reconstruction of Pleistocene Evolutionary History of the Root Vole Alexandromys oeconomus (Cricetidae, Rodentia) in Northern Asia. Diversity 2025, 17, 497. https://doi.org/10.3390/d17070497
Petrova TV, Lissovsky AA, Bodrov SY, Kuular AV, Putintsev NI, Bariushaa M, Abramson NI. Reconstruction of Pleistocene Evolutionary History of the Root Vole Alexandromys oeconomus (Cricetidae, Rodentia) in Northern Asia. Diversity. 2025; 17(7):497. https://doi.org/10.3390/d17070497
Chicago/Turabian StylePetrova, Tatyana V., Andrey A. Lissovsky, Semyon Yu. Bodrov, Aivar V. Kuular, Nikolay I. Putintsev, Munkhtsog Bariushaa, and Natalia I. Abramson. 2025. "Reconstruction of Pleistocene Evolutionary History of the Root Vole Alexandromys oeconomus (Cricetidae, Rodentia) in Northern Asia" Diversity 17, no. 7: 497. https://doi.org/10.3390/d17070497
APA StylePetrova, T. V., Lissovsky, A. A., Bodrov, S. Y., Kuular, A. V., Putintsev, N. I., Bariushaa, M., & Abramson, N. I. (2025). Reconstruction of Pleistocene Evolutionary History of the Root Vole Alexandromys oeconomus (Cricetidae, Rodentia) in Northern Asia. Diversity, 17(7), 497. https://doi.org/10.3390/d17070497