

Genetic Diversity and Structure of Geodorum eulophioides, a Plant Species with Extremely Small Populations in China

Abstract
:1. Introduction
2. Materials and Methods
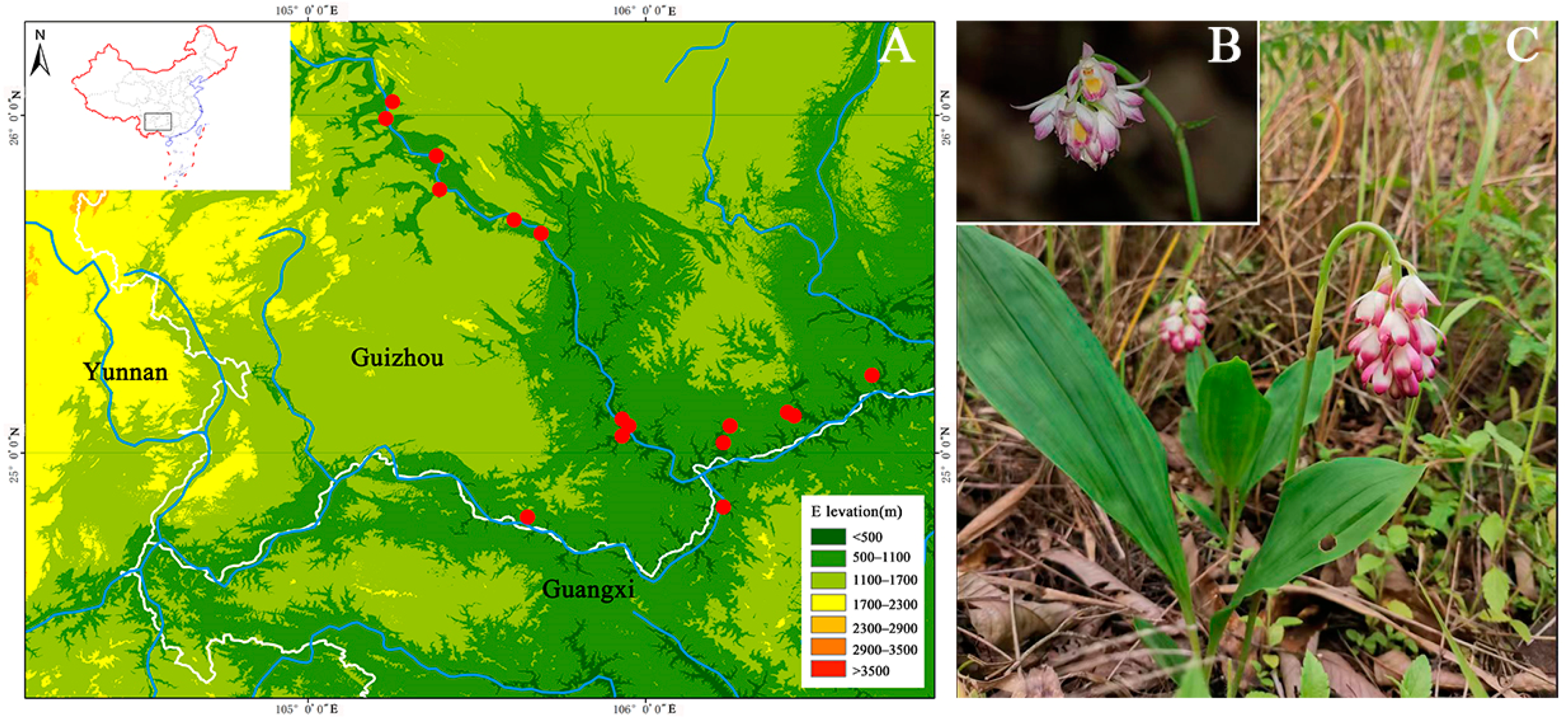
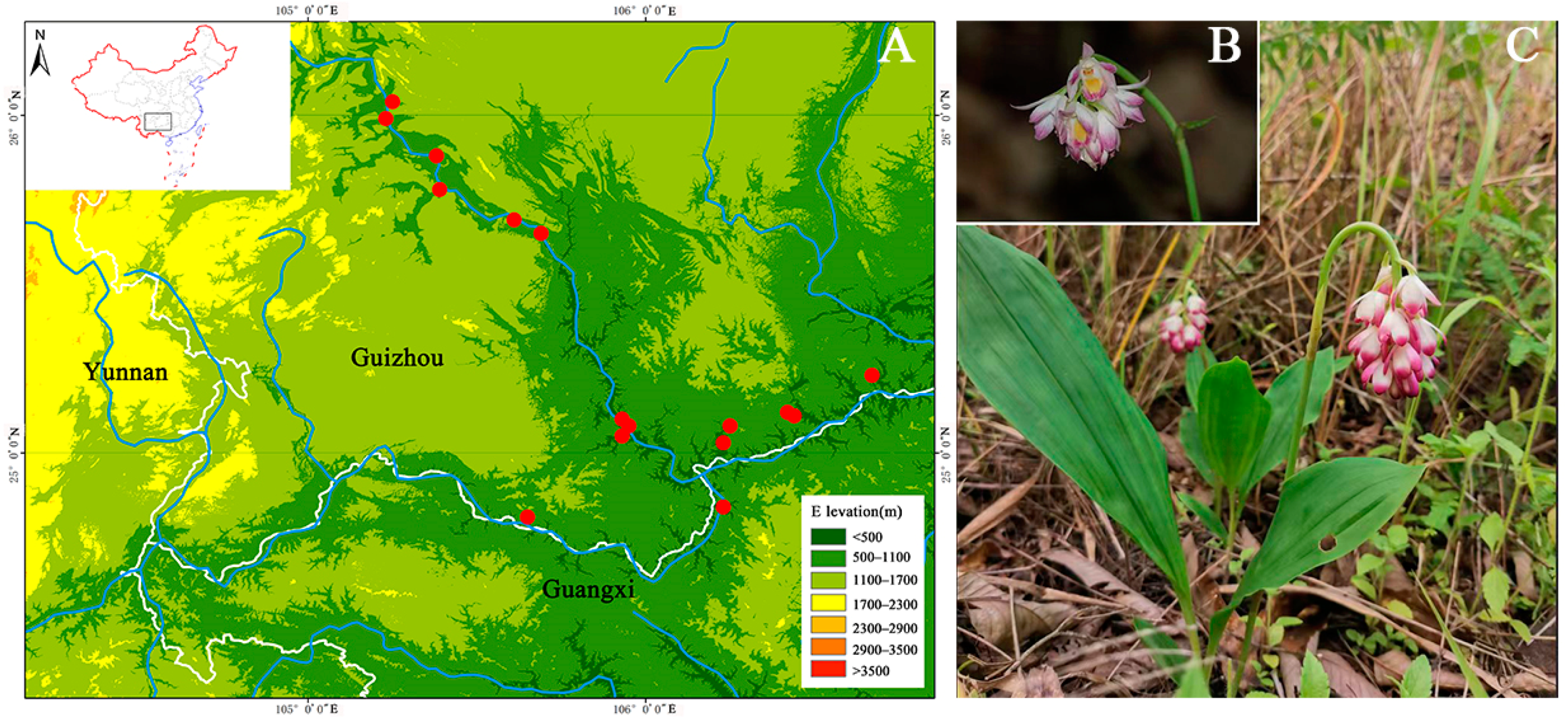
2.1. Plant Materials
2.2. Librarary Construction and SLAF Sequencing
2.3. SLAF Tag Development and SNP Calling
2.4. Diversity Analysis
2.5. Analysis of Environmental Adaptive Loci
3. Results
3.1. SNP Detection
3.2. SLAF Tags and SNP Calling
3.3. Genetic Diversity and Genetic Differentiation
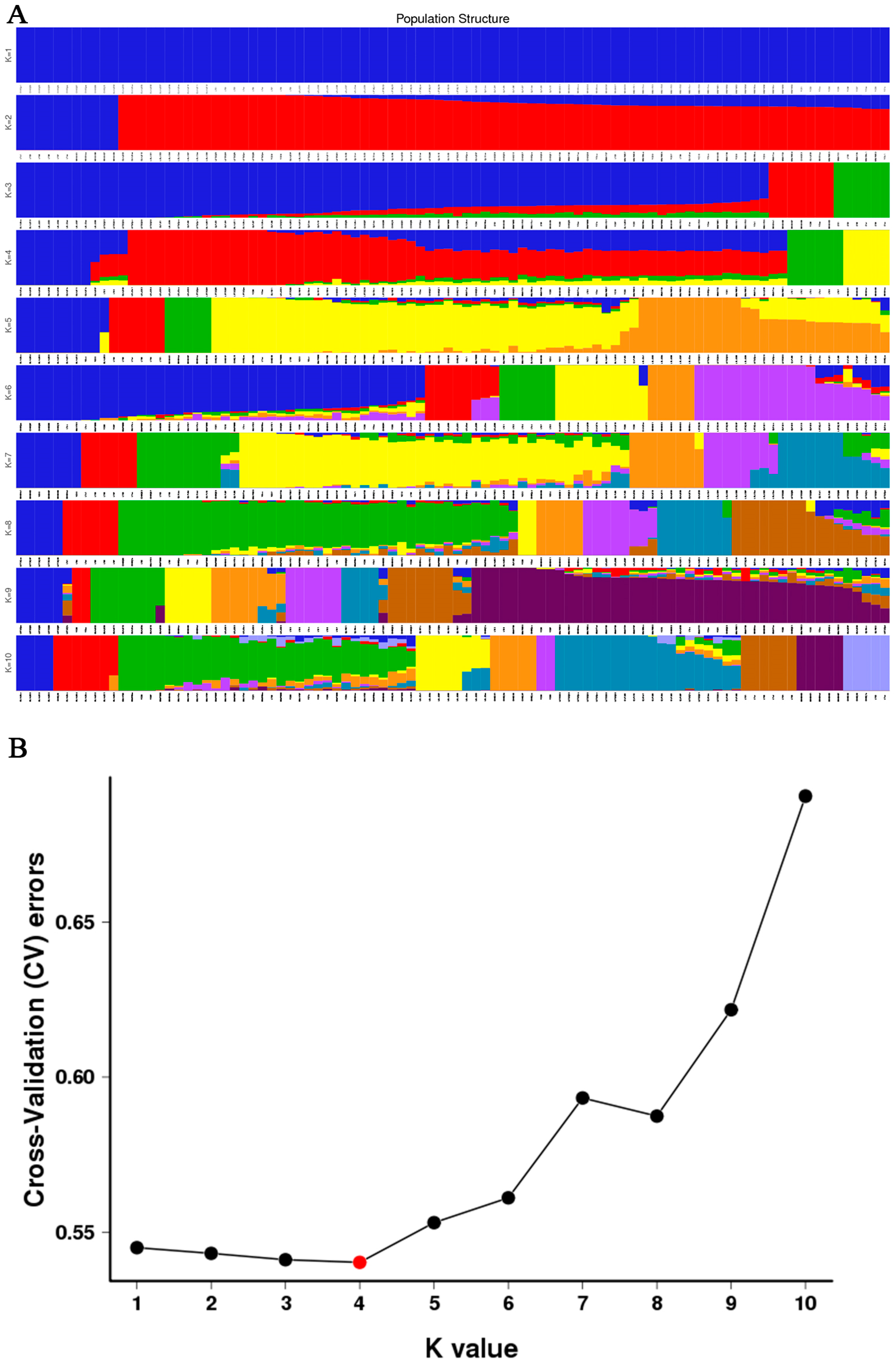
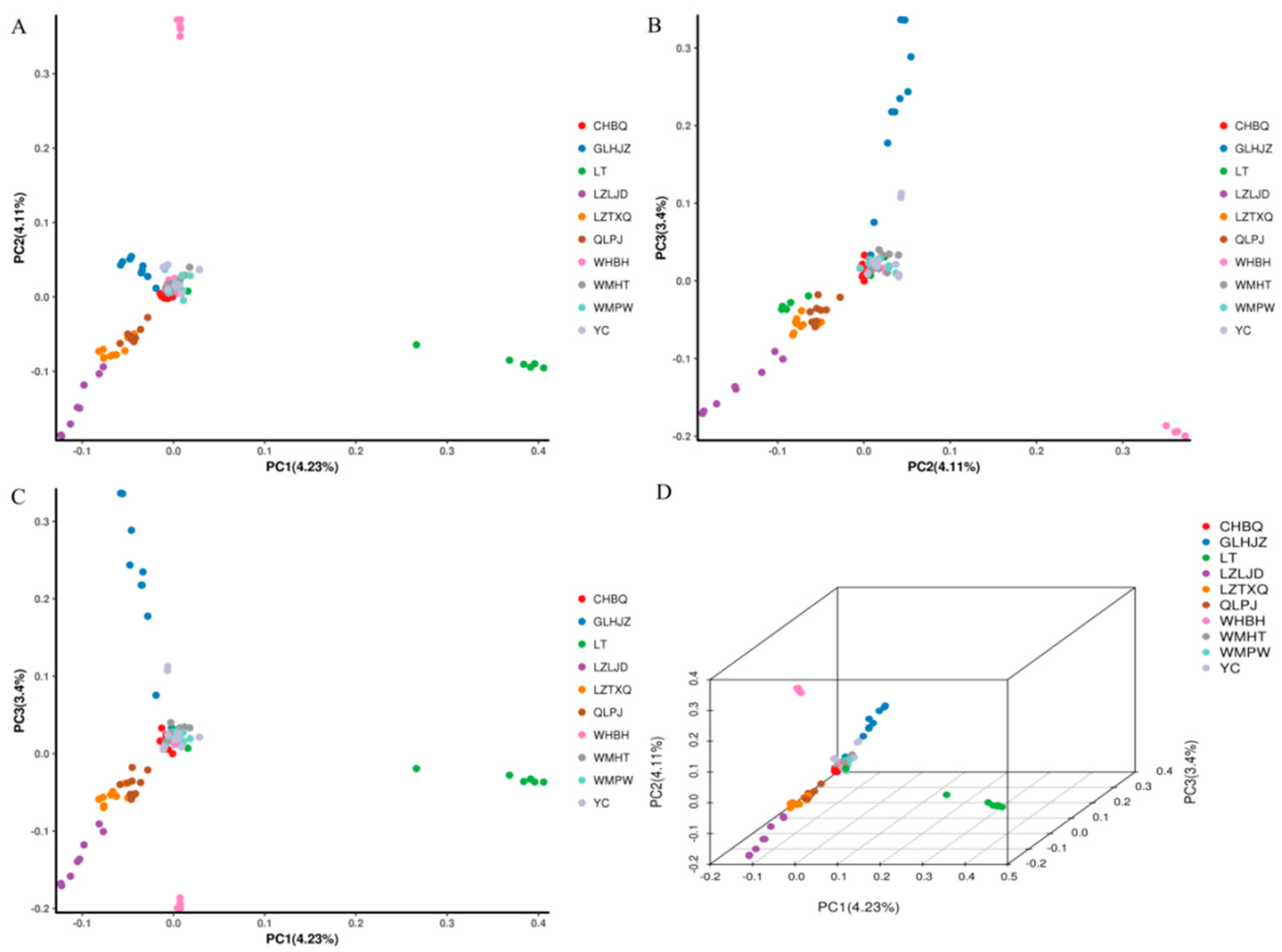
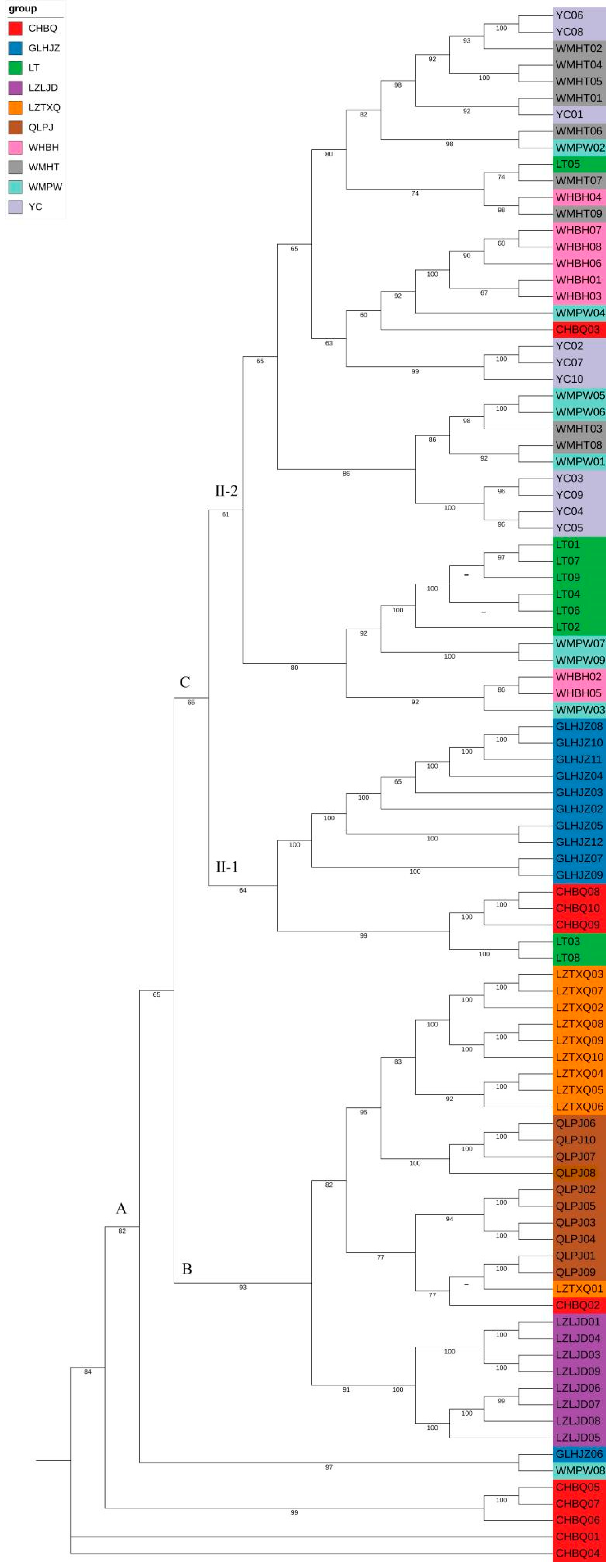
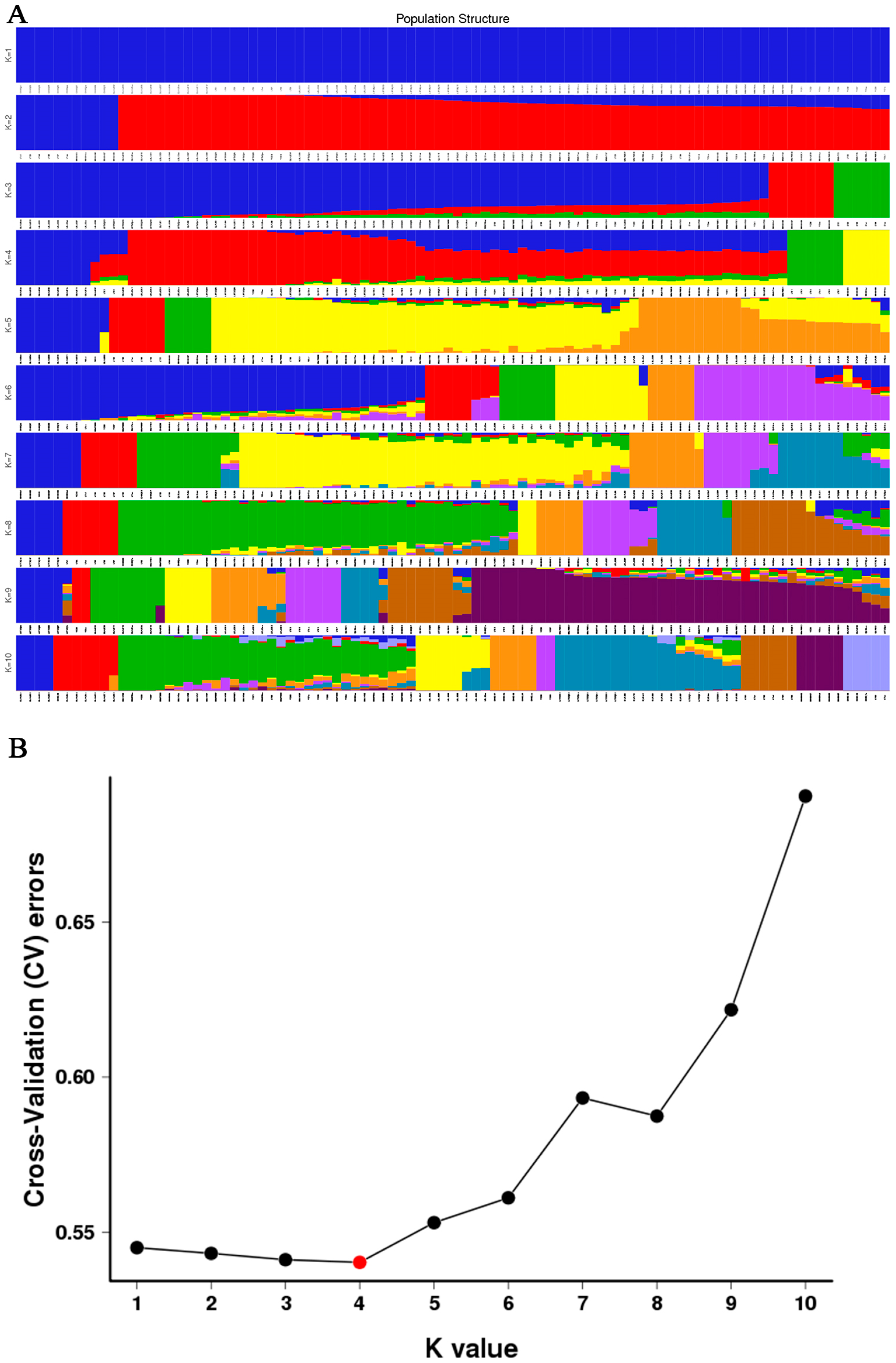
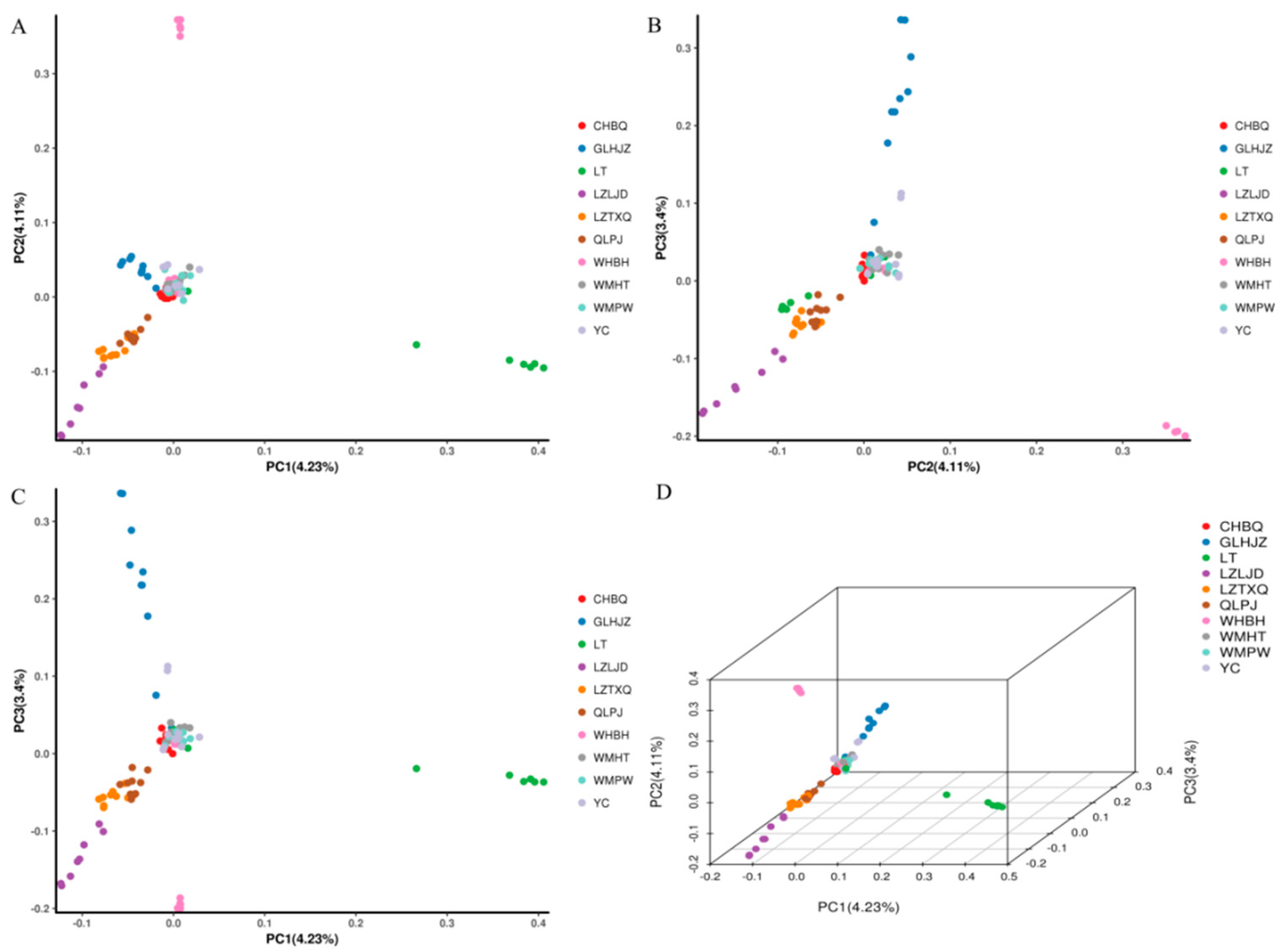
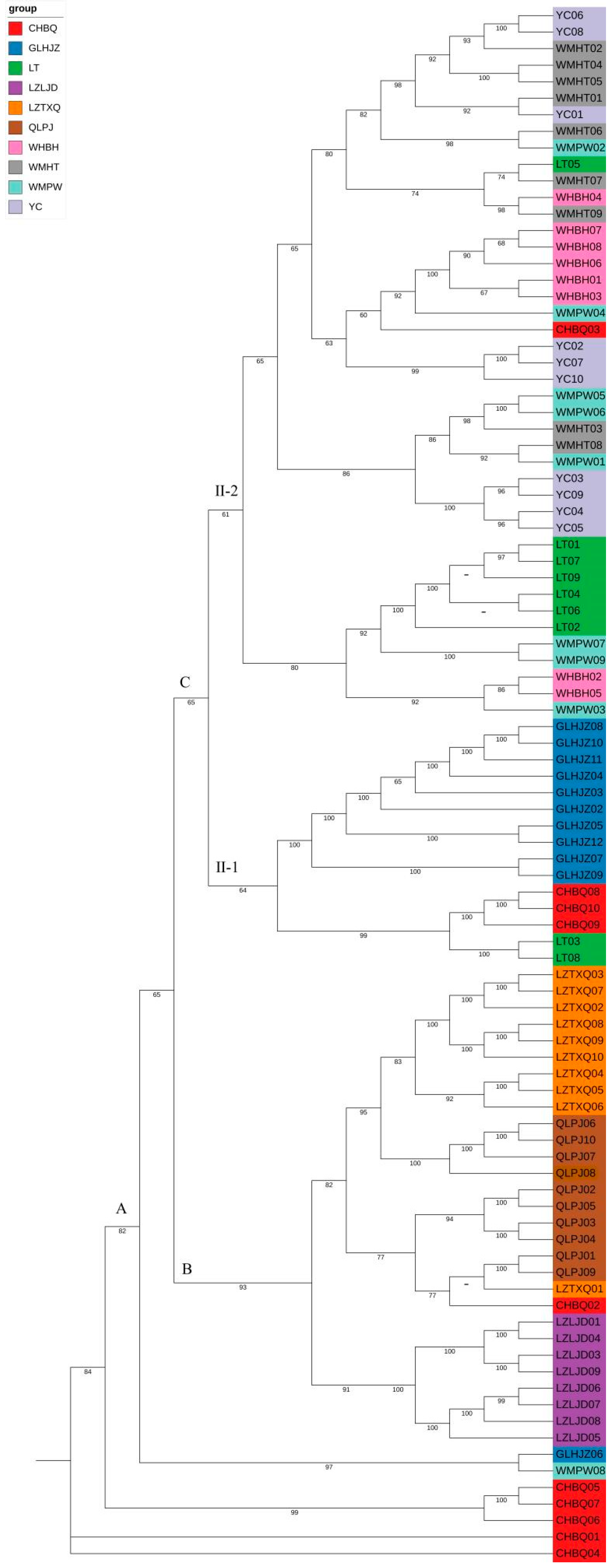
3.4. Phylogenetic Relationship and Population Structure
3.5. Association between SNP Markers and Environmental Variables
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, S.; Xie, Y. China Species Red List; Higher Education Press: Beijing, China, 2004; pp. 1–444. [Google Scholar]
- Chen, X.Q.; Liu, Z.J.; Zhu, G.H.; Lang, K.Y.; Ji, Z.H.; Luo, Y.B.; Jin, X.H.; Cribb, P.J.; Wood, J.J.; Gale, S.; et al. Flora of China; Wu, C.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China, 2009; pp. 1–506. [Google Scholar]
- Feng, C.L.; Cai, S.H.; Den, Z.H. Endangered causes and conservation strategies of Geodorum eulophioides in Yachang Forest region. For. Guangxi 2012, 9, 43–44. [Google Scholar]
- Zhang, Y.; Li, Z.; An, M.T.; Li, X.F.; Wang, W. Niche characteristics and interspecific relationship of the community of wild plant Geodorum eulophioides with extremely small population. J. Plant Resour. Environ. 2022, 31, 1–10. [Google Scholar]
- Wei, H.Y.; Li, X.F.; An, M.T.; Yang, Y.B.; Zhang, D.K. Analysis of Resource Status and Endangered Causes of the Extremely Endangered Plant Geodorum eulophioides in Guizhou Province. J. Mt. Agric. Biol. 2018, 37, 44–48. [Google Scholar]
- Luo, Y.T.; Luo, X.Y.; Lan, Y.T.; Liu, S.Y.; Xin, R.S.; Huang, L.; Wei, X.L. Study on tissue culture technology of Geodorum eulophioides Schltr. Chin. Hortic. Abstr. 2012, 28, 40–41+72. [Google Scholar]
- Lan, Y.T.; Wei, X.L.; Huang, L.; Luo, Y.T.; Xin, R.S.; Zhang, Z.B. Study on Technology of Aseptic Sowing and Proliferation of Protocorms in Wild Geodorum eulophioides. J. Anhui Agric. Sci. 2014, 42, 395–397, 418. [Google Scholar]
- Leffler, E.M.; Bullaughey, K.; Matute, D.R.; Meyer, W.K.; Ségurel, L.; Venkat, A.; Andolfatto, P.; Przeworski, M. Revisiting an old riddle: What determines genetic diversity levels within species? PLoS Biol. 2012, 10, e1001388. [Google Scholar] [CrossRef] [PubMed]
- Paetzold, C.; Wood, K.R.; Eaton, D.A.; Wagner, W.L.; Appelhans, M.S. Phylogeny of Hawaiian Melicope (Rutaceae): RAD-seq resolves species relationships and reveals ancient introgression. Front. Plant Sci. 2019, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.P.; Huang, L.; Xu, X.H.; Wang, C.Y.; Ren, M.; Wang, C.K.; Chen, M.L. Construction of a high-density SNP genetic map in flue-cured tobacco based on SLAF-seq. Mol. Breed. 2016, 36, 1–12. [Google Scholar] [CrossRef]
- Dang, Z.H.; Li, J.B.; Liu, Y.N.; Song, M.M.; Lockhart, P.J.; Tian, Y.Y.; Niu, M.M.; Wang, Q.L. RADseq-based population genomic analysis and environmental adaptation of rare and endangered recretohalophyte Reaumuria trigyna. Plant Genome, 2023; Early View. [Google Scholar] [CrossRef] [PubMed]
- Ana, O.; Pablo, V.; Mario, F.M.; Pedro, J.M.; Virginia, V.; Irene, V.M.; Andrew, L.H. A snapshot of progenitor–derivative speciation in Iberodes (Boraginaceae). Mol. Ecol. 2022, 31, 3192–3209. [Google Scholar]
- Sun, X.W.; Liu, D.Y.; Zhang, X.F.; Li, W.B.; Liu, H.; Hong, W.G.; Liu, H.; Hong, W.G.; Jiang, C.B.; Guan, N.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeqIllumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Xia, A.N.; Yang, A.A.; Meng, X.S.; Dong, G.Z.; Tang, X.J.; Lei, S.M.; Liu, Y.G. Development and application of rose (Rosa chinensis Jacq.) SNP markers based on SLAF-seq technology. Genet. Resour. Crop Evol. 2022, 69, 173–182. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Lee, T.H.; Guo, H.; Wang, X.Y.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.C.; Boyle, T. POPGENE version 1.31. In Microsoft Window-Based Freeware for Population Genetic Analysis; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Coop, G.; Witonsky, D.; Di, R.A.; Pritchard, J.K. Using environmental correlations to identify lociunderlying local adaptation. Genetics 2010, 185, 1411–1423. [Google Scholar] [CrossRef]
- Günther, T.; Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; François, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tian, L.; Zhang, J.; Huang, L.; Han, F.X.; Yan, S.R.; Wang, L.Z.; Zheng, H.K.; Sun, J.M. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genom. 2014, 15, 1086. [Google Scholar] [CrossRef]
- Luo, C.; Shu, B.; Yao, Q.; Wu, H.; Xu, W.; Wang, S. Construction of a High-Density Genetic Map Based on Large-Scale Marker Development in Mango Using Specific-Locus Amplified Fragment Sequencing (SLAF-seq). Front. Plant Sci. 2016, 7, 1310. [Google Scholar] [CrossRef]
- Ying, T. Studies on Conservation Genetic of Geodorum eulophiodes; Nanjing University: Nanjing, China, 2016; pp. 41–52. [Google Scholar]
- Zhou, H.J. Genetic Diversity and Population Structure of Natural Endangered Forest Tree Pinus bungeana in China; Northwest University: Xi’an, China, 2013; pp. 35–63. [Google Scholar]
- Tian, Q.; Liu, S.W.; Niu, S.H.; Li, W. Development of SNP molecular markers of Pinus bungeana based on SLAF-seq technology. J. Beijing For. Univ. 2021, 43, 1–8. [Google Scholar]
- He, X.D.; Zheng, J.W.; Jiao, Z.Y.; Dou, Q.Q.; Huang, L.B. Genetic diversity and structure analyses of Quercus shumardii populations based on SLAF-seq technology. J. Nanjing For. Univ. 2022, 46, 81–87. [Google Scholar]
- Lu, Y.Z. Ecological Adaptability and Endangered Mechanism of Paeonia ludlowii; Tibet University: Lhasa, China, 2021. [Google Scholar]
- Zhang, S.S.; Kang, H.M.; Yang, W.Z. Population Genetic Analysis of Nyssa yunnanensis by Reduced-representation Sequencing Technique. Bull. Bot. Res. 2019, 39, 899–907. [Google Scholar]
- Ma, Y.T. Variation and Genetic Diversity in Cultivation of Taxus cuspidate; Changchun University: Changchun, China, 2021. [Google Scholar]
- Li, M.Y. Population Genetics and Landscape Genomics of the Rare and Endangered Plant Tetrastigma hemsleyanum Based on SLAF-seq Technique; Henan Agricultural University: Zhengzhou, China, 2021. [Google Scholar]
- Boekema, E.J.; Ubbink-Kok, T.; Lolkema, J.S.; Brisson, A.; Konings, W.N. Visualization of a peripheral stalk in V-type ATPase: Evidence for the stator structure essential to rotational catalysis. Proc. Natl. Acad. Sci. USA 1997, 94, 14291–14293. [Google Scholar] [CrossRef]
- Xiao, H.J.; Zhang, Q.N.; Qin, X.J.; Xu, Y.H.; Ni, C.; Huang, J.; Zhu, L.; Zhong, F.; Liu, W.; Yao, G.; et al. Rice PPS1 encodes a DYW motif-containing pentatricopeptide repeat protein required for five consecutive RNA-editing sites of nad3 in mitochondria. New Phytol. 2018, 220, 878–892. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Location | Geographical Coordinates | Sample Size | Annual Average Temperature (°C) | Altitude (m) | Annual Rainfall (cm) |
|---|---|---|---|---|---|---|
| WHBH | Biaoxing Village, Wangmo County | 25°08′ N, 106°25′ E | 8 | 18.21 | 467 | 1198 |
| CHBQ | Banqi Village, Ceheng County | 24°81′ N, 105°65′ E | 10 | 17.81 | 508 | 1224 |
| GLHJZ | Hanjiazhai Village, Guangling County | 25°65′ N, 105°69′ E | 11 | 19.56 | 801 | 1296 |
| WMHT | Heting Village, Wangmo County | 25°11′ N, 106°44′ E | 9 | 17.33 | 517 | 1264 |
| LT | Loutuo Village, Loudian County | 25°23′ N, 106°67′ E | 9 | 17.84 | 411 | 1189 |
| QLPJ | PangjiangVillage, Qinglong County | 25°88′ N,105°38′ E | 10 | 19.96 | 558 | 1186 |
| WMPW | PingwangVillage, Wangmo County | 25°03′ N, 106°23′ E | 9 | 18.42 | 564 | 1206 |
| LZTXQ | Tianxingjiao Village, Liuzhi County | 26°04′ N, 105°25′ E | 10 | 17.39 | 704 | 1194 |
| YC | Yachang Village, Leye County | 25°08′ N, 109°95′ E | 10 | 18.23 | 434 | 1275 |
| LZZJD | Zhongjiaodi Village, Liuzhi County | 25°99′ N, 105°23′ E | 8 | 17.86 | 928 | 1253 |
| No. of Reads | GC Content (%) | Q30 (%) | No. of SLAF | No. of Depth | |
|---|---|---|---|---|---|
| Sum | 222,537,168 | 1,625,234 | |||
| Avg. | 2,367,417 | 35.55 | 91.94 | 146,035 | 13.29 |
| Population | Q30 (%) | GC Content (%) | No. of SLAFs | Sequencing Depth | No. of SNPs | Integrity of SNPs (%) | Heterozygosity of SNPs (%) |
|---|---|---|---|---|---|---|---|
| CHBQ | 91.84 | 35.39 | 113,817.22 | 11.69 | 515,665.52 | 31.60 | 7.06 |
| GLHJZ | 92.83 | 35.47 | 140,689.61 | 12.50 | 663,474.26 | 40.66 | 8.11 |
| LT | 91.69 | 35.09 | 146,362.45 | 12.56 | 668,646.08 | 40.98 | 9.52 |
| LZLJD | 92.44 | 35.33 | 157,542.14 | 12.90 | 792,674.11 | 48.59 | 10.54 |
| LZTXQ | 92.03 | 36.26 | 180,194.04 | 18.97 | 850,901.19 | 52.16 | 12.17 |
| QLPJ | 92.51 | 36.08 | 159,014.42 | 13.14 | 779,839.44 | 47.80 | 10.86 |
| WHBH | 91.09 | 35.18 | 143,828.25 | 10.72 | 705,106.05 | 43.22 | 9.83 |
| WMHT | 91.57 | 35.53 | 141,522.22 | 11.97 | 660,860.32 | 40.50 | 9.44 |
| WMPW | 91.18 | 35.03 | 133,174.34 | 14.08 | 659,608.61 | 40.43 | 9.10 |
| YC | 91.88 | 35.95 | 144,901.67 | 13.72 | 694,010.25 | 42.54 | 8.29 |
| Population | Na | Ne | He | H | Ho | PIC | I | FIS |
|---|---|---|---|---|---|---|---|---|
| LZTXQ | 1.8092 | 1.4469 | 0.2670 | 0.2817 | 0.3169 | 0.2156 | 0.4050 | 0.0365 |
| YC | 1.8342 | 1.4389 | 0.2655 | 0.2817 | 0.2464 | 0.2156 | 0.4059 | 0.2154 |
| QLPJ | 1.8179 | 1.4376 | 0.2635 | 0.2785 | 0.2910 | 0.2135 | 0.4018 | 0.2658 |
| WMHT | 1.8319 | 1.4328 | 0.2632 | 0.2810 | 0.2485 | 0.2141 | 0.4033 | 0.0525 |
| WMPW | 1.8172 | 1.4246 | 0.2584 | 0.2757 | 0.2411 | 0.2103 | 0.3960 | −0.1532 |
| CHBQ | 1.7569 | 1.4023 | 0.2436 | 0.2612 | 0.2074 | 0.1978 | 0.3720 | 0.1745 |
| GLHJZ | 1.7493 | 1.3976 | 0.2388 | 0.2517 | 0.2286 | 0.1934 | 0.3642 | 0.2463 |
| WHBH | 1.7135 | 1.4054 | 0.2385 | 0.2564 | 0.2678 | 0.1916 | 0.3601 | 0.1746 |
| LZLJD | 1.6796 | 1.4069 | 0.2383 | 0.2552 | 0.2936 | 0.1909 | 0.3571 | 0.1952 |
| LT | 1.6872 | 1.3997 | 0.2339 | 0.2499 | 0.2582 | 0.1874 | 0.3517 | −0.2376 |
| Average | 1.7697 | 1.4193 | 0.2511 | 0.2673 | 0.2600 | 0.2030 | 0.3817 | 0.0970 |
| LZTXQ | YC | QLPJ | WMHT | WMPW | CHBQ | GLHJZ | WHBH | LZLJD | |
|---|---|---|---|---|---|---|---|---|---|
| YC | 0.2651 | ||||||||
| QLPJ | 0.1526 | 0.2152 | |||||||
| WMHT | 0.2492 | 0.1123 | 0.0953 | ||||||
| WMPW | 0.3144 | 0.1482 | 0.2364 | 0.1462 | |||||
| CHBQ | 0.2652 | 0.1722 | 0.1114 | 0.1729 | 0.1867 | ||||
| GLHJZ | 0.1953 | 0.1654 | 0.1461 | 0.1683 | 0.1562 | 0.0526 | |||
| WHBH | 0.2155 | 0.1149 | 0.1927 | 0.0865 | 0.0778 | 0.1761 | 0.1712 | ||
| LZLJD | 0.2238 | 0.1426 | 0.2361 | 0.2975 | 0.1554 | 0.0665 | 0.0958 | 0.0861 | |
| LT | 0.1446 | 0.0652 | 0.0921 | 0.1342 | 0.0531 | 0.1462 | 0.0482 | 0.0755 | 0.0864 |
| Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation (%) |
|---|---|---|---|---|
| Among Populations | 8 | 42,856.51 | 185.2241 | 21.35 |
| Between Individuals | 263 | 324,657.2 | 682.1546 | 78.65 |
| total | 271 | 367,513.71 | 867.3787 | 100 |
| SNP ID | Position | log10 (BF) | qval | Altitude | Annual Rainfall | Annual Average Temperature |
|---|---|---|---|---|---|---|
| Marker14689 | 89 | 1.9869 | 0.0043 | * | ||
| Marker15991 | 111 | 0.9044 | 0.0399 | * | ||
| Marker21684 | 200 | 0.8587 | 0.0436 | * | ||
| Marker11786 | 29 | 2.3352 | 0.0020 | * | ||
| Marker45970 | 258 | 1.0176 | 0.0340 | * | ||
| Marker25449 | 234 | 1.9380 | 0.0047 | * | ||
| Marker27429 | 107 | 1.5769 | 0.0095 | * | ||
| Marker11418 | 115 | 1.4587 | 0.0107 | * | ||
| Marker20154 | 228 | 1.8169 | 0.0156 | * | ||
| Marker42084 | 161 | 1.1550 | 0.0236 | * | ||
| Marker22474 | 210 | 2.1394 | 0.0029 | * | ||
| Marker22886 | 255 | 1.8803 | 0.0055 | * | ||
| Marker35950 | 83 | 1.1650 | 0.0233 | * | ||
| Marker26182 | 176 | 1.9869 | 0.0043 | * | ||
| Marker85904 | 173 | 1.3667 | 0.0145 | * | ||
| Marker30072 | 28 | 1.1888 | 0.0225 | * | ||
| Marker17375 | 23 | 2.7439 | 0.0009 | * | ||
| Marker39371 | 109 | 3.6988 | 0.0001 | * | ||
| Marker36694 | 272 | 1.6793 | 0.0096 | * | ||
| Marker42949 | 188 | 1.6409 | 0.0075 | * | ||
| Marker18106 | 111 | 1.1037 | 0.0282 | * | ||
| Marker48494 | 114 | 1.2089 | 0.0216 | * | ||
| Marker26661 | 237 | 1.6614 | 0.0154 | * | ||
| Marker15596 | 53 | 1.1088 | 0.0274 | * | ||
| Marker20664 | 93 | 1.3559 | 0.0152 | * | ||
| Marker40285 | 189 | 1.6386 | 0.0083 | * | ||
| Marker25639 | 68 | 1.1872 | 0.0227 | * | ||
| Marker29116 | 250 | 1.1075 | 0.0277 | * | ||
| Marker60774 | 75 | 0.9688 | 0.0356 | * | ||
| Marker13404 | 212 | 2.9202 | 0.0005 | * |
| SNP ID | Position | Z Scores | log10 (BF) | p-Value | Altitude | Annual Rainfall | Annual Average Temperature |
|---|---|---|---|---|---|---|---|
| Marker28741 | 79 | 4.0527 | 4.0103 | 0.0004 | * | ||
| Marker20104 | 106 | 3.8584 | 3.6799 | 0.0002 | * | ||
| Marker30680 | 180 | 4.3837 | 4.5311 | 0.0001 | * | ||
| Marker21319 | 81 | 3.4618 | 3.0902 | 0.0008 | * | ||
| Marker17092 | 84 | 3.3938 | 2.9951 | 0.0011 | * | ||
| Marker27250 | 76 | −3.7637 | 3.5332 | 0.0003 | * | ||
| Marker21005 | 219 | 3.4004 | 3.0031 | 0.0012 | * | ||
| Marker15071 | 126 | 3.3627 | 2.9504 | 0.0011 | * | ||
| Marker13476 | 107 | 3.4414 | 3.0624 | 0.0009 | * | ||
| Marker22058 | 79 | 4.1152 | 4.0912 | 0.0001 | * | ||
| Marker39147 | 174 | −3.5212 | 3.1831 | 0.0008 | * | ||
| Marker17222 | 239 | 3.8479 | 3.6613 | 0.0002 | * | ||
| Marker91795 | 34 | −3.5982 | 3.2894 | 0.0005 | * | ||
| Marker60868 | 48 | 4.4826 | 4.6928 | 0.0001 | * | ||
| Marker17901 | 118 | 3.5719 | 3.2493 | 0.0005 | * | ||
| Marker32468 | 120 | 3.7926 | 3.5771 | 0.0002 | * | ||
| Marker12583 | 90 | 3.3224 | 2.8938 | 0.0013 | * | ||
| Marker15670 | 225 | 3.4942 | 3.1382 | 0.0007 | * |
| Variables | Marker ID | Position | Putative Genes | Origin |
|---|---|---|---|---|
| Altitude | Marker20104 Marker21684 | 106 200 | serine/threonine-protein kinase RNA-directed DNA polymerase | Brassica rapa Malus domestica |
| Annual rainfall | Marker39371 | 109 | ribonuclease H protein | Phoenix dactylifera |
| Annual average temperature | Marker60868 Marker15670 | 48 225 | pentatricopeptide repeat-containing protein | Nelumbo nucifera |
| F-box/LRR-repeat protein | Triticum urartu |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Ran, Z.; Zhang, Y.; Xiao, X.; An, M. Genetic Diversity and Structure of Geodorum eulophioides, a Plant Species with Extremely Small Populations in China. Diversity 2023, 15, 990. https://doi.org/10.3390/d15090990
Li Z, Ran Z, Zhang Y, Xiao X, An M. Genetic Diversity and Structure of Geodorum eulophioides, a Plant Species with Extremely Small Populations in China. Diversity. 2023; 15(9):990. https://doi.org/10.3390/d15090990
Chicago/Turabian StyleLi, Zhi, Zhaohui Ran, Yang Zhang, Xu Xiao, and Mingtai An. 2023. "Genetic Diversity and Structure of Geodorum eulophioides, a Plant Species with Extremely Small Populations in China" Diversity 15, no. 9: 990. https://doi.org/10.3390/d15090990
APA StyleLi, Z., Ran, Z., Zhang, Y., Xiao, X., & An, M. (2023). Genetic Diversity and Structure of Geodorum eulophioides, a Plant Species with Extremely Small Populations in China. Diversity, 15(9), 990. https://doi.org/10.3390/d15090990