
First Record of the Complete Mitochondrial Genome for the Genus Borbo (Lepidoptera, Hesperiidae): Characterization and Comparative Genomic Analysis


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Primer Design, PCR Amplification and Sequencing
2.3. Mitogenome Assembly and Bioinformatic Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
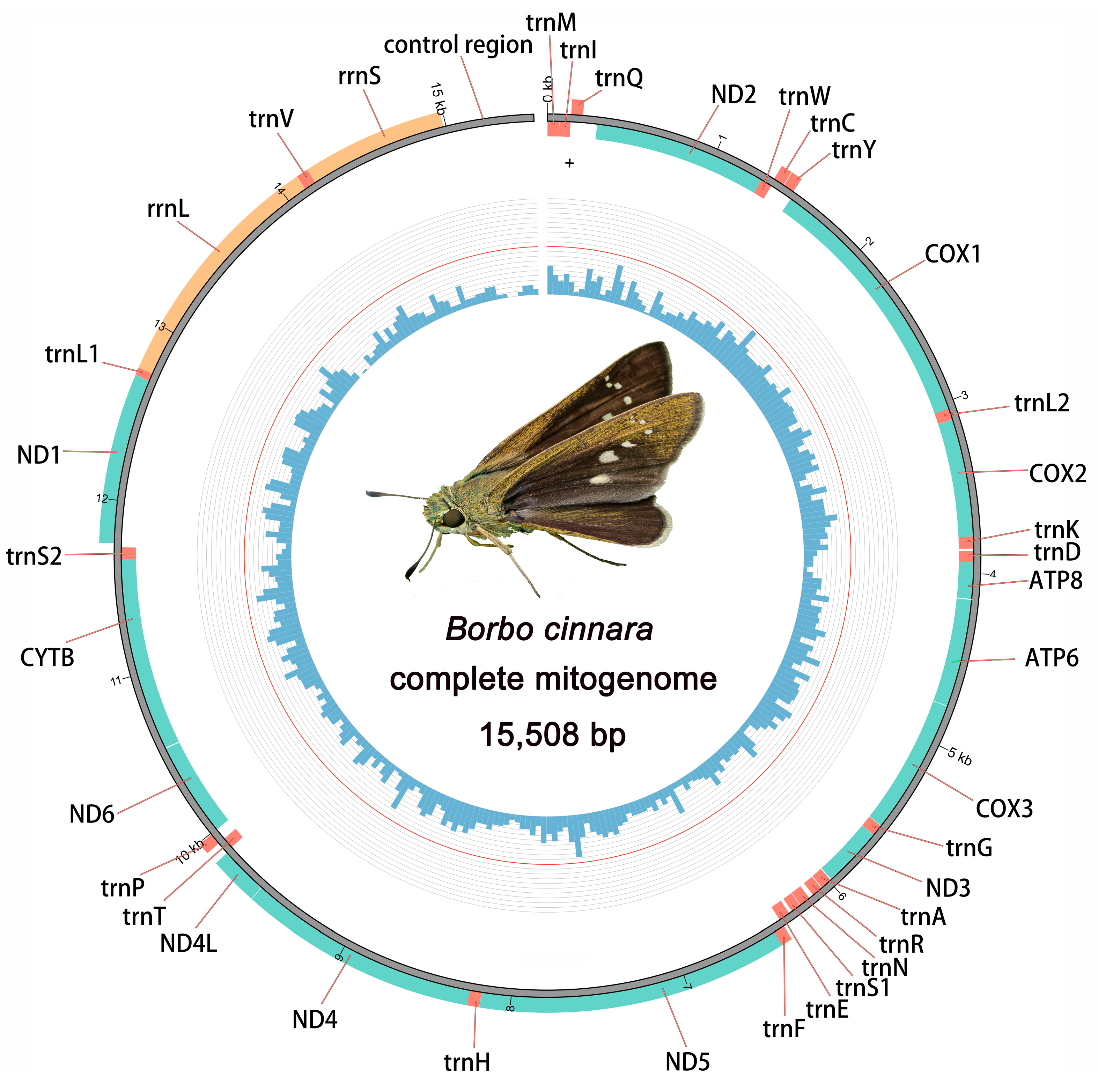
3.1. General Mitogenomic Features
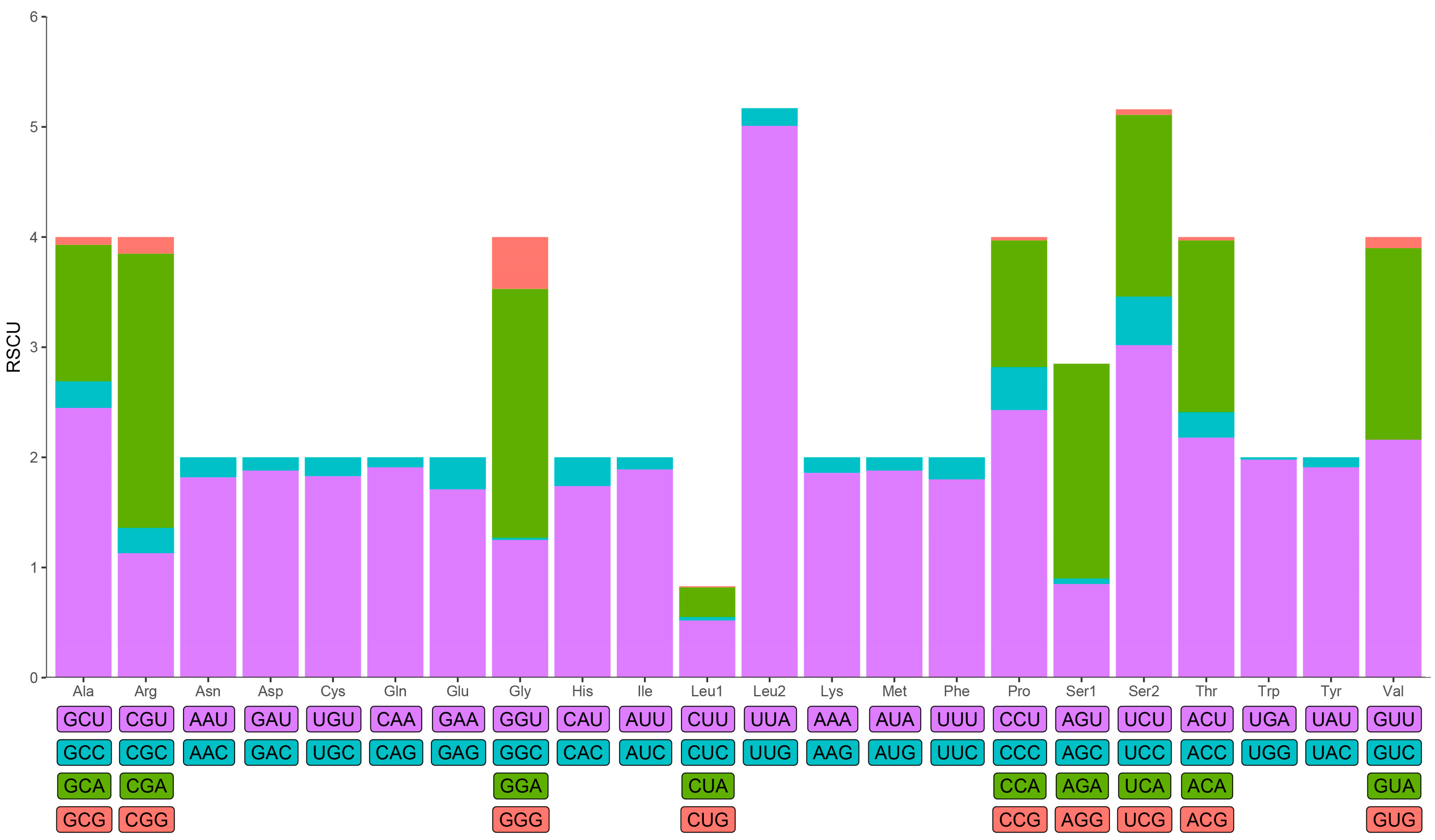
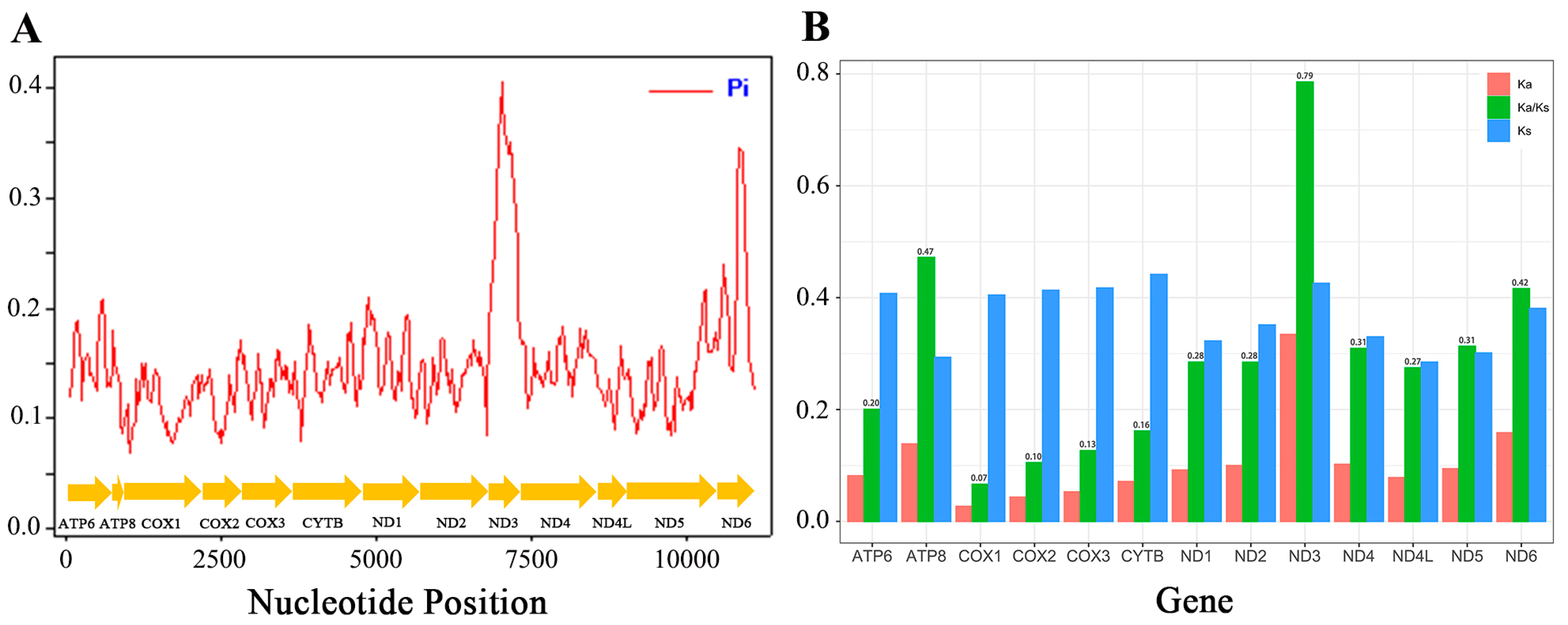
3.2. Protein-Coding Genes
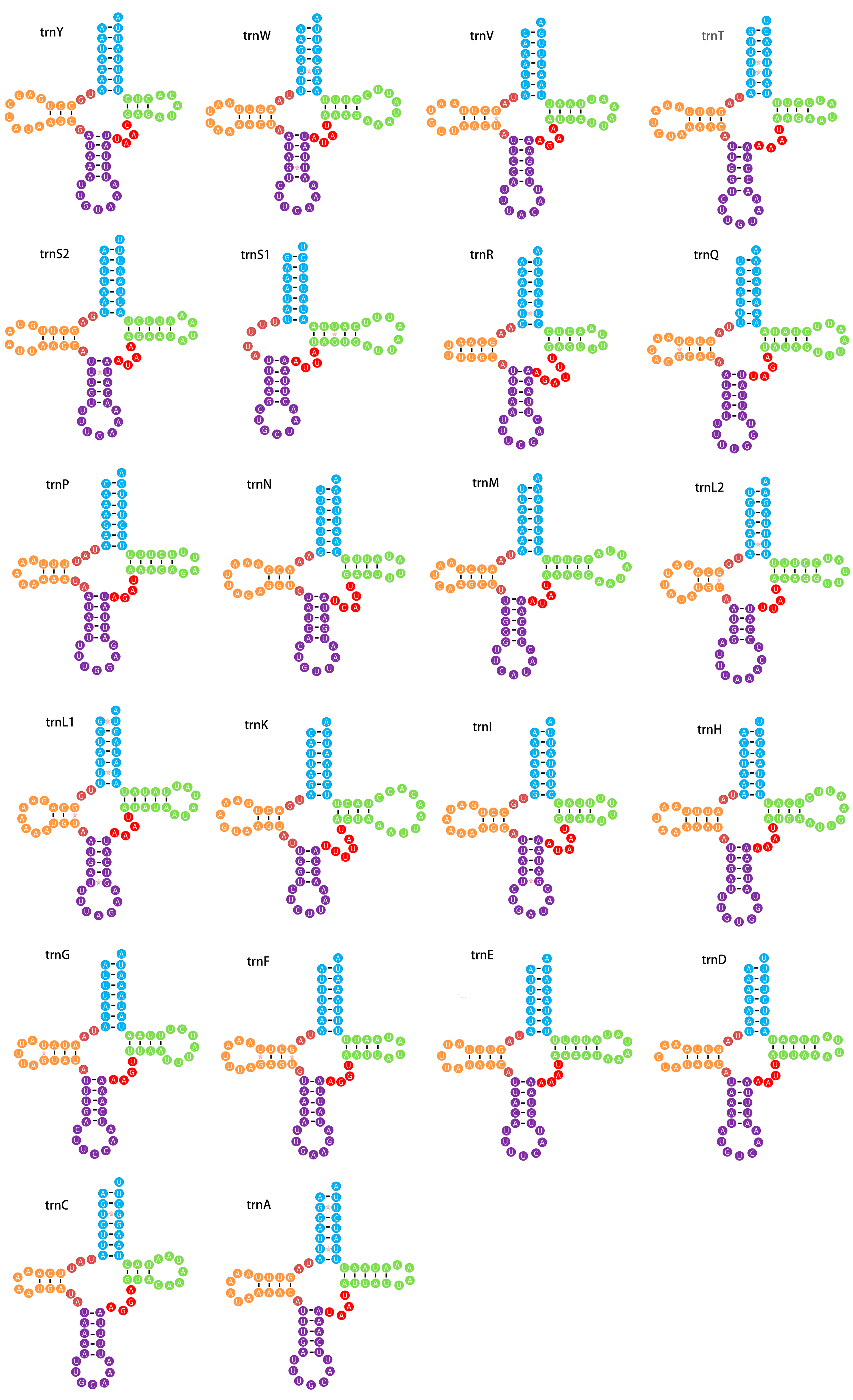
3.3. Ribosomal and Transfer RNA Genes
3.4. Non-Coding Regions
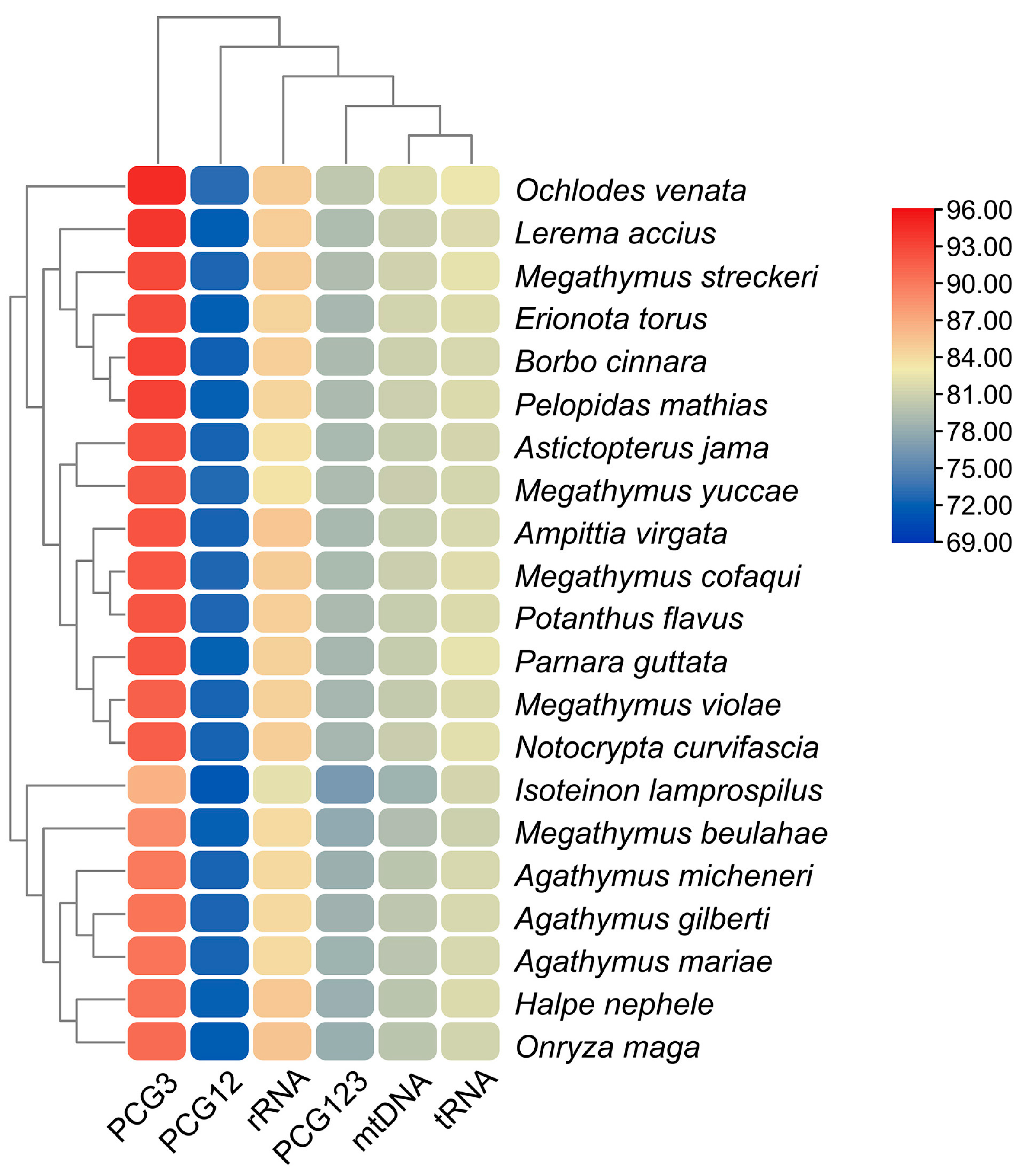
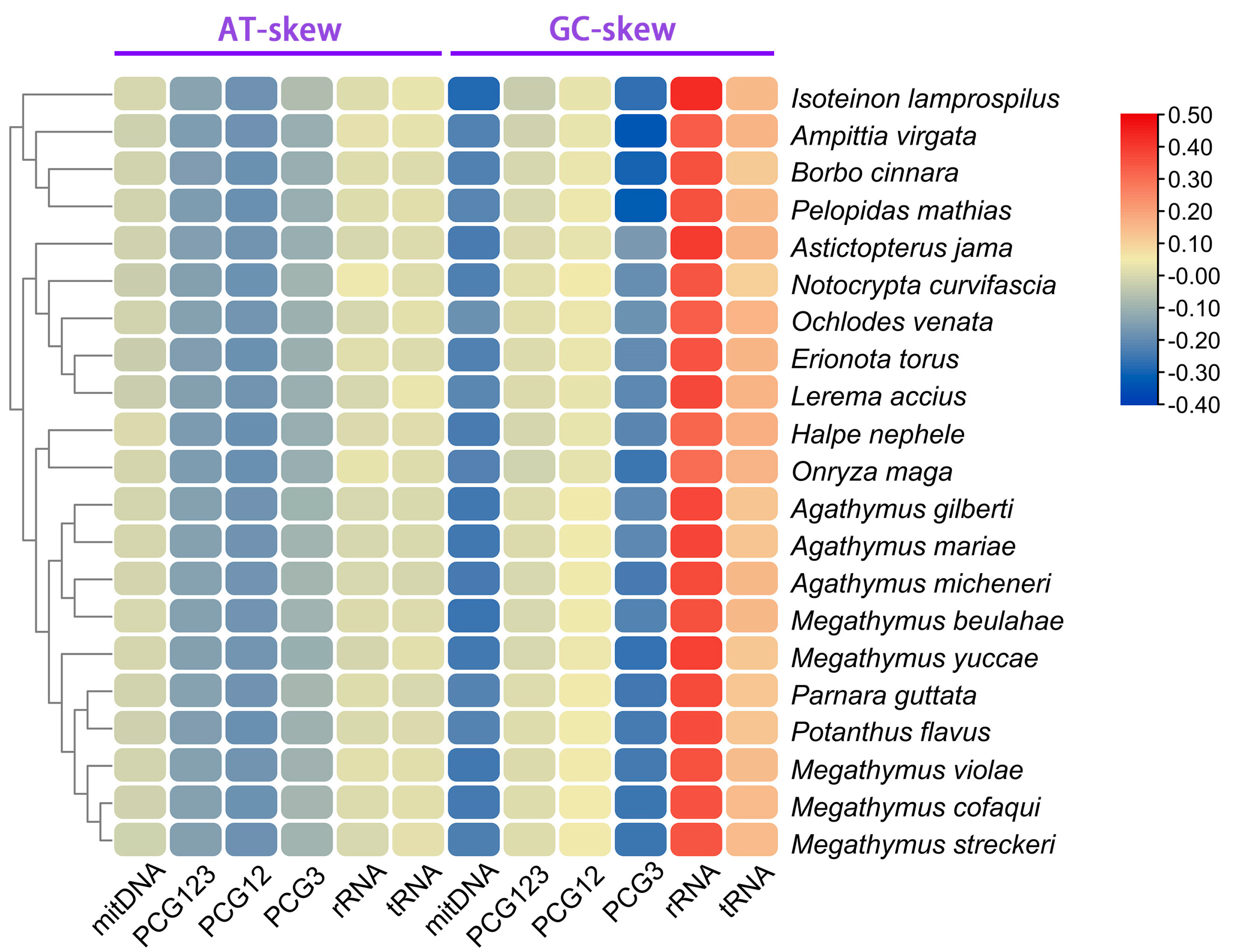
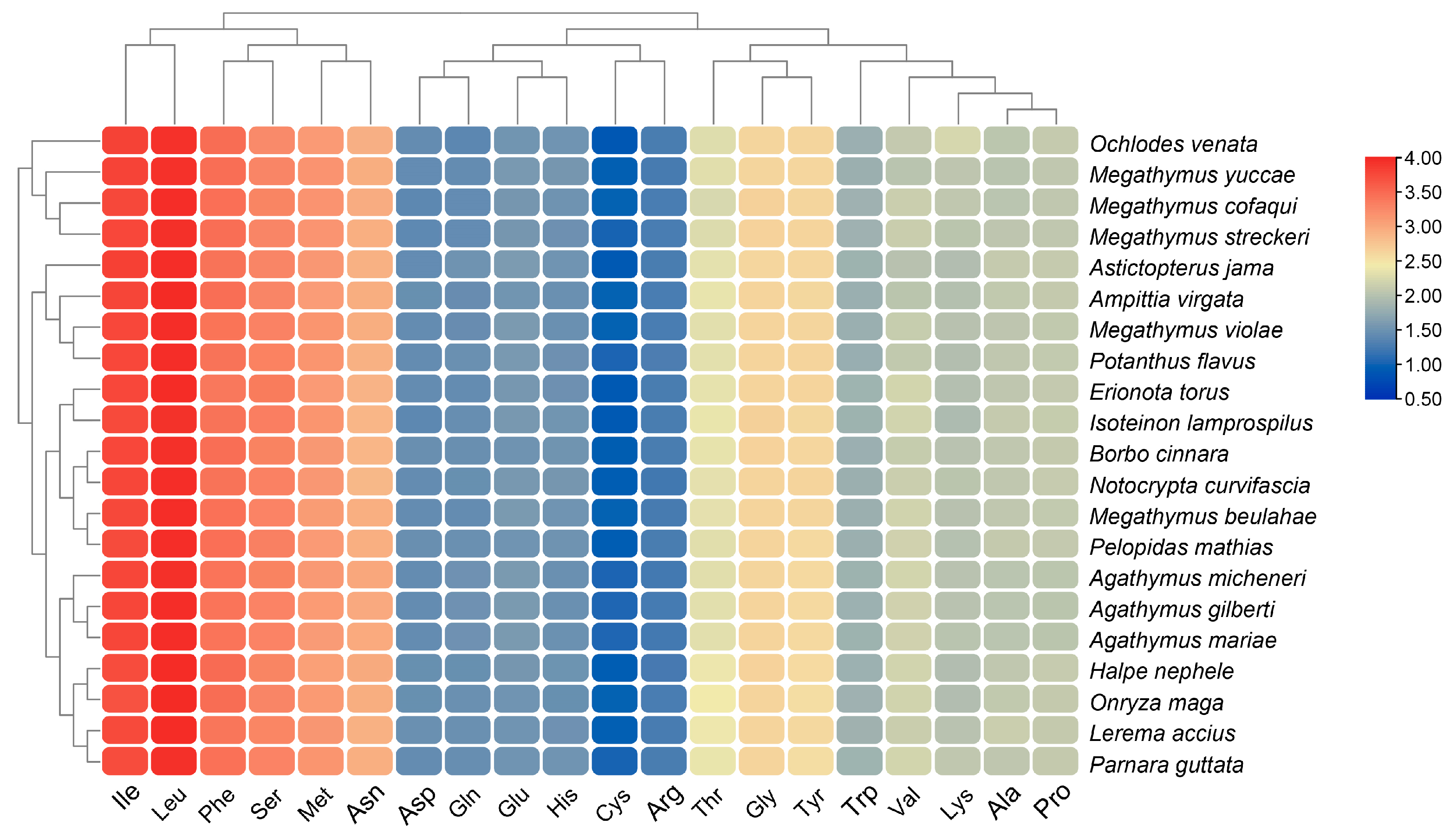
3.5. Comparative Analysis of Hesperiinae Mitogenomes
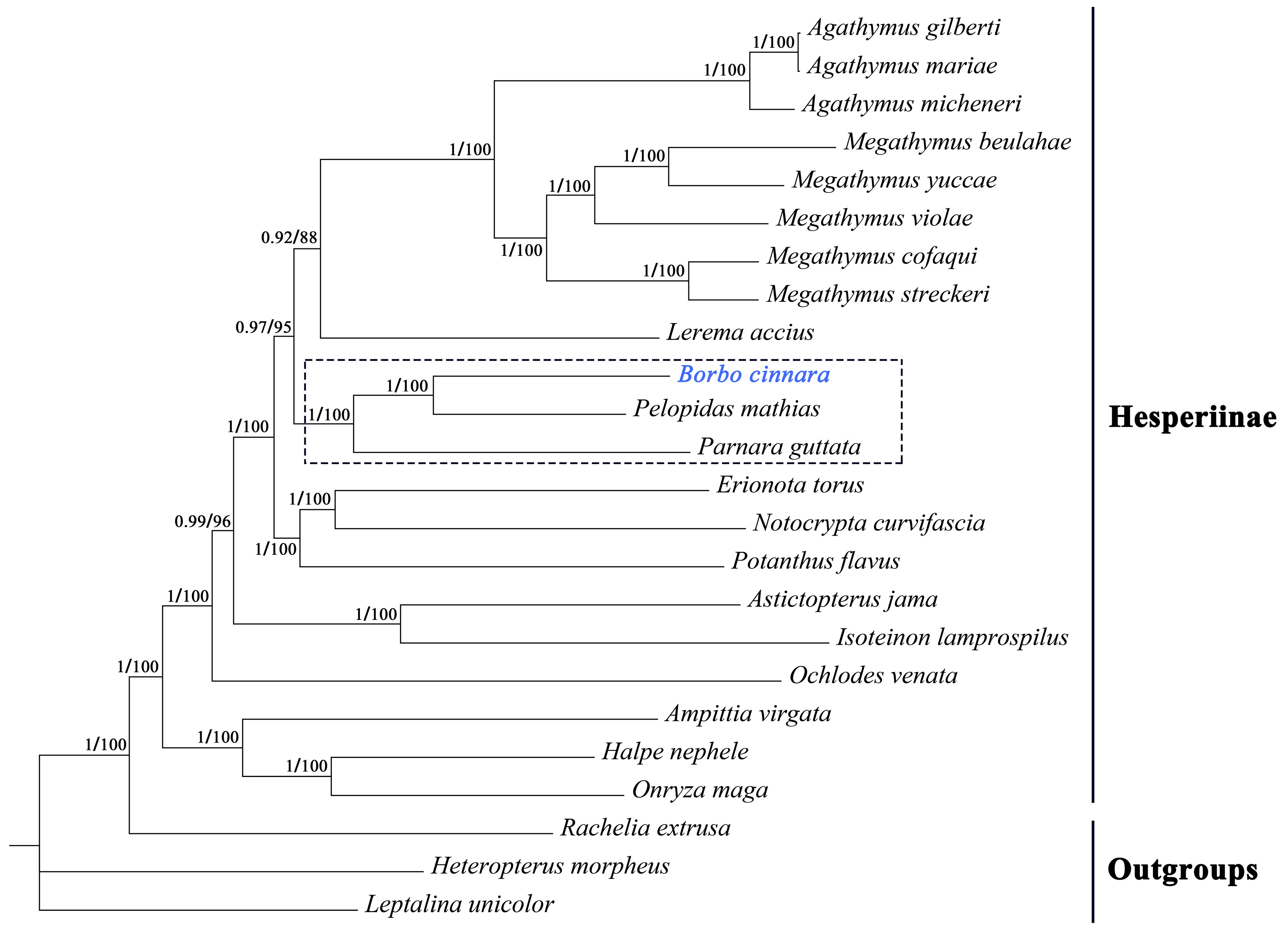
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. Drosophila mitochondrial DNA: Conserved sequences in the A+T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J. Mol. Evol. 1987, 25, 116–125. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta—Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Timmermans, M.J.; Lees, D.C.; Simonsen, T.J. Towards a mitogenomic phylogeny of Lepidoptera. Mol. Phylogenet. Evol. 2014, 79, 169–178. [Google Scholar] [CrossRef]
- Morgan, B.; Wang, T.Y.; Chen, T.Z.; Moctezuma, V.; Burgos, O.; Le, M.H.; Huang, J.P. Long-read sequencing data reveals dynamic evolution of mitochondrial genome size and the phylogenetic utility of mitochondrial DNA in Hercules beetles (Dynastes; Scarabaeidae). Genome Biol. Evol. 2022, 14, evac147. [Google Scholar] [CrossRef]
- Fan, X.L.; Chiba, H.; Huang, Z.F.; Fei, W.; Sáfián, S. Clarification of the phylogenetic framework of the tribe Baorini (Lepidoptera: Hesperiidae: Hesperiinae) inferred from multiple gene sequences. PLoS ONE 2016, 11, e0156861. [Google Scholar] [CrossRef]
- Li, Y.P.; Gao, K.; Yuan, F.; Wang, P.; Yuan, X.Q. Molecular systematics of the butterfly tribe Baorini (Lepidoptera: Hesperiidae) from China. J. Kans. Entomol. Soc. 2017, 90, 100–108. [Google Scholar] [CrossRef]
- Larsen, T.B. Two new species of African Hesperiidae: Borbo (Hepseriinae, Baorini) and Platylesches (Hepseriinae, incertae sedis). Trop. Lepid. Res. 2013, 23, 92–98. [Google Scholar]
- Toussaint, E.F.; Vila, R.; Yago, M.; Chiba, H.; Warren, A.D.; Aduse-Poku, K.; Storer, C.; Dexter, K.M.; Maruyama, K.; Lohman, D.J.; et al. Out of the Orient: Post-Tethyan transoceanic and trans-Arabian routes fostered the spread of Baorini skippers in the Afrotropics. Syst. Entomol. 2019, 44, 926–938. [Google Scholar] [CrossRef]
- Jiang, W.B.; He, H.Y.; Li, Y.Y.; Wang, Y.; Ge, C.; Zhu, J.; Yu, W. Molecular phylogeny of the butterfly tribe Baorini Doherty, 1886 (Hesperiidae, Hesperiinae) in China. Zootaxa 2019, 4565, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.; Yang, L.; Chen, X.S. Comparative analysis of twelve mitogenome of Caliscelidae (Hemiptera: Fulgoromorpha) and their phylogenetic implications. PeerJ 2021, 9, e12465. [Google Scholar] [CrossRef] [PubMed]
- Kunte, K. Butterflies of Peninsular India; Universities Press (Hyderabad) and Indian Academy of Sciences (Bangalore): Bangalore, India, 2001; 254p. [Google Scholar]
- Yuan, F.; Yuan, X.; Xue, G. Fauna Sinica: Insecta—Lepidoptera, Hesperiidae; Science Press: Beijing, China, 2015. [Google Scholar]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, Y.Q.; Shu, X.H.; Meng, L.; Li, B.P. Complete mitochondrial genomes of three Oxya grasshoppers (Orthoptera) and their implications for phylogenetic reconstruction. Genomics 2019, 112, 289–296. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. In Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 71–91. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2018, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Xia, X.H. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Castresana, J. Gblocks v. 0.91 b. Available online: https://www.biologiaevolutiva.org/jcastresana/Gblocks.html (accessed on 20 June 2023).
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree 1.4.2. Institute of Evolutionary Biology, University of Edinburgh. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 15 May 2023).
- Clary, D.O.; Wolstenholme, D.R. The ribosomal RNA genes of Drosophila mitochondrial DNA. Nucleic Acids Res. 1985, 13, 4029–4045. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, L.; Liao, C.Q.; Wang, X.; Wang, M.; Huang, G.H. Comparative mitochondrial genome analysis and phylogenetic relationship among lepidopteran species. Gene 2022, 830, 146516. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yin, A. The complete mitochondrial genome of Chibiraga houshuaii (Lepidoptera, Limacodidae) and its phylogenetic implications. Sci. Rep. 2024, 14, 7009. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.Y.; Liu, J.Q.; Chiba, H.; Xiao, J.T.; Yuan, X.Q. Complete mitochondrial genomes of three skippers in the tribe Aeromachini (Lepidoptera: Hesperiidae: Hesperiinae) and their phylogenetic implications. Ecol. Evol. 2021, 11, 8381–8393. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.Y.; Zhao, J.F.; Hao, R.W.; Zhao, Y.F.; Yuan, X.Q. Complete mitochondrial genome of the small-branded swift: Pelopidas mathias (Lepidoptera, Hesperiidae). Mitochondrial DNA Part B 2021, 6, 1599–1600. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Varani, G.; Mcclain, W.H. The G·U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Liu, L.N.; Zeng, L.; Guo, Z.X.; Yang, B.M.; Yin, K.S.; Wang, C.M.; Zheng, S.J. Complete mitochondrial genome of banana skipper Erionota torus Evans (Lepidoptera: Hesperiidae) and phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 2054–2055. [Google Scholar] [CrossRef]
- Fernández-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Maximizing transcription efficiency causes codon usage bias. Genetics 1996, 144, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ma, Z.; Zhou, C. The first two complete mitochondrial genomes of Neoephemeridae (Ephemeroptera): Comparative analysis and phylogenetic implication for Furcatergalia. Genes 2021, 12, 1875. [Google Scholar] [CrossRef]
- Zhang, L.; Li, W.H. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol. Biol. Evol. 2004, 21, 236–239. [Google Scholar] [CrossRef]
- Warren, A.D.; Ogawa, J.R.; Brower, A.V.Z. Phylogenetic relationships of subfamilies and circumscription of tribes in the family Hesperiidae (Lepidoptera: Hesperioidea). Cladistics 2008, 24, 642–676. [Google Scholar] [CrossRef]
- Sahoo, R.K.; Warren, A.D.; Wahlberg, N.; Brower, A.V.; Lukhtanov, V.A.; Kodandaramaiah, U. Ten genes and two topologies: An exploration of higher relationships in skipper butterflies (Hesperiidae). PeerJ 2016, 4, e2653. [Google Scholar] [CrossRef]
- Toussaint, E.F.; Breinholt, J.W.; Earl, C.; Warren, A.D.; Brower, A.V.; Yago, M.; Dexter, K.M.; Espeland, M.; Pierce, N.E.; Lohman, D.J.; et al. Anchored phylogenomics illuminates the skipper butterfly tree of life. BMC Evol. Biol. 2018, 18, 101. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Location | Size (bp) | IN | Anticodon | Start Codon | Stop Codon | |
|---|---|---|---|---|---|---|---|---|
| From | To | |||||||
| trnM | J | 1 | 69 | 69 | 0 | CAT | ||
| trnI | J | 71 | 135 | 65 | 1 | GAT | ||
| trnQ | N | 137 | 205 | 69 | 1 | TTG | ||
| ND2 | J | 291 | 1304 | 1014 | 85 | ATT | TAA | |
| trnW | J | 1303 | 1371 | 69 | −2 | TCA | ||
| trnC | N | 1364 | 1427 | 64 | −8 | GCA | ||
| trnY | N | 1431 | 1494 | 64 | 3 | GTA | ||
| COX1 | J | 1509 | 3039 | 1531 | 14 | CGA | T-- | |
| trnL2 | J | 3040 | 3106 | 67 | 0 | TAA | ||
| COX2 | J | 3107 | 3766 | 660 | 0 | ATG | TAA | |
| trnK | J | 3787 | 3857 | 71 | 20 | CTT | ||
| trnD | J | 3870 | 3935 | 66 | 12 | GTC | ||
| ATP8 | J | 3936 | 4103 | 168 | 0 | ATT | TAA | |
| ATP6 | J | 4097 | 4774 | 678 | −7 | ATG | TAA | |
| COX3 | J | 4774 | 5562 | 789 | −1 | ATA | TAA | |
| trnG | J | 5565 | 5631 | 67 | 2 | TCC | ||
| ND3 | J | 5632 | 5985 | 354 | 0 | ATT | TAA | |
| trnA | J | 5991 | 6057 | 67 | 5 | TGC | ||
| trnR | J | 6062 | 6124 | 63 | 4 | TCG | ||
| trnN | J | 6152 | 6217 | 66 | 27 | GTT | ||
| trnS1 | J | 6220 | 6278 | 59 | 2 | GCT | ||
| trnE | J | 6298 | 6365 | 68 | 19 | TTC | ||
| trnF | N | 6364 | 6429 | 66 | −2 | GAA | ||
| ND5 | N | 6430 | 8152 | 1723 | 0 | ATT | T-- | |
| trnH | N | 8171 | 8239 | 69 | 18 | GTG | ||
| ND4 | N | 8240 | 9578 | 1339 | 0 | ATG | T-- | |
| ND4L | N | 9581 | 9856 | 276 | 2 | ATG | TAA | |
| trnT | J | 9862 | 9927 | 66 | 5 | TGT | ||
| trnP | N | 9928 | 9993 | 66 | 0 | TGG | ||
| ND6 | J | 9996 | 10,520 | 525 | 2 | ATT | TAA | |
| CYTB | J | 10,520 | 11,671 | 1152 | −1 | ATG | TAA | |
| trnS2 | J | 11,670 | 11,736 | 67 | −2 | TGA | ||
| ND1 | N | 11,758 | 12,696 | 939 | 21 | ATG | TAA | |
| trnL1 | N | 12,697 | 12,764 | 68 | 0 | TAG | ||
| rrnL | N | 12,765 | 14,116 | 1352 | 0 | |||
| trnV | N | 14,117 | 14,183 | 67 | 0 | TAC | ||
| rrnS | N | 14,184 | 14,985 | 802 | 0 | |||
| A+T rich | J | 14,986 | 15,508 | 523 | 0 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, C.; Zhang, D.; Liu, D.; Jiao, L.; Li, R.; Yi, X. First Record of the Complete Mitochondrial Genome for the Genus Borbo (Lepidoptera, Hesperiidae): Characterization and Comparative Genomic Analysis. Diversity 2024, 16, 560. https://doi.org/10.3390/d16090560
Xue C, Zhang D, Liu D, Jiao L, Li R, Yi X. First Record of the Complete Mitochondrial Genome for the Genus Borbo (Lepidoptera, Hesperiidae): Characterization and Comparative Genomic Analysis. Diversity. 2024; 16(9):560. https://doi.org/10.3390/d16090560
Chicago/Turabian StyleXue, Chao, Dan Zhang, Dongkai Liu, Laizheng Jiao, Ran Li, and Xianfeng Yi. 2024. "First Record of the Complete Mitochondrial Genome for the Genus Borbo (Lepidoptera, Hesperiidae): Characterization and Comparative Genomic Analysis" Diversity 16, no. 9: 560. https://doi.org/10.3390/d16090560
APA StyleXue, C., Zhang, D., Liu, D., Jiao, L., Li, R., & Yi, X. (2024). First Record of the Complete Mitochondrial Genome for the Genus Borbo (Lepidoptera, Hesperiidae): Characterization and Comparative Genomic Analysis. Diversity, 16(9), 560. https://doi.org/10.3390/d16090560