Abstract
Endemic to the Indian subcontinent, the sloth bear (Melursus ursinus) is a threatened species, present in fragmented habitats across India. Field techniques such as direct observation and camera trapping alone are not sufficient and may not be explicit enough to understand a monomorphic species like the sloth bear at larger spatial scales. In this study, we looked into the genetic structure, variability and population demographics amongst the extant sloth bear populations in the highly fragmented Vidarbha landscape, using a panel of 13 microsatellite markers with a cumulative PID value of 1.48 × 10−5 PIDsibs. Our results revealed genetic clustering (K = 5) and moderate structuring amongst the study populations. Despite being geographically distant and placed in two different genetic clusters, sloth bears from the Melghat Tiger Reserve and Sahyadri Tiger Reserve shared genetic signatures, indicating connectivity, while migration was detected amongst other study areas as well. The findings from this study can serve as baseline assessment for future genetic monitoring of the species in the human-dominated landscape and assist in managerial decisions to step up protection of fragmented forest patches and reduce human–bear conflicts without compromising on the genetic connectivity.
Keywords:
Melursus ursinus; microsatellite; bear; primer; standardize; ursid; connectivity; structure 1. Introduction
Stochasticity in an ever-changing habitat and its demographics has pronounced effect on the population status of any species [1]. In a fast-paced human world, inclined towards its own development and connectivity, factors such habitat fragmentation, population decline and range contractions have become a threat to animal populations everywhere [2,3,4]. Among the animals that are most affected are large-bodied and wide-ranging species [5,6]. Large-bodied animals require larger home ranges that are contiguous. However, in the absence of such intact habitats, animals are forced to adapt to their changing surroundings in order to persist [7]. One such species is the sloth bear (Melursus ursinus), a member of the Ursidae family. Sloth bears are medium-sized omnivores with physiological adaptations to feed on insects such as ants and termites [8]. Historically, sloth bears were widely distributed throughout the Indian subcontinent, and it was only recently, in 2013, that this wide-ranging carnivore was declared extinct in Bangladesh [9]. Such an event outlined the need to pay attention to them and fill the information gaps to ensure long-term species persistence. At present, sloth bears have been given the “vulnerable” status by the IUCN, with India housing 90% of the extant population [10]. Sloth bears are found in a variety of habitats across India, with Central India and Western Ghats being strongholds of the species [11]. Protected areas, large patches of contiguous forests and dry deciduous forests support large abundances of sloth bears [11]. However, their population has declined significantly over the years due to habitat loss and fragmentation caused by human activities such as deforestation, agricultural expansion and urbanization [11,12]. These factors not only fragment landscapes but also act as impediments to genetic exchanges between animal populations. Studies have established that such isolated populations often undergo local extinction [13,14,15]. Such an exigency entails implementation of reliable molecular methods to understand and study populations of sloth bears in the wild.
Genetic monitoring methods are used extensively in other ursid species around the world [16,17] and serve as key tools for biodiversity monitoring approaches [18,19]; however, studying sloth bear populations using genetic methods is sporadic in India [20,21,22]. To date, there have been very few studies that standardized ursid microsatellite primers in sloth bears for individual identification [21,22], while no studies have used genetic tools to infer population structuring and variability in the Eastern Vidarbha landscape. Sharma et al. [21] used 7 microsatellite primers, yielding a probability of identity between siblings (PIDsibs) value of 2.15 × 10−3, but Wang et al. [23] showed that genotyping with fewer than 11 or 12 markers results in significantly changed estimates of population structure and genetic variation. Some studies have entrenched that the number of loci is more likely to enhance the power of population genetic inferences over the number of individuals being sampled [24,25]. In 2020, Thatte et al. [22] tested 13 microsatellite markers in non-invasive samples, with a PIDsibs value of 1.43 × 10−3. Given that the rough estimation of sloth bears in India is at least 20,000 individuals [26], the marker panel standardized in this study gives a better resolution and understanding of genetic structuring and variability in the focal landscape. In this paper, we aimed to (i) standardize and use microsatellite panels for genotyping sloth bears, (ii) create a baseline dataset of sloth bear genetic parameters for the eastern Vidarbha landscape and (iii) understand the pattern of genetic variability and structure in the studied populations of sloth bears.
2. Materials and Methods
2.1. Sampling Area
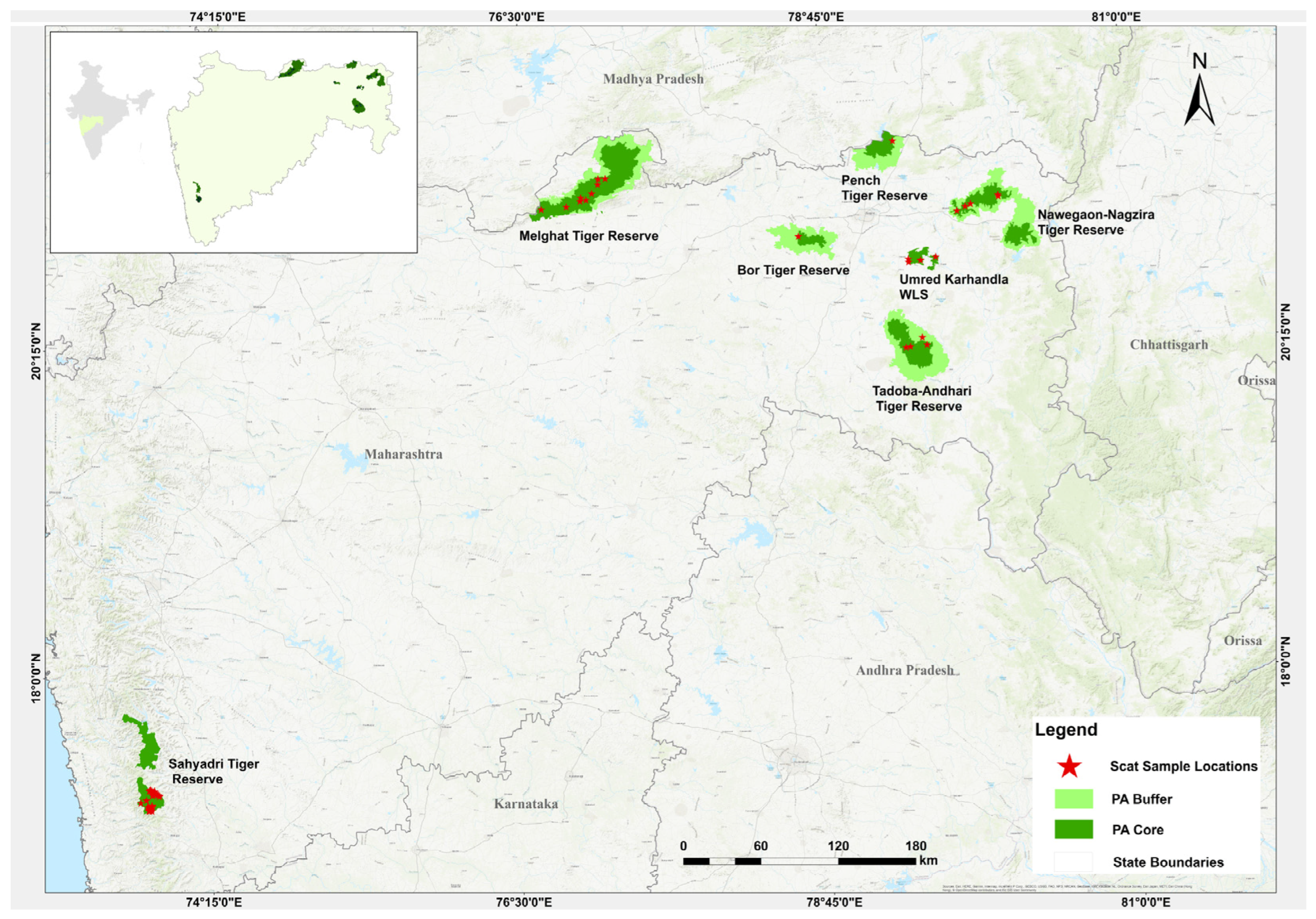
This study was conducted in the Vidarbha landscape (VL) of Maharashtra, India. The landscape has a forest cover of 22,508 sq km, accounting for areas both inside and outside of protected areas [27]. The major protected areas of the VL in which sampling was conducted include the Melghat Tiger Reserve (MTR, 2768.52 sq km), Sahyadri Tiger Reserve (STR, 1166 sq km), Tadoba Andhari Tiger Reserve (TATR, 1727.59 sq km), Bor Tiger Reserve (BTR, 816.27 sq km), Navegaon-Nagzira Tiger Reserve (NNTR, 1894.94 sq km), Pench Tiger Reserve (PTR, 741.22 sq km) and Umred-Karhandla Wildlife Sanctuary (UKWLS, 189 sq km) (Figure 1).
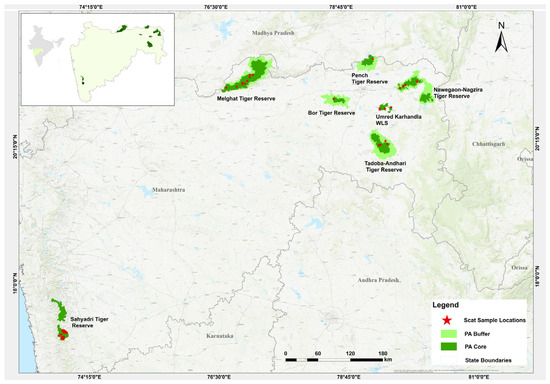
Figure 1.
Study area map indicating sampling location used (NNTR n = 82, STR n = 268, PTR n = 35, MTR n = 83, TATR n = 69, UKWLS n = 28, BTR n = 1).
2.2. Field Sampling
A total of 565 fecal samples of sloth bears were collected (NNTR n = 82, STR n = 268, PTR n = 35, MTR n = 83, TATR n = 69, UKWLS n = 28, BTR n = 1). Since only 1 sample was collected from the BTR, we did not use it for further analysis. All concerned permits for the collection and removal of fecal samples from protected areas were provided by the Maharashtra Forest Department (Permit No. 09/2016). The tissue reference samples were taken from the forensic department of the Wildlife Institute of India, Dehradun. Intensive sampling in the study sites was conducted from 2016 to 2019. Little is known about preferred defecating sites in the case of sloth bears, which made sampling both laborious and time-intensive. We focused our search mainly on animal trails, dry riverbeds, rocky plateaus, near fruiting trees or trees with beehives, trees with scratch markings and areas near bear-dug outs or termite mounds, each of which indicated the presence of bears. Sloth bear scats are remarkably different to scats of other large carnivores, lesser cats or langurs based on their size, shape, appearance and the presence of seeds, ants and termite remnants [28]. Once a scat was located, a bolus of the scat sample was collected and kept directly on a piece of butter paper and stored in an individual zip lock bag. Samples collected during fruiting season were sprayed with 99% ethanol to delay fungal growth on the undigested fruit matter. Details such as location, date, state of scat and locality-associated details such as substrata or terrain type were recorded. Upon reaching the field station, all zip lock bags were stored in a box containing silica beads to minimize chances of fungal growth until further processing.
2.3. Primer Selection
No species-specific microsatellite markers exist for sloth bears; therefore, we screened a panel of 25 different cross-species markers developed on different species of bears and canids [29,30,31,32,33,34]. Following an initial screening of the marker panels, a final marker panel of 13 tagged microsatellites was selected for the population-level study. These markers were selected based on their polymorphic information content, heterozygosity and the number of alleles per locus and rates of allelic dropouts.
2.4. DNA Extraction
All laboratory procedures and sample storage were carried out in the genetic laboratory facility of the Wildlife Institute of India, Dehradun. The tissue and the scat samples were extracted following standardized protocols. Each scat sample brought to the laboratory was swabbed twice using sterile swabs (HiMedia, India) dipped in PBS buffer. The swabs were then placed in 2 mL Eppendorf tubes until extraction. Extraction of the swabbed samples was carried out using a DNeasy kit (QIAGEN, Germany) following the instructions stated in the kit’s instruction manual with a few modifications. An overnight lysis was performed with 330 μL of InhibitEx buffer (QIAGEN) and 20 μL of Proteinase K, and DNA was extracted the following day according to the manufacturers’ instructions. In the final step, the DNA was eluted twice with 100 μL of 1×TE buffer and stored at −30 °C for long-term storage. Care was taken to include negative controls during each set of 12 extractions to monitor the chances of possible contamination during handling and extraction steps. Genomic DNA was extracted from tissue samples using the DNaesy Kit (QIAGEN, Germany) and from bone samples using the GeneiPureID DNA Isolation Kit-Bone (Merck, Germany) following the manufacturers’ protocols.
2.5. PCR Standardization and Data Validation
The selected primers were first individually standardized using the confirmed tissue samples. The individual primers were then put into multiplex panels of 2–3 primers each based on their annealing temperature, dye and PCR amplicon size (refer to Table 1). A total of 6 panels were standardized to make data generation both cost-effective and time-efficient. The multiplex panels were checked for data accuracy by replicating the PCR conditions for different cycles—40, 50, 60 cycles to ensure that the multiplex panels were consistent. The panels were then used for PCR amplifications of all study samples. PCR for each eluted sample was performed using 3.5 μL of hotstart taq mix, 3 μL of BSA, 1 μM of primer mix (fluorescent-tagged forward primer and a reverse primer) and 2 μL of DNA extract under conditions that included initial denaturation (95 °C for 15 min); 55 cycles of denaturation (94 °C for 30 s), annealing (temperature as calculated for each primer for 30 s) and extension (72 °C for 30 s) and final extension (72 °C for 10 min). Negative controls were also included during the PCR process to ensure the quality of the process and monitor the chances of any cross-species amplification. The PCR products post amplification were then prepared using GeneScan 500 LIZ (Thermo Fischer, MA, USA), for genotyping, and the plates were run on an ABI 3500xl Genetic Analyzer (ABI, MA, USA). The results of the alleles were scored using GeneMarker v3.0.1 (Softgenetics Inc., State College, PA, USA). Allelic bins for each locus were created using the data generated from the tissues and scat samples. Each sample was amplified and genotyped three times for each locus, and a consensus final dataset was used for further analysis.

Table 1.
Sequences and panels of selected ursid microsatellites.
For data validation of the scat samples, a multi-tube approach was used [35]. Two additional rounds of PCR were performed on all samples that were amplified in 50% of the loci in the panel during the initial round. Each sample was amplified and genotyped three times for each locus, and a consensus final dataset was used for quality index analysis as described in Miquel et al. [36]. In this protocol, the alleles are scored. If the repeats are identical to the first allele call, then they are assigned ‘1′, and if they are different due to non-amplification, allelic dropout or allelic slippage, then ‘0′ is given. The quality index for each locus is calculated by adding the scores of each locus and dividing each by the number of repeats for that locus. If this value was equal to or above 0.75, the sample was then used for further downstream analyses [36]. Monomorphic markers and those not conforming to the index were excluded. The quality index was calculated for each sample using the scores assigned to each repeat and dividing by the total number of repeats [37]. Allelic dropout and false alleles were calculated using MICROCHECKER V2.2.3 [38]. False allele rates were calculated manually in both homozygous and heterozygous samples as the ratio between the number of false alleles detected at a locus and the total number of amplifications, following Broquet and Petit [37].
2.6. Data Analyses
The CERVUS 3.0.7 [39] module of identity analysis was used to identify genetic recaptures (identical genotypes) by comparing the consensus data generated from all loci for each sample. The recaptures were removed from the analysis. GIMLET was used to calculate PID values and genetic variations in the loci such as expected heterozygosity, observed heterozygosity, number of alleles, false alleles and allelic dropout [40]. Genepop was used to calculate F statistics and effective population (Nm) for all the samples [41,42,43,44]. TESS was used to infer population structuring using the Bayesian approach relying on the Markov Chain Monte Carlo algorithm under the no admixture model for K = 2 to 10 with 100,000 iterations [43,45]. TESS also accounts for spatial connectivity while assigning samples to clusters. The best value of K was selected based on the Deviance Information Criterion (DIC) value. BayesAss software version 3.0.3, which utilizes genotype information from multi-locus data, was implemented to estimate the recent migration rates between the protected areas in the study landscape [46]. The run parameters for the software included 5 × 106 iterations and 105 burn-in with sampling every 2000 iterations. The results from BayesAss were visualized using the “circlize” package version 0.34 in R ver 1.2.5 [47].
3. Results
3.1. Microsatellite Genotyping
A total of 565 scat samples of wild sloth bears were collected from the study area in Maharashtra for standardization purposes. In total, 14 confirmed tissue samples were provided by Nagpur Zoo and the Forensic laboratory facility of the Wildlife Institute of India, which were used as reference samples. During standardization and trial experiments conducted to group the primers into panels, a total of 25 primers were used, out of which 13 loci generated consistent data. No large allelic dropouts were seen. Genotyping results of the tagged primers showed clean peaks and fewer stutters. These 13 loci were then used to amplify and genotype the scat samples. Only those samples that generated data for at least seven loci were taken into consideration. In total, 256 of the 565 scat samples collected from the field were amplified and yielded adequate data for analysis and individual identification. None of the loci used in the study deviated from the Hardy–Weinberg equilibrium, and no strong linkage disequilibrium was detected. The details of each locus and their measures of polymorphism are presented in Table 2. The amplification success rate of the scat samples can be attributed to their quality and the usage of InhibitEx buffer, which successfully removed PCR inhibitors in the scat samples that could be present owing to sloth bear’s diet. Following our analysis in CERVUS, one genetic recapture individual was found. The cumulative values for the 13 panel primers were found to be 9.43 × 10−14 PIDunbiased and 1.48 × 10−5 PIDsibs [48]. The number of alleles ranged from 4 to 14 alleles (Table 2).
Table 2.
Details of microsatellite markers used for sloth bears in this study.
3.2. Population Structuring and Variability
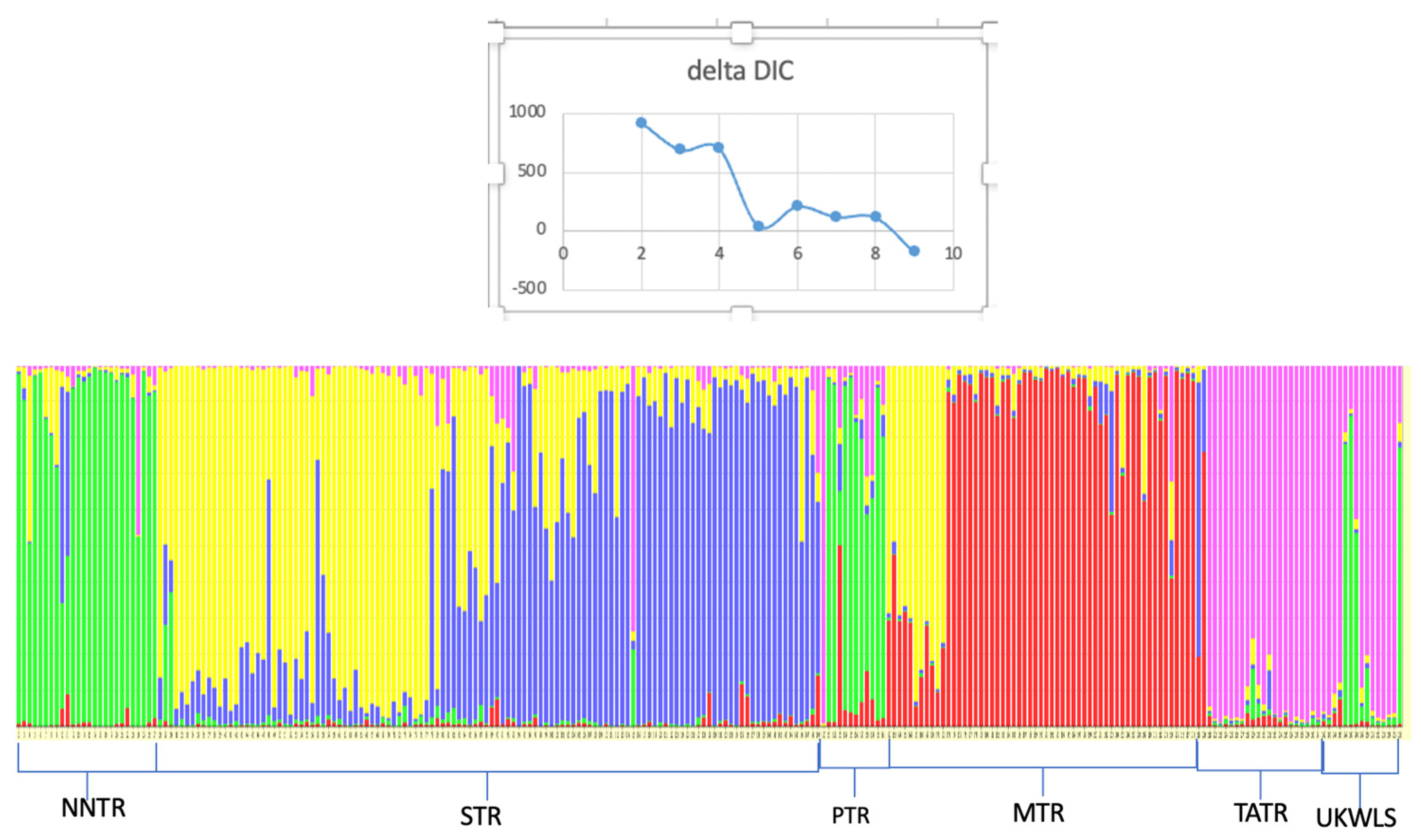
The average value for Fis was 0.095 and that for Fst was 0.09 based on 13 loci [28]. The Nm after correcting for size was 2.69. TESS assigned a cluster value of 5 for the populations of sloth bears based on the lowest value of DIC before reaching a plateau (Figure 2). The TESS results showed that neighboring populations of the TATR and UKWLS were assigned to one genetic cluster, with UKWLS samples sharing some genetic signature with the adjacent NNTR population (Figure 2). The populations of the NNTR and PTR formed the next cluster, with PTR samples showing some links to the MTR population. The majority of samples from the MTR were assigned to a third genetic cluster, while a small part of the MTR population grouped with samples from the STR into a common cluster, and the remaining part of the STR population was assigned to a separate group.
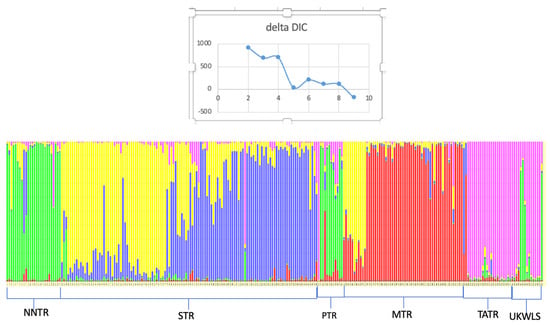
Figure 2.
TESS assignment plot of sloth bears across the Vidarbha landscape, India, assigning the samples to 5 distinct populations.
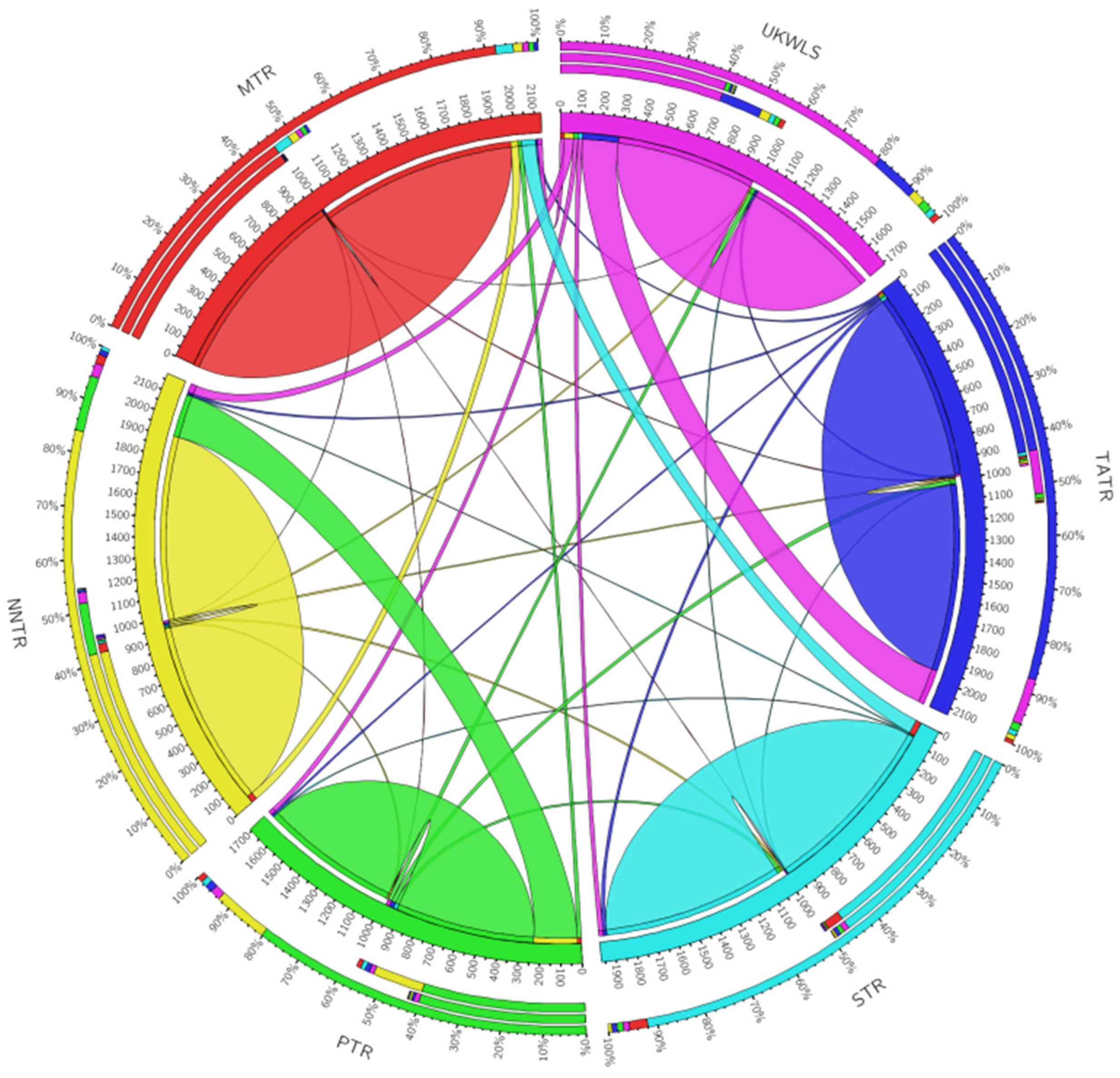
The circus plot based on BayesAss analysis shows the pairwise migration rates obtained between each of the seven protected areas of the landscape. The plot depicts the source of migration for each migration movement (Figure 3). The thickness of the ribbons is proportional to the rate of flow of genes from the source population. The contemporary rate of migration was estimated to be 0.098 (range 0.009–0.21). The rate of migration represented by the proportion of individuals migrating was found to be the highest from the NNTR to PTR (0.214) and from TATR to UKWLS (0.177).
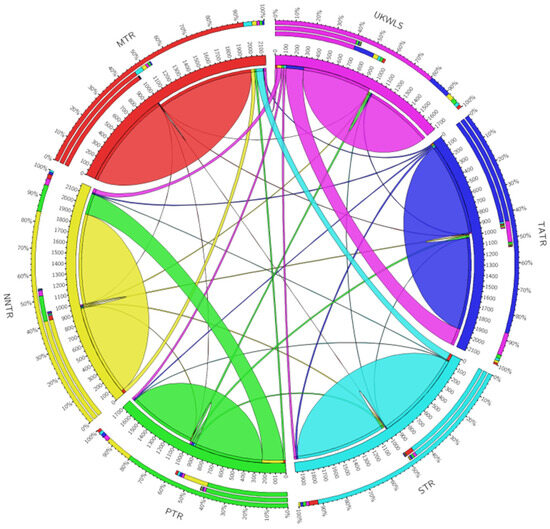
Figure 3.
Circus plot visualization of migration rates between different protected areas of the Vidarbha landscape, India.
4. Discussion
4.1. Primer Standardization and Data Analysis
In this paper, we have tested and standardized a panel of 13 microsatellite tagged primers to infer population structuring and genetic variability. Using a larger number of primers in the initial stage allowed us to test for appropriate panels to identify individuals with robust statistical bolstering. We have taken care to include both dinucleotide and tetranucleotide markers to balance stuttering from peaks and higher amplification success from degraded samples [29,33]. Based on reproducibility and consistency, 13 markers were selected for this study. The final panel was divided into six multiplex reactions to make the entire process cost-effective and efficient. For genetic estimation of the population size in each area, it is recommended that the PIDsibs value be double the expected population size [42]. Using 13 microsatellites, the PIDsibs value obtained in this study (1.48 × 10−5) should be sufficient to conduct a large-spatial-scale study and is better compared to the values published in studies prior to this [20,21,22]. The amplification success of freshly collected samples has been well established [49]. For this study, however, we collected both fresh and old fecal samples to ensure extensive sampling. A few samples that were either very old or very dry did not yield any data. It was also noticed that samples that might not have been adequately sprayed with alcohol had fungal growth, which may have also degraded sample quality. Therefore, it needs to be highlighted that the data generated in this study were from the 45.3% of the total scat samples collected that were reliable. Only one genetic recapture was found in the samples. This low recapture rate could potentially be an offshoot of our sampling strategy wherein we focused on collecting samples from as many individuals as possible to capture the genetic variability in the landscape. The difference in study design and number of markers used for studying sloth bear populations is an obstacle to comparing our study with other sloth bear studies conducted by Dutta et al. [20] and Thatte et al. [22]. This study used 13 microsatellite primers in six panel combinations to obtain a PIDsibs value of 1.48 × 10−5, which is better than the studies previously conducted and has the scope to be used for future population studies and monitoring.
4.2. Population Structuring and Variability
The result from this study showed that the observed heterozygosity for most loci was greater than the expected heterozygosity (see Table 2) [41]. The population structure visualized in TESS divided the sloth bear populations into five distinct genetic clusters. The value of K was selected as the lowest value in DIC before reaching consistency. The plot shows moderate structuring in the six populations of sloth bears that were sampled (Figure 2). The UKWLS, which is a known connecting link between [50] the PTR and TATR, was not assigned any distinct genetic cluster and showed mixed genetics signals from two major populations (NNTR and TATR). It was interesting to see that the MTR and STR, although grouped in different genetic clusters, shared some genetic signatures as well. This could be attributed to two plausible explanations. Firstly, owing to the higher dispersal capability of sloth bears, the genetic differentiation is not very high since the exchange of genes is facilitated [22]. Similar trends have also been seen in other long-ranging carnivore species such as tigers [50,51,52] and leopards [53] of the same landscape. Secondly, it is also possible that the STR and MTR had migrating populations through other forest patches such as the present day Yawal Wildlife Sanctuary and Gautala Autramghat Sanctuary before the landscape was fragmented. Studies using mtDNA may give insight into this. The BayesAss plot shows migration from one population to another [46]. The most prominent migration occurs from the PTR to NNTR and from the UKWLS to TATR. The low Fis value of 0.095 and Fst value of 0.09 [40] show that the sloth bear populations are not inbred and they exhibit moderate genetic differentiation. In their paper, Thatte et al. (2020) pointed out that since sloth bears were the most widely distributed species, their genetic distribution would be low based on the fact that long-ranging species, like the sloth bear, would have higher gene flow when compared to short-dispersal species [22]. Although our study does show moderate levels of genetic differentiation, there are no baseline data to compare our results to. The findings from this study may be used as the baseline data to understand and compare demographic trends for future studies.
The most fascinating, and to some extent, daunting explanation for the genetic clustering and the moderate genetic differentiation that has been obtained from this study is the absence of contiguous forests. Such that, as a result of adaptive behavior and in order to persist, smaller patches of forests are used as stepping stones to facilitate movement. The moderate genetic variation observed also supports the fact that corridors used to be functional, but with ongoing developments and alterations in the landscape, the persistence of such functionality comes under question [52]. In the study landscape of eastern Vidarbha, patches of forests are often intercepted by developmental and anthropogenic factors such as linear infrastructure and upcoming townships or developmental projects, which curb movement and dispersal of species. About 567 barriers with 30 linkages were enumerated in a study focused on a larger landscape that also included the EVL [54], and such barriers are potential impediments to movement and connectivity. In such a scenario, one of the plausible modes of movement would be that of using stepping-stone patches of forests and other similar habitats. Compared to other long-ranging ursid species such as polar bears or brown bears, whose movements have been widely studied, very few studies have investigated the movement and range of sloth bears. On average, the home range of sloth bears ranges between 2 sq kms and more than 100 sq kms [11,55,56] based on VHF collaring data, whereas dispersal studies have not been conducted. Studies have shown that stepping stones are capable of the dispersal of long-ranging species and can aid in connectivity [57], and the role of such fragments in the Vidarbha landscape cannot be overlooked. The best possible way to establish the use of corridors or stepping-stone patches of forests is to combine molecular studies with telemetry. However, this is beyond the scope of this paper. This study, however, shows genetic exchange between sloth bear populations, which is enough to establish the need to identify and protect corridors and intact forest patches. Other large-bodied bear species like the polar bear, the brown bear and the Asiatic black bear have suffered the brunt of shrinking habitats, causing severe inbreeding and population decline [19,58,59,60]. But the sloth bear is much less studied, and therefore, more studies focused on the species, investigating the changing demographics across landscapes and the effect of fragmentation, are warranted.
5. Conclusions
This study is the first in the Vidarbha landscape to use genetic tools to understand population structuring and genetic variability. Landscapes that are the stronghold of some large-bodied species, especially in the tropics, have been undergoing rapid alterations and degradation, making it imperative to understand the ill-effects of these changes on the species inhabiting these landscapes [61]. At a larger spatial scale, especially for unmarked species, the utilization of non-invasive genetic methods to understand genetic variation and population dynamics is one of the most robust and reliable methods. The results from this study can be used as a baseline for subsequent studies to understand the population dynamics over time and adopt appropriate conservation measures for the protection or restoration of fragmented forest patches.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/d16020074/s1. Table S1, Sample Details of Sloth Bear Scats of Vidarbha Landscape.
Author Contributions
L.G.: sampling, experiments, formal analysis, investigation, data curation, writing—original draft, visualization. S.M.: sampling, guidance. P.N.: editing, supervision. B.H.: project conceptualization, editing, supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Maharashtra State Forest Department (09/2016). The funders had no role in the conceptualization of study design, data collection and analysis or preparation of the manuscript.
Data Availability Statement
Data is contained within the Supplementary Material.
Acknowledgments
We acknowledge the forest officials and staff of Maharashtra for their assistance in fieldwork. We thank the Faculty of Wildlife Sciences, and WII, for their continuous support in this research. We also thank Madhanraj and Ankit for their guidance in data generation and analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ouborg, N. Isolation, Population Size and Extinction: The Classical and Metapopulation Approaches Applied to Vascular Plants along the Dutch Rhine-System. Oikos 1993, 66, 298–308. [Google Scholar] [CrossRef]
- Pacifici, M.; Rondinini, C.; Rhodes, J.; Vurbidge, A.; Cristiano, A.; Watson, J.; Woinarski, J.C.Z.; Di Marco, M. Global correlates of range contractions and expansions in terestrial mammals. Nat. Commun. 2020, 11, 2840. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Ripple, W. Range Contractions of the world’s large carnivores. R. Soc. Open Sci. 2017, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ripple, W.; Newsome, T.; Wolf, C.; Dirzo, R.; Everatt, K.; Galetti, M.; Hayward, M.W.; Kerley, G.I.H.; Levi, T.; Lindsey, P.A.; et al. Collapse of World’s largest herbivores. Sci. Adv. 2015, 1, e1400103. [Google Scholar] [CrossRef]
- Fisher, D.; Owens, I. The comparative methods in conservation biology. Trends Ecol. Evol. 2004, 6, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Crooks, K.; Burdett, C.; Theobald, D.; Rondinini, C.; Boitani, L. Global patterns of fragmentation and connectivity of mammalian carnivore habitat. Philos. Trans. R. Soc. B: Biol. Sci. 2011, 366, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.; Durant, S.; Woodroffe, R. What wild dogs want: Habitat selection differs across life stages and orders of selection in a wide-ranging carnivore. BMC Zool. 2020, 5, 2–11. [Google Scholar] [CrossRef]
- Sacco, T.; Valkenburgh, B. V Ecomorphological indicators of feeding behaviour in the bears (Carnivora: Ursidae). J. Zool. 2006, 263, 41–54. [Google Scholar] [CrossRef]
- Islam, M.; Uddin, M.; Aziz, M.; Muzaffar, S.; Chakma, S.; Chowdhury, U.; Chowdhury, G.W.; Rashid, M.A.; Samiul, M.; Jahan, I.; et al. Status of bears in Bangladesh: Going, going, gone? Ursus 2013, 24, 83–90. [Google Scholar] [CrossRef]
- Dhraiya, N.; Baragli, H.; Sharp, T. Melursus ursinus (Amended Version of 2016 Assessment). 2020. Available online: https://www.iucnredlist.org/species/13143/166519315#geographic-range (accessed on 25 November 2023).
- Yoganand, K.; Rice, C.; Seidensticker, J.; Johnsingh, A. Is the sloth bear in India secure? A preliminary report on distribution, threats and conservation requirements. J. Bombay Nat. Hist. Soc. 2006, 103, 172–181. [Google Scholar]
- Shankar, K.; Murthy, R.S. Assessment of Bear-Man Conflict in North Bilaspur Forest Division, Bilaspur, Madhya Pradesh; Wildlife Institute of India: Dehradun, India, 1995.
- Frankham, R. Genetics and Extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Frankham, R Genetic rescue of small inbred populations: Meta-analysis reveals large and consistent benefits of gene flow. Mol. Ecol. 2015, 24, 2610–2618. [CrossRef] [PubMed]
- Ralls, K.; Ballou, J.; Dudash, M.; Elridge, M.; Fenster, C.; Lacy, R.; Sunucks, R.; Frankham, R. Call for a paradigm shift in the genetic management of fragmented populations: Genetic management. Conserv. Lett. 2018, 11, 1–6. [Google Scholar] [CrossRef]
- Tallmon, D.A.; Bellemain, E.; Swenson, J.E.; Taberlet, P. Genetic monitoring of Scandinavian brown bear effective population size and immigration. J. Wildl. Manag. 2009, 68, 960–965. [Google Scholar] [CrossRef]
- Kruckenhauser, L.; Rauer, G.; Daubl, B.; Haring, E. Genetic monitoring of a founder population of brown bears (Ursus arctos) in central Austria. Conserv. Genet. 2008, 10, 1223–1233. [Google Scholar] [CrossRef]
- Carroll, E.; Bruford, M.; DeWoody, J.; Leroy, G.; Strand, A.; Waits, L.; Wang, J. Genetic and genomic monitoring with minimally invasive sampling methods. Evol. Appl. 2018, 11, 1094–1119. [Google Scholar] [CrossRef] [PubMed]
- Procter, M.; Kasworm, W.; Teisberg, J.; Servheen, C.; Radandt, T.; Lamb, C.T.; Kendall, K.C.; Mace, R.D.; Paetkau, D.; Boyce, M. American black bear population fragmentation detected with pedigrees in the transborder Canada–United States region. Ursus 2020, 31, 1–15. [Google Scholar] [CrossRef]
- Dutta, T.; Sharma, S.; Maldonado, J.; Seidensticker, J. Genetic Variation, Structure, and Gene Flow in a Sloth Bear (Melursus ursinus) Meta-Population in the Satpura-Maikal Landscape of Central India. PLoS ONE 2020, 10, e0123384. [Google Scholar] [CrossRef]
- Sharma, S.; Dutta, T.; Maldonando, J.E.; Wood, T.C. Selection of microsatellite loci for genetic monitoring of sloth bears. Ursus 2013, 24, 164–169. [Google Scholar] [CrossRef]
- Thatte, P.; Chandramouli, A.; Tyagi, A.; Patel, K.; Baro, P.; Chhattani, H.; Ramakrishnan, U. Human footprint differentially impacts genetic connectivity of four wide-ranging mammals in a fragmented landscape. Divers. Distrib. 2020, 26, 299–314. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Wang, H.; Xiao, H. Impact of different numbers of microsatellite markers on population genetic results using SLAF-seq data for Rhododendron species. Sci. Rep. 2021, 11, 8597. [Google Scholar] [CrossRef] [PubMed]
- Landguth, E.; Fedy, B.; Oyler-McCance, S.J.; Garey, A.L.; Emel, S.L.; Mumma, M.; Wagner, H.H.; Fortin, M.J. Effects of sample size, number of markers, and allelic richness on the detection of spatial genetic pattern. Mol. Ecol. Resour. 2011, 12, 276–284. [Google Scholar] [CrossRef]
- Hale, M.; Burg, T.; Steeves, T. Sampling for Microsatellite-Based Population Genetic Studies: 25 to 30 Individuals per Population Is Enough to Accurately Estimate Allele Frequencies. PLoS ONE 2012, 7, e45170. [Google Scholar] [CrossRef] [PubMed]
- Sathyakumar, S.; Kaul, R.; Ashraf, N.; Mookherjee, A.; Menon, V. National Bear Conservation and Welfare Action Plan; Wildlife Institute of India and Wildlife Trust of India: Dehradun, India, 2012.
- Forest Survey of India. Indian State Forest Report. 2021. Available online: https://fsi.nic.in/forest-report-2021-details (accessed on 3 January 2023).
- Laurie, A.; Seidensticker, J. Behavioural ecology of the Sloth Bear (Melursus ursinus). J. Zool. 1977, 182, 187–204. [Google Scholar] [CrossRef]
- Paetkau, D.; Strobeck, C.S.; Calvert, W. Microsatellite analysis of population-structure in Canadian Polar Bears. Mol. Ecol. 1995, 4, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Camarra, J.; Grifin, S.; Uhres, E.; Hanotte, O.; Waits, L.; Dubois-Paganon, D.; Burke, T.; Bouvet, J. Noninvasive genetic tracking of the endangered Pyrenean brown bear population. Mol. Ecol. 1997, 6, 869–876. [Google Scholar] [CrossRef]
- Kitahara, E.; Isagi, Y.I.; Saitoh, T. Polymorphic microsatellite DNA markers in the asiatic black bear Ursus thibetanus. Mol. Ecol. 2000, 9, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Huang, C.; Li, S.; Hwang, M. Ten novel tetranucleotide microsatellite DNA markers from Asiatic black bear, Ursus thibetanus. Conserv. Genet. 2009, 10, 1845–1847. [Google Scholar] [CrossRef]
- Kleven, O.; Hallstrom, B.; Hailer, F.; Janke, A.; Hagen, S.; Kopatz, A.; Eiken, H. Identification and evaluation of novel di- and tetranucleotide microsatellite markers from the brown bear (Ursus arctos). Conserv. Genet. Resour. 2012, 4, 737–741. [Google Scholar] [CrossRef]
- Meredith, E.; Rodzen, J.; Banks, J.D.; Jones, K. Characterization of 29 tetranucleotide microsatellite loci in black bear (Ursus americanus) for use in forensic and population applications. Conserv. Genet. 2008, 10, 693–696. [Google Scholar] [CrossRef]
- Mondol, S.; Karanth, K.; Samba Kumar, N.; Gopalswamy, A.; Andheria, A.; Ramakrishnan, U. Evaluation of non-invasive genetic sampling methods for estimating tiger population size. Biol. Conserv. 2009, 142, 2350–2360. [Google Scholar] [CrossRef]
- Miquel, C.; Bellemain, E.; Poillot, C.; Bessière, J.; Durand, A.; Taberlet, P. Quality indexes to assess the reliability of genotypes in studies using noninvasive sampling and multiple-tube approach. Mol. Ecol. Notes 2006, 6, 985–988. [Google Scholar] [CrossRef]
- Broquet, T. Petit, Quantifying genotyping errors in noninvasive population genetics. Mol. Ecol. 2004, 13, 3601–3608. [Google Scholar] [CrossRef]
- Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Kalinowski, S.; Taper, M.; Marshall, T. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Valiere, N. Gimlet: A computer program for analysing genetic individual identification data. Mol. Ecol. Resour. 2002, 2, 377–379. [Google Scholar] [CrossRef]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef]
- Nei, M. F-statistics and analysis of gene diversity in subdivided populations. Ann. Hum. Genet. 1977, 41, 225–233. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Caye, K.; Deist, T.M.; Martins, H.; Michel, O.; François, O. TESS3: Fast inference of spatial population structure and genome scans for selection. Mol. Ecol. Resour. 2016, 16, 540–548. [Google Scholar] [CrossRef]
- Walsh, P.S.; Fildes, N.J.; Reynolds, R. Sequence analysis and characterization of stutter products at the tetranucleotide repeat locus. Nucleic Acids Res. 1996, 24, 2807–2812. [Google Scholar] [CrossRef]
- Rannala. BayesAss Edition 3.0 User’s Manual; University of California: Davis, CA, USA, 2007. [Google Scholar]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize: Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Waits, L.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: Cautions and guidelines. Mol. Ecol. 2001, 10, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, L.; Holloway, J.; Patterson, B.; White, B. An improved field method to obtain DNA for individual identification from wold scat. J. Wildl. Manag. 2009, 73, 1430–1435. [Google Scholar] [CrossRef]
- Mondal, I.; Habib, B.; Talukdar, G.; Nigam, P. Triage of means: Options for conserving tiger corridors beyond designated protected lands in India. Front. Ecol. Evol. 2016, 4, 2–7. [Google Scholar] [CrossRef]
- Reddy, P.A.; Gour, D.S.; Bhavanishankar, M.; Jaggi, K.; Hussain, S.M.; Harika, K.; Shivaju, S. Spatial genetic analysis reveals high connectivity of tiger (Panthera tigris) populations in the Satpura–Maikal landscape of Central India. PLoS ONE 2012, 3, 48–60. [Google Scholar]
- Sharma, S.; Dutta, T.; Maldonado, J.E.; Wood, T.C.; Panwar, H.S.; Seidenstick, J. Genetic Evidence of Tiger Population Structure and Migration within an Isolated and Fragmented Landscape in Northwest India. PLoS ONE 2013, 3, 48–60. [Google Scholar]
- Yumnam, B.; Jhala, Y.; Qureshi, Q.; Maldonado, J.; Gopal, R.; Saini, S.; Srinivas, Y.; Fleischer, R. Prioritizing tiger conservation through landscape genetics and habitat linkages. PLoS ONE 2014, 9, e111207. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Sharma, S.; DeFries, R. Targeting restoration sites to improve connectivity in a tiger conservation landscape in India. PeerJ 2018, 6, 587. [Google Scholar] [CrossRef]
- Dutta, T.; Sharma, S.; Maldonado, J.; Wood, T.; Panwar, H.; Seidensticker, J. Fine-scale population geneic structure in a wide-ranging carnivore, the leopard (Panthera pardus fusca) in central India. Divers. Distrib. 2012, 19, 760–771. [Google Scholar] [CrossRef]
- Ratnayeke, S.; Van Manen, F.T.; Padmalal, U.K.G.K. Home ranges and habitat use of sloth bears Melursus ursinus inornatus in Wasgomuwa National Park. Sri Lanka. Wildl. Biol. 2007, 13, 272–284. [Google Scholar] [CrossRef]
- Joshi, A.R.; Garshelis, D.L.; Smith, J.L.D. Home Ranges of Sloth bears in Nepal: Implications for Conservation. J. Wildl. Manag. 1995, 59, 204–214. [Google Scholar] [CrossRef]
- Diniz, M.F.; Coehlo, M.T.P.; Sousa, F.G.; Hasui, E.; Loyola, R. The underestimated role of small fragments for carnivore dispersal in the Atlantic Forest. Perspect. Ecol. Conserv. 2021, 19, 81–89. [Google Scholar] [CrossRef]
- Murphy, S.; Augustine, B.; Ulrey, W.; Guthrie, J.; McCown, J.; Cox, J. Consequences of severe habitat fragmentation on density, genetics, and spatial capture-recapture analysis of a small bear population. PLoS ONE 2017, 12, e0181849. [Google Scholar] [CrossRef]
- Maduna, S.; Aars, J.; Fløystad, I.; Klütsch, C.; Zeyl Fiskebeck, E.; Wiig, O.; Ehrich, D.; Anderson, M.; Bachmann, L.; Derocher, A.E.; et al. Sea ice reduction drives genetic differentiation among Barents Sea polar bears. Proc. R. Soc. B Biol. Sci. 2021, 288, 1–9. [Google Scholar] [CrossRef]
- Crooks, K.R.; Burdett, C.L.; Theobald, D.M.; King, S.R.; Di Marco, M.; Rondinini, C.; Boitani, L. Quantification of habitat fragmentation reveals extinction risk in terrestrial mammals. Proc. Natl. Acad. Sci. USA 2017, 114, 7635–7640. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).