
Phylogeography and Past Distribution of Peripheral Individuals of Large Hairy Armadillo Chaetophractus villosus

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Collection of Material from Peripheral Populations
2.2. MtDNA Extraction
2.3. Genetic Diversity and Genealogical Analysis of C. villosus Sequences
2.4. Historical Demographic Analysis
2.5. Potential Geographic Distribution Modeling
3. Results
3.1. Sequence Variability
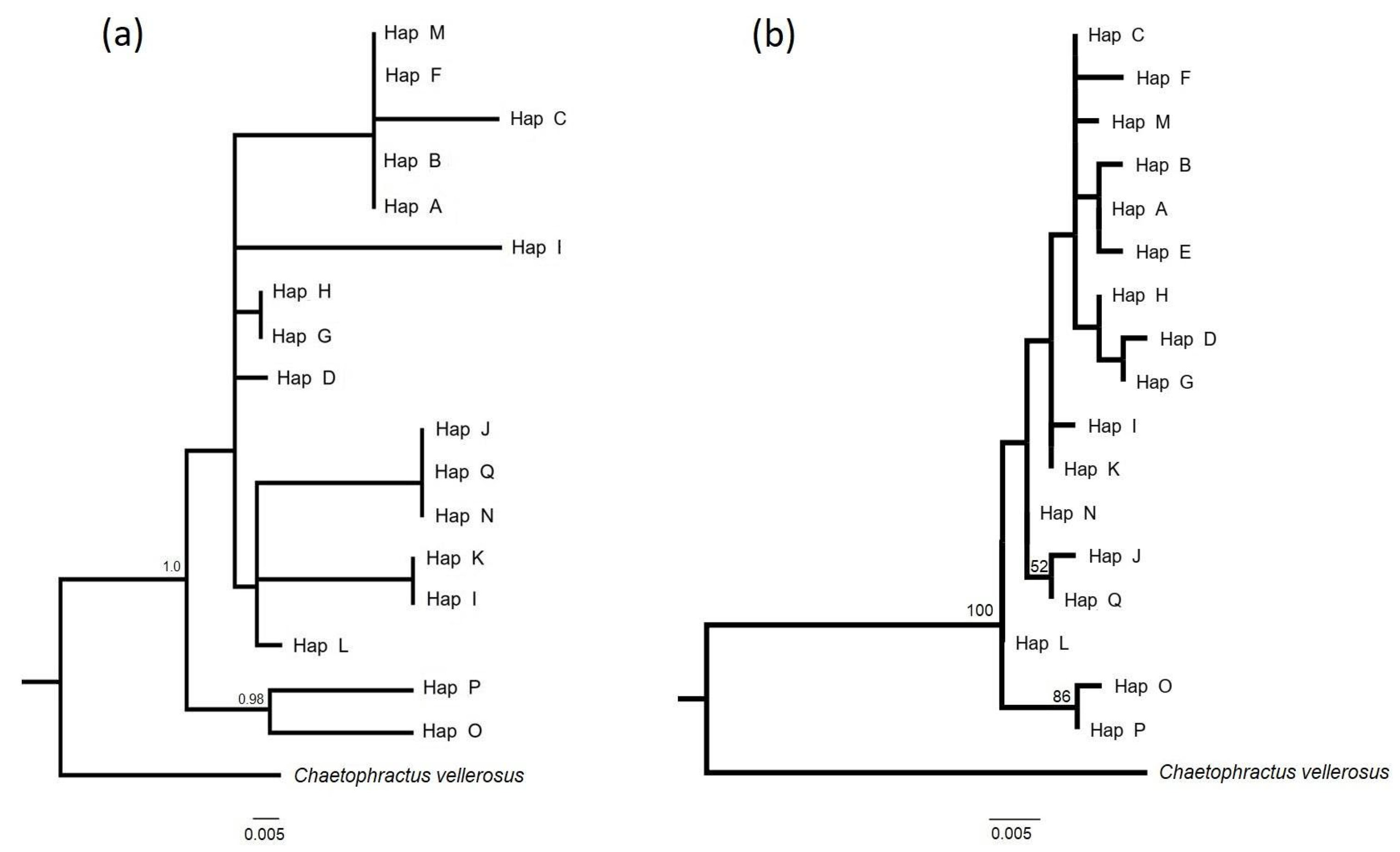
3.2. Relationships Between Haplotypes
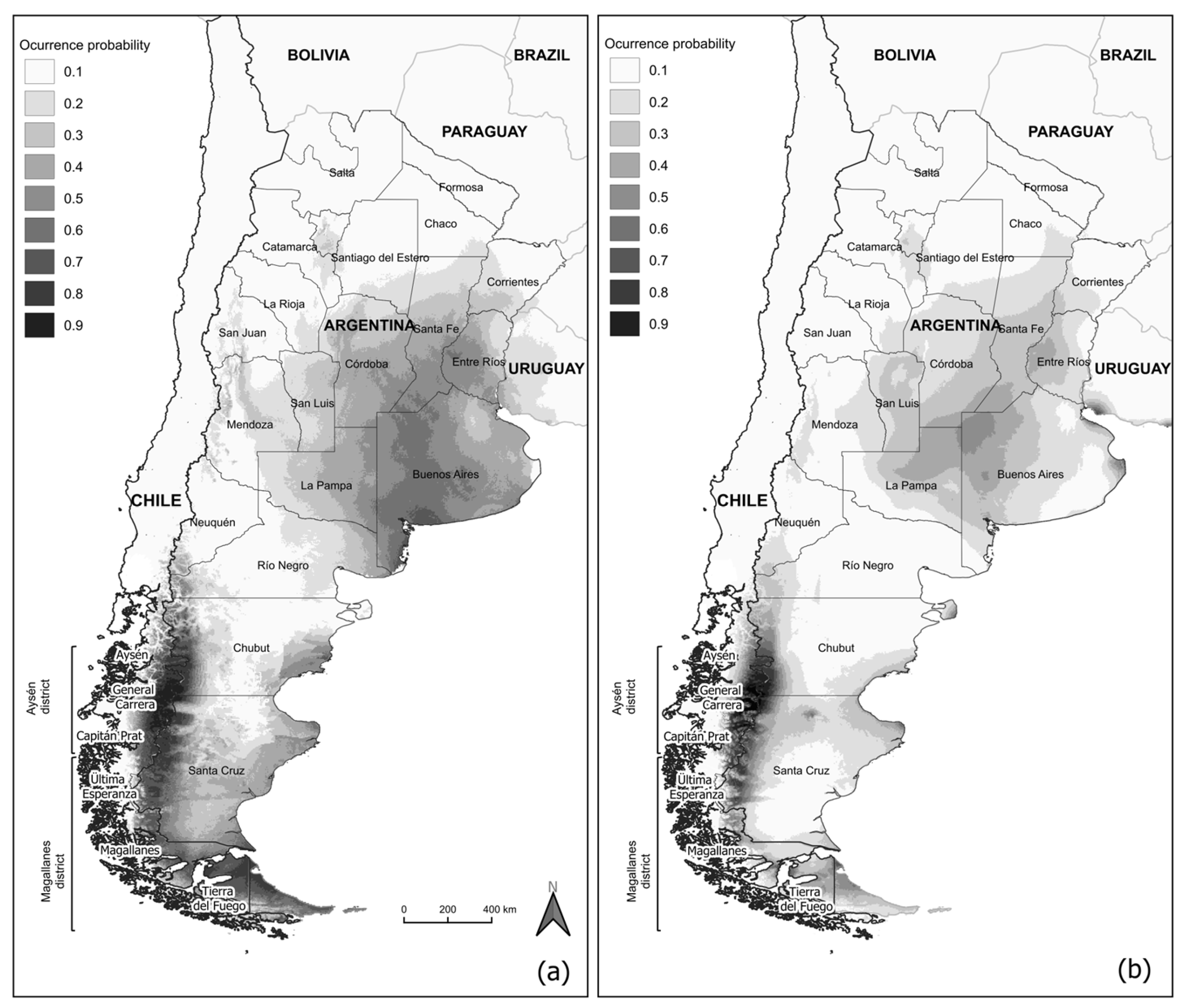
3.3. Past Geographic Distribution of C. villosus
4. Discussion
4.1. Low Haplotype Diversity in Chilean Peripheral Populations
4.2. Relationships and Geographic Distribution of Haplotypes
4.3. Past Geographic Distribution of C. villosus
5. Final Considerations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rabassa, J. Late Cenozoic glaciations in Patagonia and Tierra del Fuego. In The late Cenozoic of Patagonia and Tierra del Fuego; Rabassa, J., Ed.; Elsevier: Oxford, UK, 2008; pp. 151–204. [Google Scholar]
- Rabassa, J.; Coronato, J.; Salemme, M. Chronology of the Late Cenozoic Patagonian glaciations and their correlation with biostratigraphic units of the Pampean Region (Argentina). J. S. Am. Earth Sci. 2005, 20, 81–103. [Google Scholar] [CrossRef]
- Cosacov, A.; Sérsic, A.; Sosa, V.; Johnson, L.; Cocucci, A. Multiple Periglacial Refugia in the Patagonian Steppe and Post-Glacial Colonization of the Andes: The Phylogeography of Calceolaria Polyrhiza. J. Biogeogr. 2010, 37, 1463–1477. [Google Scholar] [CrossRef]
- Markgraf, V. Late and postglacial vegetational and paleoclimatic changes in subantarctic, temperate, and arid environments in Argentina. Palynology 1983, 7, 43–70. [Google Scholar] [CrossRef]
- Scillato-Yané, G. Los Dasypodidae (Mammalia, Edentata) del Plioceno y Pleistoceno de Argentina. Ph.D. Thesis, Universidad Nacional de La Plata, La Plata, Argentina, 1982; p. 159. [Google Scholar]
- Poljak, S.; Confalonieri, V.; Fasanella, M.; Gabrielli, M.; Lizarralde, M. Phylogeography of the armadillo Chaetophractus villosus (Dasypodidae Xenarthra): Post-glacial range expansion from Pampas to Patagonia (Argentina). Mol. Phylogenet. Evol. 2010, 55, 38–46. [Google Scholar] [CrossRef]
- Abba, A.; Poljak, S.; Gabrielli, M.; Teta, P.; Pardiñas, U. Armored invaders in Patagonia: Recent southward dispersion of armadillos (Cingulata, Dasypodidae). Mastozool. Neotrop. 2014, 21, 311–318. [Google Scholar]
- Wetzel, R. Taxonomy and distribution of armadillos, Dasypodidae. In The Ecology and Evolution of Armadillos, Sloths, and Vermilinguas; Montgomery, G., Ed.; Smithsonian Institution Press: Washington, DC, USA, 1985; pp. 23–46. [Google Scholar]
- Osgood, W. Mammals of Chile. Pub. Field Mus. Nat. Hist. Zool. Ser. 1943, 30, 1–268. [Google Scholar]
- Tamayo, M. Los armadillos de Chile. Situación de Euphractus sexcinctus (Linnaeus, 1758). Not. Mens. Del Mus. Nac. Hist. Nat. 1973, 203–204, 3–6. [Google Scholar]
- Arriagada, A.; Baessolo, L.; Saucedo, C.; Crespo, J.; Cerda, J.; Parra, L.; Aldridge, D.; Ojeda, J.; Hernández, A. Hábitos alimenticios de poblaciones periféricas de Zaedys pichiy y Chaetophractus villosus (Cingulata, Chlamyphoridae) en la Patagonia chilena. Iheringia Ser. Zool. 2017, 107, e2017013. [Google Scholar] [CrossRef]
- Poljak, S.; Escobar, J.; Deferrari, G.; Lizarralde, M. Un nuevo mamífero introducido en la Tierra Del Fuego: El Peludo Chaetophractus villosus (Mammalia, Dasypodidae) en Isla Grande. Rev. Chil. Hist. Nat. 2007, 80, 285–294. [Google Scholar] [CrossRef]
- Lesica, P.; Allendorf, F. When are peripheral populations valuable for conservation? when are peripheral populations valuable for conservation? Conserv. Biol. 1995, 9, 753–760. [Google Scholar] [CrossRef]
- Parmesan, C.; Gaines, S.; Gonzalez, L.; Kaufman, D.; Kingsolver, J.; Peterson, T.; Sagarin, R. Empirical perspectives on species borders: From traditional biogeography to global change. Oikos 2005, 108, 58–75. [Google Scholar] [CrossRef]
- Haak, A.; Williams, J.; Neville, H.; Dauwalter, D.; Colyer, W. Conserving peripheral trout populations: The values and risks of life on the edge. Fisheries 2010, 35, 530–549. [Google Scholar] [CrossRef]
- Garner, T.; Pearman, P.; Angelone, S. Genetic diversity across a vertebrate species range: A test of the central-peripheral hypothesis. Mol. Ecol. 2004, 13, 1047–1053. [Google Scholar] [CrossRef]
- Kirkpatrick, M.; Barton, N. Evolution of a species’ range. Am. Nat. 1997, 150, 1–23. [Google Scholar] [CrossRef]
- Lenormand, T. Gene flow and the limits to natural selection. Trends Ecol. Evol. 2002, 17, 183–189. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nakajima, M.; Taniguchi, N. Loss of genetic variation and increased population differentiation in geographically peripheral populations of japanese char Salvelinus leucomaenis. Aquaculture 2010, 308, S20–S27. [Google Scholar] [CrossRef]
- Hampe, A.; Petit, R. Conserving biodiversity under climate change: The rear edge matters. Ecol. Lett. 2005, 8, 461–467. [Google Scholar] [CrossRef]
- Volis, S.; Ormanbekova, D.; Yermekbaayev, K.; Song, M.; Shugilna, I. The conservation value of peripheral populations and a relationship between quantitative trait and molecular variation. Evol. Biol. 2015, 43, 26–36. [Google Scholar] [CrossRef]
- Strasburg, J.; Kearney, M.; Moritz, C.; Templeton, A. Combining phylogeography with distribution modeling: Multiple pleistocene range expansions in a parthenogenetic gecko from the Australian arid zone. PLoS ONE 2007, 2, e760. [Google Scholar] [CrossRef]
- John, S.; Weitzner, G.; Rozen, R.; Scriver, C. A rapid procedure for extracting genomic DNA from leukocytes. Nucleic Acids Res. 1991, 19, 408. [Google Scholar] [CrossRef]
- Sambrock, J.; Fritsch, E.; Maniatis, T. Molecular Cloning, a Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Roland, N.; Hofreiter, M. Ancient DNA extraction from bones and teeth. Nat. Protoc. 2007, 2, 1756–1762. [Google Scholar] [CrossRef]
- Vilá, C.; Amorin, I.; Leonard, J. Mitochondrial DNA phylogeography and population history of the grey Wolf Canis lupus. Mol. Ecol. 1999, 8, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Salzburger, W.; Ewing, G.; von Haeseler, A. The performance of phylogenetic algorithms in estimating haplotype genealogies with migration. Mol. Ecol. 2011, 20, 1952–1963. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of Phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Fu, X. Statistical test of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Tajima, F. Measurement of DNA polymorphism. In Mechanisms of Molecular Evolution. Introduction to Molecular Paleopopulation Biology; Takahata, N., Clark, A., Eds.; Japan Scientific Societies Press: Tokyo, Japan; Sinauer Associates, Inc.: Sunderland, MA, USA, 1993; pp. 37–59. [Google Scholar]
- Rozas, J.; Ferre-Mata, A.; Sanchez-Del Barrio, J.; Guiao-Rico, S.; Librado, P.; Ramos-Onsins, S.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence polymorphism analysis. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Elith, J.; Phillips, S.; Hastie, T.; Dudik, M.; Chee, Y.; Yates, C. A statistical explanation of MaxEnt for ecologists. Divers. Distrib. 2010, 17, 43–57. [Google Scholar] [CrossRef]
- Hijmans, R.; Cameron, S.; Parra, J.; Jones, P.; Jarvos, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Brown, J.L. 2014. SD Mtoolbox: A python-based GIS toolkit for landscape genetic, biogeographic and species distribution model analyses. Methods Ecol. Evol. 2014, 5, 694–700. [Google Scholar] [CrossRef]
- Hirzel, A.; Lay, G.; Helfer, V.; Randin, C.; Guisan, A. Evaluating the ability of habitat suitability models to predict species presences. Ecological Modelling 2006, 199, 142–152. [Google Scholar] [CrossRef]
- Peterson, A.; Soberón, J.; Pearson, R.; Anderson, R.; Martínez-Meyer, E.; Nakamura, M.; Araujo, M. Ecological Niches and Geographic Distributions; Princeton University Press: Princeton, NJ, USA, 2011. [Google Scholar]
- Pearson, R.; Raxworthy, C.; Nakamura, M.; Peterson, T. Predicting species distributions from small numbers of occurrence records: A test case using cryptic geckos in Madagascar. J. Biogeogr. 2007, 34, 102–117. [Google Scholar] [CrossRef]
- Boyce, M. Relating populations to habitats using resource selection functions. Ecol. Model. 2002, 157, 281–300. [Google Scholar] [CrossRef]
- Almalki, M.; Alrashidi, M. ¸O’Connell, M.; Shobrak, M.; Szekely, T. Modelling the distribution of wetland birds on the Red Sea coast in the Kingdom of Saudi Arabia. Appl. Ecol. Environ. Res. 2015, 13, 7–84. [Google Scholar]
- Eckert, C.; Samis, K.; Lougheed, S. Genetic variation across species geographical ranges: The central-marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef]
- Dai, Q.; Fu, J. When central populations exhibit more genetic diversity than peripheral populations: A simulation study. Chin. Sci. Bull. 2011, 56, 2531–2540. [Google Scholar] [CrossRef]
- Quiroga, M.; Premoli, A. El rol de las poblaciones marginales en la conservación del acervo genético de la única conífera del Sur de Yungas en Argentina y Bolivia, Podocarpus parlatorei (Podocarpaceae). Ecología en Bolivia 2013, 48, 4–16. [Google Scholar]
- Cuéllar, E. Biology and ecology of armadillos in the Bolivian Chaco. In The Biology of the Xenarthra; Vizcaíno, S., Loughry, W., Eds.; University of Florida Press: Gainesville, FL, USA, 2008. [Google Scholar]
- Superina, M.; Abba, A. Family Chlamyphoridae (Chlamyphorid armadillos). In Handbook of the Mammals of the World; Wilson, D.E., Mittermeier, R.A., Eds.; Lynx Edicions: Barcelona, Spain, 2018. [Google Scholar]
- Mcdonough, C.; Delaney, M.; Quoc Le, P.; Blackmore, M.; Loughry, W. Burrow characteristics and habitat associations of armadillos in Brazil and the United States of America. Rev. Biol. Trop. 2000, 48, 109–120. [Google Scholar]
- Pilot, M.; Jedrzejewski, W.; Branicki, W.; Sidorovich, V.; Jedrzejewska, B.; Stachura, K.; Funk, S. Ecological factors influence population genetic structure of European grey wolves. Mol. Ecol. 2006, 15, 4533–4553. [Google Scholar] [CrossRef] [PubMed]
- Niedziałkowska, M.; Jędrzejewska, B.; Honnen, A.; Otto, T.; Sidorovich, V.; Perzanowski, K.; Skog, A.; Hartl, G.; Borowik, T.; Bunevich, A.; et al. Molecular biogeography of red deer Cervus elaphus from Eastern Europe: Insights from mitochondrial DNA sequences. Acta Theriol. 2011, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Silva, F. Caracterización y propiedades de los suelos de la Patagonia Occidental (Aysén). Boletín INIA 2014, 298, 35–52. [Google Scholar]
- Markraf, V.; Whitlock, C.; Haberle, S. Vegetation and fire history during the last 18,000 cal yr B.P. in Southern Patagonia: Mallín Pollux, Coyhaique, Province Aisén (45°41′30″ S, 71°50′30″ W, 640 m elevation). Palaeogeogr. Palaeoclimatol. Palaeoecol. 2007, 254, 492–507. [Google Scholar] [CrossRef]
- Iglesias, V.; Haberle, S.; Holz, A.; Whitlock, C. Holocene dynamics of temperate rainforests in West-Central Patagonia. Front. Ecol. Evol. 2018, 5, 177. [Google Scholar] [CrossRef]
- Iglesias, V.; Whitlock, C.; Bianchi, M.; Villarosa, G.; Outes, V. Holocene climate variability and environmental history at the Patagonian forest/steppe ecotone: Lago Mosquito (42°29′37.89′’S, 71°24′14.57′’W) and Laguna del Cóndor (42°20′47.22′’S, 71°17′07.62′’W). Holocene 2011, 22, 1297–1307. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Province | Locality | Latitude | Longitude | Sample |
|---|---|---|---|---|---|
| TAP01 | Coyhaique | Tapera | 44°39′13.576″ S | 71°43′6.272″ W | carcass tissue |
| BNU01 | Baño Nuevo | 45°15′5.764″ S | 71°36′54.238″ W | carcass tissue | |
| CAL01 | Coyhaique Alto | 45°18′16.693″ S | 71°23′10.9″ W | blood | |
| LAT01 | Lago Atravesado | 45°43′27.134″ S | 72°13′10.175″ W | carcass tissue | |
| BAL01 | Balmaceda | 45°59′28.216″ S | 71°45′35.748″ W | carcass tissue | |
| BAL02 | Balmaceda | 46°1′0.293″ S | 71°46′7.111″ W | carcass tissue | |
| BAL03 | Balmaceda | 46°7′55.718″ S | 72°12′22.943″ W | carcass tissue | |
| CCA02 | General Carrera | Cerro Castillo | 46°6′29.005″ S | 72°3′8.258″ W | carcass tissue |
| CCA01 | Cerro Castillo | 46°6′42.217″ S | 72°9′3.128″ W | blood | |
| VIB01 | Valle Ibañez | 46°8′19.662″ S | 72°12′35.752″ W | carcass tissue | |
| VIB02 | Valle Ibañez | 46°9′40.115″ S | 72°21′22.81″ W | carcass tissue | |
| CIB01 | Valle Ibañez | 46°10′7.435″ S | 72°3′36.173″ W | carcass tissue | |
| SRI01 | Río Ibañez | 46°14′27.892″ S | 71°59′21.692″ W | carcass tissue | |
| PIB01 | Puerto Ibañez | 46°17′30.214″ S | 71°49′16.414″ W | carcass tissue | |
| CCH01 | Chile Chico | 46°34′3.774″ S | 71°47′40.157″ W | carcass tissue | |
| CJE01 | Sector Jeinimeni | 46°42′59.069″ S | 71°43′12.032″ W | carcass tissue | |
| RNJ01 | Sector Jeinimeni | 46°49′27.53″ S | 71°57′47.844″ W | carcass tissue | |
| ALE01 | Capitán Prat | La Alegría | 47°8′42.72″ S | 72°55′17.227″ W | carcass tissue |
| NEF01 | Río Nef | 47°8′47.389″ S | 73°2′5.244″ W | carcass tissue | |
| PNA01 | Última Esperanza | Puerto Natales | 51°42′54.076″ S | 72°27′20.372″ W | carcass tissue |
| LBA01 | Lago Balmaceda | 52°1′3.256″ S | 72°19′59.761″ W | carcass tissue | |
| PUA01 | Magallanes | Punta Arenas | 53°5′20.256″ S | 70°54′50.807″ W | carcass tissue |
| Sample Origin | n | h | S | Hd | ∏ |
|---|---|---|---|---|---|
| Coyhaique * | 7 | 1 | 0 | 0 | 0 |
| General Carrera * | 10 | 1 | 0 | 0 | 0 |
| Capitán Prat * | 2 | 1 | 0 | 0 | 0 |
| Última Esperanza * | 2 | 1 | 0 | 0 | 0 |
| Magallanes * | 1 | 1 | 0 | 0 | 0 |
| Argentine population | 76 | 17 | 12 | 0.88 | 0.004 |
| Argentina + Chile | 98 | 17 | 12 | 0.71 | 0.003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arriagada, A.; Canales-Aguirre, C.B.; Fuentes, N.; Saucedo, C.; Colihueque, N. Phylogeography and Past Distribution of Peripheral Individuals of Large Hairy Armadillo Chaetophractus villosus. Diversity 2025, 17, 390. https://doi.org/10.3390/d17060390
Arriagada A, Canales-Aguirre CB, Fuentes N, Saucedo C, Colihueque N. Phylogeography and Past Distribution of Peripheral Individuals of Large Hairy Armadillo Chaetophractus villosus. Diversity. 2025; 17(6):390. https://doi.org/10.3390/d17060390
Chicago/Turabian StyleArriagada, Aldo, Cristian B. Canales-Aguirre, Norka Fuentes, Cristián Saucedo, and Nelson Colihueque. 2025. "Phylogeography and Past Distribution of Peripheral Individuals of Large Hairy Armadillo Chaetophractus villosus" Diversity 17, no. 6: 390. https://doi.org/10.3390/d17060390
APA StyleArriagada, A., Canales-Aguirre, C. B., Fuentes, N., Saucedo, C., & Colihueque, N. (2025). Phylogeography and Past Distribution of Peripheral Individuals of Large Hairy Armadillo Chaetophractus villosus. Diversity, 17(6), 390. https://doi.org/10.3390/d17060390